Immune Checkpoint Inhibitors: Fundamental Mechanisms, Current Status and Future Directions

Abstract
:1. Introduction
2. Immune Checkpoint Pathways
3. CTLA-4 Physiological Role
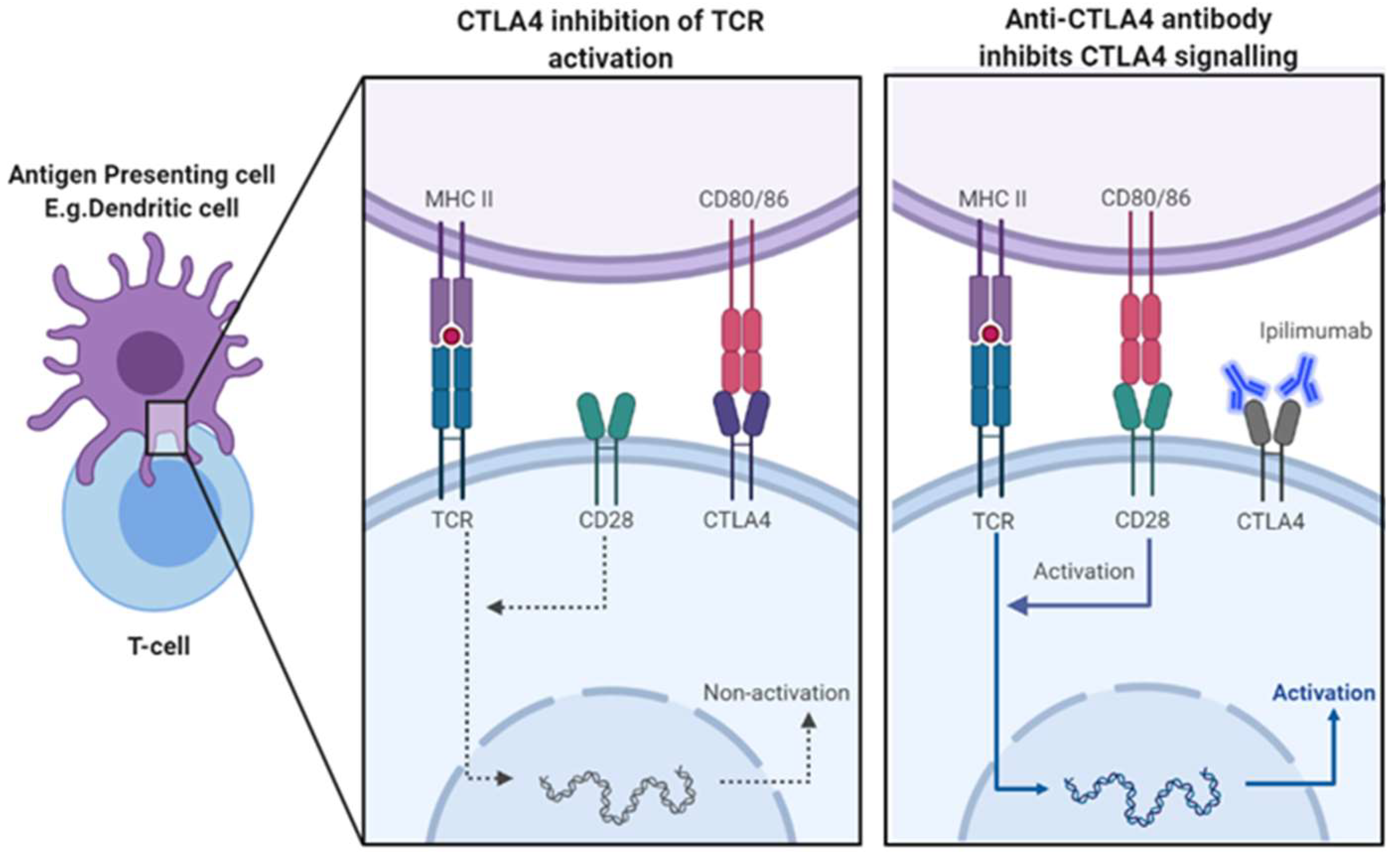
3.1. Mechanism of CTLA-4 Blockade-Induced Tumour Rejection
3.2. Efficacy of Ipilimumab in Treatment of Advanced Melanoma
4. PD-1/PD-L1 Physiological Role
4.1. Mechanism of PD-1/PD-L1 Blockade-Induced Tumour Rejection
4.2. Efficacy of Anti-PD-1/PD-L1 Agents
4.2.1. Pembrolizumab
4.2.2. Atezolizumab
5. LAG-3 Physiological Role
5.1. Mechanism of LAG-3 Blockade-Induced Tumour Rejection
5.2. Efficacy of Relatimab Plus Nivolumab in the Treatment of Advanced Melanoma
6. Adverse Effects of Immune Checkpoint Inhibitors
7. Predictive Biomarkers
8. Future Directions
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer Immunotherapy Comes of Age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Smith, M.R.; Campbell, K.S. Immunotherapy of Cancer. Eur. J. Pharmacol. 2009, 625, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, J. Current Status and Future Directions of Cancer Immunotherapy. J. Cancer 2018, 9, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Voena, C.; Chiarle, R. Advances in Cancer Immunology and Cancer Immunotherapy. Discov. Med. 2016, 21, 125–133. [Google Scholar] [PubMed]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune Checkpoint Inhibitors: Recent Progress and Potential Biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Kates, M.; Sopko, N.A.; Matsui, H.; Drake, C.G.; Hahn, N.M.; Bivalacqua, T.J. Immune Checkpoint Inhibitors: A New Frontier in Bladder Cancer. World J. Urol. 2016, 34, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Camacho, L.H. CTLA-4 Blockade with Ipilimumab: Biology, Safety, Efficacy, and Future Considerations. Cancer Med. 2015, 4, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Barbee, M.S.; Ogunniyi, A.; Horvat, T.Z.; Dang, T.O. Current Status and Future Directions of the Immune Checkpoint Inhibitors Ipilimumab, Pembrolizumab, and Nivolumab in Oncology. Ann. Pharmacother. 2015, 49, 907–937. [Google Scholar] [CrossRef]
- Paik, J. Nivolumab Plus Relatlimab: First Approval. Drugs 2022, 82, 925–931. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; Van Der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in Patients with Locally Advanced and Metastatic Urothelial Carcinoma Who Have Progressed Following Treatment with Platinum-Based Chemotherapy: A Single-Arm, Multicentre, Phase 2 Trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of Cancer Immunity and the Cancer-Immune Set Point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The Determinants of Tumour Immunogenicity. Nat. Rev. Cancer 2012, 12, 307–313. [Google Scholar] [CrossRef]
- Fancello, L.; Gandini, S.; Pelicci, P.G.; Mazzarella, L. Tumor Mutational Burden Quantification from Targeted Gene Panels: Major Advancements and Challenges. J. Immunother. Cancer 2019, 7, 183. [Google Scholar] [CrossRef] [PubMed]
- Kourie, H.R.; Klastersky, J. Immune Checkpoint Inhibitors Side Effects and Management. Immunotherapy 2016, 8, 799–807. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, R.E.; Cao, X. Co-Stimulatory and Co-Inhibitory Pathways in Cancer Immunotherapy. Adv. Cancer Res. 2019, 143, 145–194. [Google Scholar] [CrossRef]
- Nirschl, C.J.; Drake, C.G. Molecular Pathways: Coexpression of Immune Checkpoint Molecules: Signaling Pathways and Implications for Cancer Immunotherapy. Clin. Cancer Res. 2013, 19, 4917–4924. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed]
- Juneja, V.R.; McGuire, K.A.; Manguso, R.T.; LaFleur, M.W.; Collins, N.; Nicholas Haining, W.; Freeman, G.J.; Sharpe, A.H. PD-L1 on Tumor Cells Is Sufficient for Immune Evasion in Immunogenic Tumors and Inhibits CD8 T Cell Cytotoxicity. J. Exp. Med. 2017, 214, 895–904. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel Immune Checkpoint Targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. Cancer Clin. Trials 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Sansom, D.M. CD28, CTLA-4 and Their Ligands: Who Does What and to Whom? Immunology 2000, 101, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic Self-Tolerance Maintained by Cd25+Cd4+Regulatory T Cells Constitutively Expressing Cytotoxic T Lymphocyte–Associated Antigen 4. J. Exp. Med. 2000, 192, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Fields, P.E.; Gajewski, T.F. B7-1 Engagement of Cytotoxic T Lymphocyte Antigen 4 Inhibits T Cell Activation in the Absence of CD28. J. Exp. Med. 1998, 188, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative Disorders with Early Lethality in Mice Deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Camacho, L.H.; Antonia, S.; Sosman, J.; Kirkwood, J.M.; Gajewski, T.F.; Redman, B.; Pavlov, D.; Bulanhagui, C.; Bozon, V.A.; Gomez-Navarro, J.; et al. Phase I/II Trial of Tremelimumab in Patients with Metastatic Melanoma. J. Clin. Oncol. 2009, 27, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Kefford, R.; Marshall, M.A.; Punt, C.J.A.; Haanen, J.B.; Marmol, M.; Garbe, C.; Gogas, H.; Schachter, J.; Linette, G.; et al. Phase III Randomized Clinical Trial Comparing Tremelimumab with Standard-of-Care Chemotherapy in Patients with Advanced Melanoma. J. Clin. Oncol. 2013, 31, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A. Combination of CTLA-4 and PD-1 Blockers for Treatment of Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in Patients with Metastatic DNA Mismatch Repair-Deficient or Microsatellite Instability-High Colorectal Cancer (CheckMate 142): An Open-Label, Multicentre, Phase 2 Study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.-W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-Line Nivolumab plus Ipilimumab in Unresectable Malignant Pleural Mesothelioma (CheckMate 743): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef]
- Chau, I.; Doki, Y.; Ajani, J.A.; Xu, J.; Wyrwicz, L.; Motoyama, S.; Ogata, T.; Kawakami, H.; Hsu, C.-H.; Adenis, A.; et al. Nivolumab (NIVO) plus Ipilimumab (IPI) or NIVO plus Chemotherapy (Chemo) versus Chemo as First-Line (1L) Treatment for Advanced Esophageal Squamous Cell Carcinoma (ESCC): First Results of the CheckMate 648 Study. J. Clin. Oncol. 2021, 39, LBA4001. [Google Scholar] [CrossRef]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133.e17. [Google Scholar] [CrossRef]
- Wherry, E.J. T Cell Exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-Dependent Depletion of Tumor-Infiltrating Regulatory t Cells Co-Defines the Efficacy of Anti-CTLA-4 Therapy against Melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Selby, M.J.; Engelhardt, J.J.; Quigley, M.; Henning, K.A.; Chen, T.; Srinivasan, M.; Korman, A.J. Anti-CTLA-4 Antibodies of IgG2a Isotype Enhance Antitumor Activity through Reduction of Intratumoral Regulatory T Cells. Cancer Immunol. Res. 2013, 1, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Bulliard, Y.; Jolicoeur, R.; Windman, M.; Rue, S.M.; Ettenberg, S.; Knee, D.A.; Wilson, N.S.; Dranoff, G.; Brogdon, J.L. Activating Fc γ Receptors Contribute to the Antitumor Activities of Immunoregulatory Receptor-Targeting Antibodies. J. Exp. Med. 2013, 210, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Tang, F.; Liu, M.; Su, J.; Zhang, Y.; Wu, W.; Devenport, M.; Lazarski, C.A.; Zhang, P.; Wang, X.; et al. A Reappraisal of CTLA-4 Checkpoint Blockade in Cancer Immunotherapy. Cell Res. 2018, 28, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on Both Effector and Regulatory T Cell Compartments Contributes to the Antitumor Activity of Anti-CTLA-4 Antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Ramagopal, U.A.; Liu, W.; Garrett-Thomson, S.C.; Bonanno, J.B.; Yan, Q.; Srinivasan, M.; Wong, S.C.; Bell, A.; Mankikar, S.; Rangan, V.S.; et al. Structural Basis for Cancer Immunotherapy by the First-in-Class Checkpoint Inhibitor Ipilimumab. Proc. Natl. Acad. Sci. USA 2017, 114, E4223–E4232. [Google Scholar] [CrossRef] [PubMed]
- Altekruse, S.F.; Kosary, C.L.; Krapcho, M.; Neyman, N.; Aminou, R.; Waldron, W.; Ruhl, J.; Howlader, N.; Tatalovich, Z.; Cho, H.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2007; Based on November 2009 SEER Data Submission, Posted to the SEER Web Site; National Cancer Institute: Bethesda, MD, USA, 2010. [Google Scholar]
- Tarhini, A.; Lo, E.; Minor, D.R. Releasing the Brake on the Immune System: Ipilimumab in Melanoma and Other Tumors. Cancer Biother. Radiopharm 2010, 25, 601–613. [Google Scholar] [CrossRef]
- Peggs, K.S.; Quezada, S.A.; Korman, A.J.; Allison, J.P. Principles and Use of Anti-CTLA4 Antibody in Human Cancer Immunotherapy. Curr. Opin. Immunol. 2006, 18, 206–213. [Google Scholar] [CrossRef]
- Hersh, E.M.; O’Day, S.J.; Powderly, J.; Khan, K.D.; Pavlick, A.C.; Cranmer, L.D.; Samlowski, W.E.; Nichol, G.M.; Yellin, M.J.; Weber, J.S. A Phase II Multicenter Study of Ipilimumab with or without Dacarbazine in Chemotherapy-Naïve Patients with Advanced Melanoma. Investig. New Drugs 2011, 29, 489–498. [Google Scholar] [CrossRef]
- Hersh, E.; Weber, J.; John, P., II; Pavlik, A.; Nichol, G.; Yellin, M.; Cranmer, L.; Urba, W.; O’Day, S. Long-term survival of patients (pts) with advanced melanoma treated with ipilimumab with or without dacarbazine. J. Clin. Oncol. 2009, 27 (Suppl. S15), 9038. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Neyns, B.; Linette, G.; Negrier, S.; Lutzky, J.; Thomas, L.; Waterfield, W.; Schadendorf, D.; Smylie, M.; Guthrie, T.; et al. Ipilimumab Monotherapy in Patients with Pretreated Advanced Melanoma: A Randomised, Double-Blind, Multicentre, Phase 2, Dose-Ranging Study. Lancet Oncol. 2010, 11, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Hodi, F.S.; Weber, J.S.; Allison, J.P.; Urba, W.J.; Robert, C.; O’Day, S.J.; Hoos, A.; Humphrey, R.; Berman, D.M.; et al. Development of Ipilimumab: A Novel Immunotherapeutic Approach for the Treatment of Advanced Melanoma. Ann. N. Y. Acad. Sci. 2013, 1291, 1–13. [Google Scholar] [CrossRef]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.-T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef]
- Dong, Y.; Sun, Q.; Zhang, X. PD-1 and Its Ligands Are Important Immune Checkpoints in Cancer. Oncotarget 2017, 8, 2171–2186. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed Cell Death 1 Forms Negative Costimulatory Microclusters That Directly Inhibit T Cell Receptor Signaling by Recruiting Phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef]
- Riella, L.V.; Paterson, A.M.; Sharpe, A.H.; Chandraker, A. Role of the PD-1 Pathway in the Immune Response. Am. J. Transplant. 2012, 12, 2575–2587. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of Lupus-like Autoimmune Diseases by Disruption of the PD-1 Gene Encoding an ITIM Motif-Carrying Immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Xiang, X.; Yu, P.C.; Long, D.; Liao, X.L.; Zhang, S.; You, X.M.; Zhong, J.H.; Li, L.Q. Prognostic Value of PD -L1 Expression in Patients with Primary Solid Tumors. Oncotarget 2018, 9, 5058–5072. [Google Scholar] [CrossRef]
- PD-1/PD-L1 Landscape Analysis—Cancer Research Institute (CRI). Available online: https://www.cancerresearch.org/scientists/immuno-oncology-landscape/pd-1-pd-l1-landscape (accessed on 23 March 2020).
- Loke, P.; Allison, J.P. PD-L1 and PD-L2 Are Differentially Regulated by Th1 and Th2 Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 5336–5341. [Google Scholar] [CrossRef]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J.; Sharpe, H. PD-L1 Interacts Specifically with B7-1 to Inhibit T Cell Proliferation. Immunity 2009, 27, 111–122. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-Year Survival Outcomes for Patients with Advanced Melanoma Treated with Pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef]
- Herbst, R.S.; Garon, E.B.; Kim, D.-W.; Cho, B.C.; Perez-Gracia, J.L.; Han, J.-Y.; Arvis, C.D.; Majem, M.; Forster, M.D.; Monnet, I.; et al. Long-Term Outcomes and Retreatment Among Patients With Previously Treated, Programmed Death-Ligand 1–Positive, Advanced Non–Small-Cell Lung Cancer in the KEYNOTE-010 Study. J. Clin. Oncol. 2020, 38, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.Y.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and Clinical Activity of Pembrolizumab for Treatment of Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (KEYNOTE-012): An Open-Label, Multicentre, Phase 1b Trial. Lancet Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef]
- Chen, R.; Zinzani, P.L.; Fanale, M.A.; Armand, P.; Johnson, N.A.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 2125–2132. [Google Scholar] [CrossRef] [PubMed]
- Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Vuky, J.; Powles, T.; Plimack, E.R.; Hahn, N.M.; de Wit, R.; Pang, L.; et al. First-Line Pembrolizumab in Cisplatin-Ineligible Patients with Locally Advanced and Unresectable or Metastatic Urothelial Cancer (KEYNOTE-052): A Multicentre, Single-Arm, Phase 2 Study. Lancet Oncol. 2017, 18, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.; Marabelle, A.; Kim, T.W.; Geva, R.; Cutsem, E.V.; André, T.; Ascierto, P.A.; Maio, M.; Delord, J.-P.; Gottfried, M.; et al. Efficacy of Pembrolizumab in Phase 2 KEYNOTE-164 and KEYNOTE-158 Studies of Microsatellite Instability High Cancers. Ann. Oncol. 2017, 28, v128–v129. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Kang, Y.-K.; Catenacci, D.V.; Muro, K.; Fuchs, C.S.; Geva, R.; Hara, H.; Golan, T.; Garrido, M.; Jalal, S.I.; et al. Pembrolizumab Alone or in Combination with Chemotherapy as First-Line Therapy for Patients with Advanced Gastric or Gastroesophageal Junction Adenocarcinoma: Results from the Phase II Nonrandomized KEYNOTE-059 Study. Gastric Cancer 2019, 22, 828–837. [Google Scholar] [CrossRef]
- Chung, H.C.; Ros, W.; Delord, J.-P.; Perets, R.; Italiano, A.; Shapira-Frommer, R.; Manzuk, L.; Piha-Paul, S.A.; Xu, L.; Zeigenfuss, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Cervical Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2019, 37, 1470–1478. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib (KEYNOTE-224): A Non-Randomised, Open-Label Phase 2 Trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Nghiem, P.; Bhatia, S.; Lipson, E.J.; Sharfman, W.H.; Kudchadkar, R.R.; Brohl, A.S.; Friedlander, P.A.; Daud, A.; Kluger, H.M.; Reddy, S.A.; et al. Durable Tumor Regression and Overall Survival in Patients With Advanced Merkel Cell Carcinoma Receiving Pembrolizumab as First-Line Therapy. J. Clin. Oncol. 2019, 37, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Shah, M.A.; Muro, K.; Francois, E.; Adenis, A.; Hsu, C.-H.; Doi, T.; Moriwaki, T.; Kim, S.-B.; Lee, S.-H.; et al. Randomized Phase III KEYNOTE-181 Study of Pembrolizumab Versus Chemotherapy in Advanced Esophageal Cancer. J. Clin. Oncol. 2020, 38, 4138–4148. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. KEYNOTE-355: Randomized, Double-Blind, Phase III Study of Pembrolizumab + Chemotherapy versus Placebo + Chemotherapy for Previously Untreated Locally Recurrent Inoperable or Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2020, 38, 1000. [Google Scholar] [CrossRef]
- Grob, J.-J.; Gonzalez, R.; Basset-Seguin, N.; Vornicova, O.; Schachter, J.; Joshi, A.; Meyer, N.; Grange, F.; Piulats, J.M.; Bauman, J.R.; et al. Pembrolizumab Monotherapy for Recurrent or Metastatic Cutaneous Squamous Cell Carcinoma: A Single-Arm Phase II Trial (KEYNOTE-629). J. Clin. Oncol. 2020, 38, 2916–2925. [Google Scholar] [CrossRef]
- O’Malley, D.M.; Bariani, G.M.; Cassier, P.A.; Marabelle, A.; Hansen, A.R.; De Jesus Acosta, A.; Miller, W.H.; Safra, T.; Italiano, A.; Mileshkin, L.; et al. Pembrolizumab in Patients With Microsatellite Instability–High Advanced Endometrial Cancer: Results From the KEYNOTE-158 Study. J. Clin. Oncol. 2022, 40, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H.; Lao, C.D.; et al. Nivolumab versus Chemotherapy in Patients with Advanced Melanoma Who Progressed after Anti-CTLA-4 Treatment (CheckMate 037): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Younes, A.; Santoro, A.; Zinzani, P.L.; Timmerman, J.; Ansell, S.M.; Armand, P.; Fanale, M.A.; Ratanatharathorn, V.; Kuruvilla, J.; Cohen, J.B.; et al. Checkmate 205: Nivolumab (Nivo) in Classical Hodgkin Lymphoma (cHL) after Autologous Stem Cell Transplant (ASCT) and Brentuximab Vedotin (BV)—A Phase 2 Study. J. Clin. Oncol. 2016, 34, 7535. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Retz, M.; Siefker-Radtke, A.; Baron, A.; Necchi, A.; Bedke, J.; Plimack, E.R.; Vaena, D.; Grimm, M.-O.; Bracarda, S.; et al. Nivolumab in Metastatic Urothelial Carcinoma after Platinum Therapy (CheckMate 275): A Multicentre, Single-Arm, Phase 2 Trial. Lancet Oncol. 2017, 18, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Ready, N.; Farago, A.F.; De Braud, F.; Atmaca, A.; Hellmann, M.D.; Schneider, J.G.; Spigel, D.R.; Moreno, V.; Chau, I.; Hann, C.L.; et al. Third-Line Nivolumab Monotherapy in Recurrent SCLC: CheckMate 032. J. Thorac. Oncol. 2019, 14, 237–244. [Google Scholar] [CrossRef]
- Cho, B.C.; Kato, K.; Takahashi, M.; Okada, M.; Lin, C.-Y.; Chin, K.; Kadowaki, S.; Ahn, M.-J.; Hamamoto, Y.; Doki, Y.; et al. Nivolumab versus Chemotherapy in Advanced Esophageal Squamous Cell Carcinoma (ESCC): The Phase III ATTRACTION-3 Study. Ann. Oncol. 2019, 30, v873–v874. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Stratigos, A.J.; Sekulic, A.; Peris, K.; Bechter, O.; Prey, S.; Kaatz, M.; Lewis, K.D.; Basset-Seguin, N.; Chang, A.L.S.; Dalle, S.; et al. Cemiplimab in Locally Advanced Basal Cell Carcinoma after Hedgehog Inhibitor Therapy: An Open-Label, Multi-Centre, Single-Arm, Phase 2 Trial. Lancet Oncol. 2021, 22, 848–857. [Google Scholar] [CrossRef]
- Sezer, A.; Kilickap, S.; Gümüş, M.; Bondarenko, I.; Özgüroğlu, M.; Gogishvili, M.; Turk, H.M.; Cicin, I.; Bentsion, D.; Gladkov, O.; et al. Cemiplimab Monotherapy for First-Line Treatment of Advanced Non-Small-Cell Lung Cancer with PD-L1 of at Least 50%: A Multicentre, Open-Label, Global, Phase 3, Randomised, Controlled Trial. Lancet 2021, 397, 592–604. [Google Scholar] [CrossRef]
- Khoja, L.; Butler, M.O.; Kang, S.P.; Ebbinghaus, S.; Joshua, A.M. Pembrolizumab. J. Immunother. Cancer 2015, 3, 36. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R.; Weber, J.S.; Joshua, A.M.; Hwu, W.J.; Gangadhar, T.C.; et al. Anti-Programmed-Death-Receptor-1 Treatment with Pembrolizumab in Ipilimumab-Refractory Advanced Melanoma: A Randomised Dose-Comparison Cohort of a Phase 1 Trial. Lancet 2014, 384, 1109–1117. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma (KEYNOTE-006): Post-Hoc 5-Year Results from an Open-Label, Multicentre, Randomised, Controlled, Phase 3 Study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Hellmann, M.D.; Rizvi, N.A.; Carcereny, E.; Leighl, N.B.; Ahn, M.J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Five-Year Overall Survival for Patients with Advanced Non-Small-Cell Lung Cancer Treated with Pembrolizumab: Results from the Phase i KEYNOTE-001 Study. J. Clin. Oncol. 2019, 37, 2518–2527. [Google Scholar] [CrossRef] [PubMed]
- Atezolizumab for Urothelial Carcinoma|FDA. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/atezolizumab-urothelial-carcinoma (accessed on 9 April 2020).
- Hoffman-Censits, J.H.; Grivas, P.; Van Der Heijden, M.S.; Dreicer, R.; Loriot, Y.; Retz, M.; Vogelzang, N.J.; Perez-Gracia, J.L.; Rezazadeh, A.; Bracarda, S.; et al. IMvigor 210, a Phase II Trial of Atezolizumab (MPDL3280A) in Platinum-Treated Locally Advanced or Metastatic Urothelial Carcinoma (mUC). J. Clin. Oncol. 2016, 34, 355. [Google Scholar] [CrossRef]
- Balar, A.V.; Galsky, M.D.; Rosenberg, J.E.; Powles, T.; Petrylak, D.P.; Bellmunt, J.; Loriot, Y.; Necchi, A.; Hoffman-Censits, J.; Perez-Gracia, J.L.; et al. Atezolizumab as First-Line Treatment in Cisplatin-Ineligible Patients with Locally Advanced and Metastatic Urothelial Carcinoma: A Single-Arm, Multicentre, Phase 2 Trial. Lancet 2017, 389, 67–76. [Google Scholar] [CrossRef]
- Atezolizumab (TECENTRIQ)|FDA. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/atezolizumab-tecentriq (accessed on 9 April 2020).
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus Docetaxel for Patients with Previously Treated Non-Small-Cell Lung Cancer (POPLAR): A Multicentre, Open-Label, Phase 2 Randomised Controlled Trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus Docetaxel in Patients with Previously Treated Non-Small-Cell Lung Cancer (OAK): A Phase 3, Open-Label, Multicentre Randomised Controlled Trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Hematology/Oncology (Cancer) Approvals & Safety Notifications|FDA. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/hematologyoncology-cancer-approvals-safety-notifications (accessed on 9 April 2020).
- FDA Approves Atezolizumab for PD-L1 Positive Unresectable Locally Advanced or Metastatic Triple-Negative Breast Cancer|FDA. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-atezolizumab-pd-l1-positive-unresectable-locally-advanced-or-metastatic-triple-negative (accessed on 9 April 2020).
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Clinical Trials Using Atezolizumab—National Cancer Institute. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/atezolizumab (accessed on 9 April 2020).
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, Vemurafenib, and Cobimetinib as First-Line Treatment for Unresectable Advanced BRAFV600 Mutation-Positive Melanoma (IMspire150): Primary Analysis of the Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Chen, A.P.; Sharon, E.; O’sullivan-Coyne, G.; Moore, N.; Foster, J.C.; Hu, J.S.; Van Tine, B.A.; Conley, A.P.; Read, W.L.; Riedel, R.F.; et al. Atezolizumab for Advanced Alveolar Soft Part Sarcoma. N. Engl. J. Med. 2023, 389, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Russell, J.S.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Milella, M.; Brownell, I.; et al. Updated Efficacy of Avelumab in Patients with Previously Treated Metastatic Merkel Cell Carcinoma after ≥1 Year of Follow-up: JAVELIN Merkel 200, a Phase 2 Clinical Trial. J. Immunother. Cancer 2018, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Ellerton, J.; Infante, J.R.; Agrawal, M.; Gordon, M.; Aljumaily, R.; Britten, C.D.; Dirix, L.; Lee, K.-W.; Taylor, M.; et al. Avelumab in Metastatic Urothelial Carcinoma after Platinum Failure (JAVELIN Solid Tumor): Pooled Results from Two Expansion Cohorts of an Open-Label, Phase 1 Trial. Lancet Oncol. 2018, 19, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Park, S.H.; Voog, E.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Kalofonos, H.; Radulović, S.; Demey, W.; Ullén, A.; et al. Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2020, 383, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; O’Donnell, P.H.; Massard, C.; Arkenau, H.-T.; Friedlander, T.W.; Hoimes, C.J.; Lee, J.L.; Ong, M.; Sridhar, S.S.; Vogelzang, N.J.; et al. Efficacy and Safety of Durvalumab in Locally Advanced or Metastatic Urothelial Carcinoma: Updated Results From a Phase 1/2 Open-Label Study. JAMA Oncol. 2017, 3, e172411. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; De Wit, M.; et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab plus Platinum–Etoposide versus Platinum–Etoposide in First-Line Treatment of Extensive-Stage Small-Cell Lung Cancer (CASPIAN): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.-Y.; Ruth He, A.; Qin, S.; Chen, L.-T.; Okusaka, T.; Vogel, A.; Kim, J.W.; Suksombooncharoen, T.; Ah Lee, M.; Kitano, M.; et al. Durvalumab plus Gemcitabine and Cisplatin in Advanced Biliary Tract Cancer. NEJM Evid. 2022, 1, EVIDoa2200015. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chan, S.L.; Kudo, M.; Lau, G.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Dao, T.V.; De Toni, E.N.; et al. Phase 3 Randomized, Open-Label, Multicenter Study of Tremelimumab (T) and Durvalumab (D) as First-Line Therapy in Patients (Pts) with Unresectable Hepatocellular Carcinoma (uHCC): HIMALAYA. J. Clin. Oncol. 2022, 40, 379. [Google Scholar] [CrossRef]
- MacLachlan, B.J.; Mason, G.H.; Greenshields-Watson, A.; Triebel, F.; Gallimore, A.; Cole, D.K.; Godkin, A. Molecular Characterization of HLA Class II Binding to the LAG-3 T Cell Co-Inhibitory Receptor. Eur. J. Immunol. 2021, 51, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Huard, B.; Mastrangeli, R.; Prigent, P.; Bruniquel, D.; Donini, S.; El-Tayar, N.; Maigret, B.; Dréano, M.; Triebel, F. Characterization of the Major Histocompatibility Complex Class II Binding Site on LAG-3 Protein. Proc. Natl. Acad. Sci. USA 1997, 94, 5744–5749. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e12. [Google Scholar] [CrossRef] [PubMed]
- Kouo, T.; Huang, L.; Pucsek, A.B.; Cao, M.; Solt, S.; Armstrong, T.; Jaffee, E. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol. Res. 2015, 3, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, S.A.; Rehman, A.U.; Gabr, M.T. Discovery of First-in-Class Small Molecule Inhibitors of Lymphocyte Activation Gene 3 (LAG-3). ACS Med. Chem. Lett. 2023, 14, 629–635. [Google Scholar] [CrossRef]
- Grosso, J.F.; Kelleher, C.C.; Harris, T.J.; Maris, C.H.; Hipkiss, E.L.; Marzo, A.D.; Anders, R.; Netto, G.; Getnet, D.; Bruno, T.C.; et al. LAG-3 Regulates CD8+ T Cell Accumulation and Effector Function in Murine Self- and Tumor-Tolerance Systems. J. Clin. Investig. 2007, 117, 3383–3392. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhang, A.; Qiu, C.; Wang, W.; Yang, Y.; Qiu, C.; Liu, A.; Zhu, L.; Yuan, S.; Hu, H.; et al. The Upregulation of LAG-3 on T Cells Defines a Subpopulation with Functional Exhaustion and Correlates with Disease Progression in HIV-Infected Subjects. J. Immunol. 2015, 194, 3873–3882. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and Cellular Insights into T Cell Exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Maruhashi, T.; Okazaki, I.-M.; Sugiura, D.; Takahashi, S.; Maeda, T.K.; Shimizu, K.; Okazaki, T. LAG-3 Inhibits the Activation of CD4+ T Cells That Recognize Stable pMHCII through Its Conformation-Dependent Recognition of pMHCII. Nat. Immunol. 2018, 19, 1415–1426. [Google Scholar] [CrossRef]
- Huard, B.; Prigent, P.; Tournier, M.; Bruniquel, D.; Triebel, F. CD4/Major Histocompatibility Complex Class II Interaction Analyzed with CD4- and Lymphocyte Activation Gene-3 (LAG-3)-Ig Fusion Proteins. Eur. J. Immunol. 1995, 25, 2718–2721. [Google Scholar] [CrossRef]
- Graydon, C.G.; Mohideen, S.; Fowke, K.R. LAG3’s Enigmatic Mechanism of Action. Front. Immunol. 2021, 11, 615317. [Google Scholar] [CrossRef] [PubMed]
- Workman, C.J.; Dugger, K.J.; Vignali, D.A.A. Cutting Edge: Molecular Analysis of the Negative Regulatory Function of Lymphocyte Activation Gene-3. J. Immunol. 2002, 169, 5392–5395. [Google Scholar] [CrossRef]
- Huang, R.-Y.; Eppolito, C.; Lele, S.; Shrikant, P.; Matsuzaki, J.; Odunsi, K. LAG3 and PD1 Co-Inhibitory Molecules Collaborate to Limit CD8+ T Cell Signaling and Dampen Antitumor Immunity in a Murine Ovarian Cancer Model. Oncotarget 2015, 6, 27359–27377. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Francois, A.; McGray, A.R.; Miliotto, A.; Odunsi, K. Compensatory Upregulation of PD-1, LAG-3, and CTLA-4 Limits the Efficacy of Single-Agent Checkpoint Blockade in Metastatic Ovarian Cancer. OncoImmunology 2017, 6, e1249561. [Google Scholar] [CrossRef]
- Yap, T.A.; LoRusso, P.M.; Wong, D.J.; Hu-Lieskovan, S.; Papadopoulos, K.P.; Holz, J.-B.; Grabowska, U.; Gradinaru, C.; Leung, K.-M.; Marshall, S.; et al. A Phase 1 First-in-Human Study of FS118, a Tetravalent Bispecific Antibody Targeting LAG-3 and PD-L1 in Patients with Advanced Cancer and PD-L1 Resistance. Clin. Cancer Res. 2023, 29, 888–898. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Z. The Effect of Immune Microenvironment on the Progression and Prognosis of Colorectal Cancer. Med. Oncol. 2014, 31, 82. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, N.A.; Becht, E.; Pagès, F.; Skliris, G.; Verkarre, V.; Vano, Y.; Mejean, A.; Saint-Aubert, N.; Lacroix, L.; Natario, I.; et al. Orchestration and Prognostic Significance of Immune Checkpoints in the Microenvironment of Primary and Metastatic Renal Cell Cancer. Clin. Cancer Res. 2015, 21, 3031–3040. [Google Scholar] [CrossRef]
- He, Y.; Yu, H.; Rozeboom, L.; Rivard, C.J.; Ellison, K.; Dziadziuszko, R.; Suda, K.; Ren, S.; Wu, C.; Hou, L.; et al. LAG-3 Protein Expression in Non–Small Cell Lung Cancer and Its Relationship with PD-1/PD-L1 and Tumor-Infiltrating Lymphocytes. J. Thorac. Oncol. 2017, 12, 814–823. [Google Scholar] [CrossRef]
- Burugu, S.; Gao, D.; Leung, S.; Chia, S.K.; Nielsen, T.O. LAG-3+ Tumor Infiltrating Lymphocytes in Breast Cancer: Clinical Correlates and Association with PD-1/PD-L1+ Tumors. Ann. Oncol. 2017, 28, 2977–2984. [Google Scholar] [CrossRef]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.; Okazaki, T. LAG-3: From Molecular Functions to Clinical Applications. J. Immunother. Cancer 2020, 8, e001014. [Google Scholar] [CrossRef]
- Grosso, J.F.; Goldberg, M.V.; Getnet, D.; Bruno, T.C.; Yen, H.-R.; Pyle, K.J.; Hipkiss, E.; Vignali, D.A.A.; Pardoll, D.M.; Drake, C.G. Functionally Distinct LAG-3 and PD-1 Subsets on Activated and Chronically Stimulated CD8 T Cells. J. Immunol. 2009, 182, 6659–6669. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Fu, Y.; Cui, Z.; Abidin, Z.; Yuan, J.; Zhang, X.; Li, R.; Zhao, C. Relatlimab: A Novel Drug Targeting Immune Checkpoint LAG-3 in Melanoma Therapy. Front. Pharmacol. 2024, 14, 1349081. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The Promising Immune Checkpoint LAG-3: From Tumor Microenvironment to Cancer Immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar] [CrossRef]
- Huang, C.-T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in Regulatory T Cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Ohashi, P.S. Clinical Blockade of PD1 and LAG3—Potential Mechanisms of Action | Nature Reviews Immunology. Nat. Rev. Immunol. 2015, 15, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Lipson, E.J.; Dummer, R.; Larkin, J.; Long, G.V.; Sanborn, R.E.; Chiarion-Sileni, V.; Dréno, B.; Dalle, S.; Schadendorf, D.; et al. Nivolumab and Relatlimab in Patients With Advanced Melanoma That Had Progressed on Anti–Programmed Death-1/Programmed Death Ligand 1 Therapy: Results From the Phase I/IIa RELATIVITY-020 Trial. J. Clin. Oncol. 2023, 41, 2724–2735. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Gutiérrez, E.C.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, S.C.; Pisetsky, D.S. Mechanisms of Immune-Related Adverse Events during the Treatment of Cancer with Immune Checkpoint Inhibitors. Rheumatol. Oxf. Engl. 2019, 58, vii59–vii67. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Zang, X.Y.; Wang, J.C.; Huang, S.S.; Xu, J.; Zhang, P. Diagnosis and Management of Immune Related Adverse Events (irAEs) in Cancer Immunotherapy. Biomed. Pharmacother. 2019, 120, 109437. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Das, S.; Johnson, D.B. Immune-Related Adverse Events and Anti-Tumor Efficacy of Immune Checkpoint Inhibitors. J. Immunother. Cancer 2019, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Andrews, A. Treating with Checkpoint Inhibitors-Figure $1 Million per Patient. Am. Health Drug Benefits 2015, 8, 9. [Google Scholar] [PubMed]
- Arora, S.; Velichinskii, R.; Lesh, R.W.; Ali, U.; Kubiak, M.; Bansal, P.; Borghaei, H.; Edelman, M.J.; Boumber, Y. Existing and Emerging Biomarkers for Immune Checkpoint Immunotherapy in Solid Tumors. Adv. Ther. 2019, 36, 2638–2678. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.A.; Patel, V.G. The Role of PD-L1 Expression as a Predictive Biomarker: An Analysis of All US Food and Drug Administration (FDA) Approvals of Immune Checkpoint Inhibitors. J. Immunother. Cancer 2019, 7, 278. [Google Scholar] [CrossRef]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Schlötterer, C.; Harr, B.; Schlo, C. Microsatellite Instability. In Encyclopedia of Life Sciences; Nature Publishing Group: London, UK, 2017; pp. 1–4. [Google Scholar] [CrossRef]
- Drescher, K.M.; Sharma, P.; Watson, P.; Gatalica, Z.; Thibodeau, S.N.; Lynch, H.T. Lymphocyte Recruitment into the Tumor Site Is Altered in Patients with MSI-H Colon Cancer. Fam. Cancer 2009, 8, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Duruisseaux, M.; Martínez-Cardús, A.; Calleja-Cervantes, M.E.; Moran, S.; Castro de Moura, M.; Davalos, V.; Piñeyro, D.; Sanchez-Cespedes, M.; Girard, N.; Brevet, M.; et al. Epigenetic Prediction of Response to Anti-PD-1 Treatment in Non-Small-Cell Lung Cancer: A Multicentre, Retrospective Analysis. Lancet Respir. Med. 2018, 6, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-J.; Wu, E. The Role of Gut Microbiota in Immune Homeostasis and Autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef]
- Zitvogel, L.; Ayyoub, M.; Routy, B.; Kroemer, G. Microbiome and Anticancer Immunosurveillance. Cell 2016, 165, 276–287. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut Microbiome Modulates Response to Anti–PD-1 Immunotherapy in Melanoma Patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The Evolving Landscape of Biomarkers for Checkpoint Inhibitor Immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive Biomarkers for Checkpoint Inhibitor-Based Immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [PubMed]
- Sauer, N.; Janicka, N.; Szlasa, W.; Skinderowicz, B.; Kołodzińska, K.; Dwernicka, W.; Oślizło, M.; Kulbacka, J.; Novickij, V.; Karłowicz-Bodalska, K. TIM-3 as a Promising Target for Cancer Immunotherapy in a Wide Range of Tumors. Cancer Immunol. Immunother. 2023, 72, 3405–3425. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 Ligand Galectin-9 Negatively Regulates T Helper Type 1 Immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, N.; Rinne, M.L.; Sun, H.; Stein, A.M. Sabatolimab (MBG453) Model-Informed Drug Development for Dose Selection in Patients with Myelodysplastic Syndrome/Acute Myeloid Leukemia and Solid Tumors. CPT Pharmacomet. Syst. Pharmacol. 2023, 12, 1653–1665. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.M.; Esteve, J.; Porkka, K.; Knapper, S.; Traer, E.; Scholl, S.; Garcia-Manero, G.; Vey, N.; Wermke, M.; Janssen, J.; et al. Efficacy and Safety of Sabatolimab (MBG453) in Combination with Hypomethylating Agents (HMAs) in Patients (Pts) with Very High/High-Risk Myelodysplastic Syndrome (vHR/HR-MDS) and Acute Myeloid Leukemia (AML): Final Analysis from a Phase Ib Study. Blood 2021, 138, 244. [Google Scholar] [CrossRef]
- Crescioli, S.; Kaplon, H.; Chenoweth, A.; Wang, L.; Visweswaraiah, J.; Reichert, J.M. Antibodies to Watch in 2024. mAbs 2024, 16, 2297450. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Giagounidis, A.; Sekeres, M.A.; Xiao, Z.; Sanz, G.F.; Hoef, M.V.; Ma, F.; Hertle, S.; Santini, V. STIMULUS-MDS2 Design and Rationale: A Phase III Trial with the Anti-TIM-3 Sabatolimab (MBG453) + Azacitidine in Higher Risk MDS and CMML-2. Future Oncol. 2023, 19, 631–642. [Google Scholar] [CrossRef]
- Zhao, J.; Li, L.; Yin, H.; Feng, X.; Lu, Q. TIGIT: An Emerging Immune Checkpoint Target for Immunotherapy in Autoimmune Disease and Cancer. Int. Immunopharmacol. 2023, 120, 110358. [Google Scholar] [CrossRef]
- Chu, X.; Tian, W.; Wang, Z.; Zhang, J.; Zhou, R. Co-Inhibition of TIGIT and PD-1/PD-L1 in Cancer Immunotherapy: Mechanisms and Clinical Trials. Mol. Cancer 2023, 22, 93. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Oh, D.-Y.; Pelster, M.; Wainberg, Z.A.; Sison, E.A.R.; Scott, J.R.; Ronayne, J.; Wishengrad, D.; Rhee, J.; Nuyten, D.S.A.; et al. EDGE-Gastric Arm A1: Phase 2 Study of Domvanalimab, Zimberelimab, and FOLFOX in First-Line (1L) Advanced Gastroesophageal Cancer. J. Clin. Oncol. 2023, 41, 433248. [Google Scholar] [CrossRef]
- Johnson, M.L.; Fox, W.; Lee, Y.-G.; Lee, K.H.; Ahn, H.K.; Kim, Y.-C.; Lee, K.-Y.; Lee, J.-S.; He, X.; Park, C.; et al. ARC-7: Randomized Phase 2 Study of Domvanalimab + Zimberelimab ± Etrumadenant versus Zimberelimab in First-Line, Metastatic, PD-L1-High Non-Small Cell Lung Cancer (NSCLC). J. Clin. Oncol. 2022, 40, 397600. [Google Scholar] [CrossRef]
- Chouaid, C.; Ho, G.F.; Runglodvatana, Y.; He, X.; Ahlers, C.M.; Pomponio, D.; Todd, T.; Dang, T.; Naidoo, J. ARC-10: A Phase 3 Study to Evaluate Zimberelimab + Domvanalimab versus Pembrolizumab in Front-Line, PD-L1-High, Locally Advanced or Metastatic Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2023, 41, TPS9148. [Google Scholar] [CrossRef]
- Seliger, B. Combinatorial Approaches with Checkpoint Inhibitors to Enhance Anti-Tumor Immunity. Front. Immunol. 2019, 10, 999. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.; Toor, S.M.; Khalaf, S.; Elkord, E. Breast Cancer Cells and PD-1/PD-L1 Blockade Upregulate the Expression of PD-1, CTLA-4, TIM-3 and LAG-3 Immune Checkpoints in CD4+ T Cells. Vaccines 2019, 7, 149. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Formenti, S.C.; Demaria, S. Toward Precision Radiotherapy for Use with Immune Checkpoint Blockers. Clin. Cancer Res. 2018, 24, 259–265. [Google Scholar] [CrossRef]
- Rudqvist, N.P.; Pilones, K.A.; Lhuillier, C.; Wennerberg, E.; Sidhom, J.W.; Emerson, R.O.; Robins, H.S.; Schneck, J.; Formenti, S.C.; Demaria, S. Radiotherapy and CTLA-4 Blockade Shape the Tcr Repertoire of Tumor-Infiltrating t Cells. Cancer Immunol. Res. 2018, 6, 139–150. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ipilimumab | |||
|---|---|---|---|
| Cancer Type | FDA Approval Year | Key Clinical Trial (Phase) | Monotherapy/Combination Therapy |
| Melanoma | 2011 | MDX010-20 (Phase 3) [13] | Monotherapy |
| 2015 | CheckMate-067 (Phase 3) [34] | Combination with nivolumab | |
| Renal Cell Carcinoma | 2018 | CheckMate-214 (Phase 3) [12] | Combination with nivolumab |
| Colorectal Cancer—Microsatellite instability-high/mismatch repair-deficient (MSI-H/dMMR) | 2018 | CheckMate 142 (Phase 2) [35] | Combination with nivolumab |
| Hepatocellular Carcinoma | 2020 | CheckMate 040 (Phase 1/2) [36] | Combination with nivolumab |
| Non-small-cell lung carcinoma | 2020 | CheckMate 227 (Phase 3) [37] | Combination with nivolumab |
| Pleural Mesothelioma | 2020 | CheckMate 743 (Phase 3) [38] | Combination with nivolumab |
| Oesophageal Squamous Cell Carcinoma | 2021 | CheckMate 648 (Phase 3) [39] | Combination with nivolumab |
| ANTI-PD-1 Immune Checkpoint Inhibitors | ||||
|---|---|---|---|---|
| Drug | Cancer Type | FDA Approval Year | Key Trial (Phase) | Monotherapy/Combination Therapy |
| Pembrolizumab | Melanoma | 2014 | KEYNOTE-006 (Phase 3) [65] | Monotherapy |
| Non-Small-Cell Lung Carcinoma | 2015 | KEYNOTE-010 (Phase 2/3) [66] | Monotherapy | |
| Squamous Cell Carcinoma ofHead and Neck | 2016 | KEYNOTE-012 (Phase 1b) [67] | Monotherapy | |
| Hodgkin’s Lymphoma | 2017 | KEYNOTE-087 (Phase 2) [68] | Monotherapy | |
| Urothelial Carcinoma | 2017 | KEYNOTE-052 (Phase 2) [69] | Monotherapy | |
| Colorectal cancer (MSI-H/dMMR) | 2017 | KEYNOTE-164 (Phase 2) [70] | Monotherapy | |
| Gastric/Gastroesophageal junction (GEJ) Carcinoma | 2017 | KEYNOTE-059 (Phase 2) [71] | Monotherapy or Combination with chemotherapy | |
| Cervical Carcinoma | 2018 | KEYNOTE-158 (Phase 2) [72] | Monotherapy | |
| Hepatocellular Carcinoma | 2018 | KEYNOTE-224 (Phase 2) [73] | Monotherapy | |
| Merkel Cell Carcinoma | 2018 | KEYNOTE-017 (Phase 2) [74] | Monotherapy | |
| Renal Cell Carcinoma | 2019 | KEYNOTE-426 (Phase 3) [75] | Combination with axitinib | |
| Oesophageal Carcinoma | 2019 | KEYNOTE-181 (Phase 3) [76] | Monotherapy | |
| Triple-Negative Breast Carcinoma | 2020 | KEYNOTE-355 (Phase 3) [77] | Combination with chemotherapy | |
| Cutaneous Squamous Cell Carcinoma | 2020 | KEYNOTE-629 (Phase 2) [78] | Monotherapy | |
| Endometrial Carcinoma (MSI-H/dMMR) | 2022 | KEYNOTE-158 (Phase 2) [79] | Monotherapy | |
| Biliary Tract Carcinoma | 2023 | KEYNOTE-966 (Phase 3) [78] | Combination with chemotherapy | |
| Nivolumab | Melanoma | 2014 | CheckMate-037 (Phase 3) [80] | Monotherapy |
| 2015 | CheckMate-067 (Phase 3) [34] | Combination with ipilimumab | ||
| 2017 | CheckMate-238 (Phase 3) [81] | Adjuvant treatment | ||
| Non-Small-Cell Lung Carcinoma | 2015 | CheckMate-057 (Phase 3) [14] | Monotherapy | |
| 2020 | CheckMate-227 (Phase 3) [37] | Combination with ipilimumab | ||
| Renal Cell Carcinoma | 2015 | CheckMate-025 (Phase 3) [82] | Monotherapy | |
| 2018 | CheckMate-214 (Phase 3) [12] | Combination with ipilimumab | ||
| Classical Hodgkin Lymphoma | 2016 | CheckMate-205 (Phase 2) [83] | Monotherapy | |
| Squamous Cell Carcinoma of Head and Neck Carcinoma | 2016 | CheckMate-141 (Phase 3) [84] | Monotherapy | |
| Urothelial Carcinoma | 2017 | CheckMate-275 (Phase 2) [85] | Monotherapy | |
| Colorectal Carcinoma (MSI-H/dMMR) | 2017 | CheckMate-142 (Phase 2) [34] | Combination with ipilimumab | |
| Hepatocellular carcinoma | 2020 | CheckMate-040 (Phase 1/2) [36] | Combination with ipilimumab | |
| Small-Cell Lung Carcinoma | 2018 | CheckMate-032 (Phase 1/2) [86] | Monotherapy | |
| Oesophageal Squamous Cell Carcinoma | 2020 | ATTRACTION-3 (Phase 3) [87] | Monotherapy | |
| 2020 | CheckMate-648 (Phase 3) [39] | Combination with Ipilimumab or Chemotherapy | ||
| Pleural Mesothelioma | 2020 | CheckMate-743 (Phase 3) [38] | Combination with Ipilimumab | |
| Gastric/GEJ Carcinoma | 2021 | CheckMate-649 (Phase 3) | Combination with Chemotherapy | |
| Cemiplimab | Cutaneous Squamous Cell Carcinoma | 2018 | EMPOWER-CSCC-1 (Phase 2) [88] | Monotherapy |
| Basal Cell Carcinoma | 2021 | Study-1620 (Phase 2) [89] | Monotherapy | |
| Non-Small-Cell Lung Cancer | 2021 | EMPOWER-Lung 1 (Phase 3) [90] | Monotherapy | |
| ANTI-PD-L1 Immune Checkpoint Inhibitors | ||||
|---|---|---|---|---|
| Drug | Cancer Type | FDA Approval Year | Key Trials | Monotherapy/Combination |
| Atezolizumab | Urothelial Carcinoma | 2016 | IMvigor210 (Phase 2) [97] | Monotherapy |
| Non-Small-Cell Lung Carcinoma | 2016 | OAK (Phase 3) [100] | Monotherapy | |
| Small-Cell Lung Carcinoma | 2019 | IMpower133 (Phase 3) [105] | Combination with chemotherapy | |
| Triple-Negative Breast Cancer | 2019 | IMpassion130 (Phase 3) [103] | Combination with chemotherapy | |
| Hepatocellular Carcinoma | 2020 | IMbrave150 (Phase 3) [106] | Combination with bevacizumab | |
| Melanoma (BRAF V600 mutation-positive) | 2020 | IMspire150 (Phase 3) [107] | Combination with cobimetinib and vemurafenib | |
| Alveolar Soft Part Sarcoma | 2022 | Study ML39345 (Phase 2) [108] | Monotherapy | |
| Avelumab | Merkel Cell Carcinoma | 2017 | JAVELIN Merkel 200 (Phase 2) [109] | Monotherapy |
| Urothelial Carcinoma | 2017 | JAVELIN Solid Tumor (Phase 1) [110] | Monotherapy | |
| 2020 | JAVELIN Bladder 100 (Phase 3) [111] | 1st line maintenance: Combination with best supportive care | ||
| Renal Cell Carcinoma | 2019 | JAVELIN Renal 101 (Phase 3) [112] | Combination with Axitinib | |
| Durvalumab | Urothelial Carcinoma | 2017 | Study 1108 (Phase 1/2) [113] | Monotherapy |
| Non-Small-Cell Lung Carcinoma | 2018 | PACIFIC (Phase 3) [114] | Monotherapy | |
| Small-Cell Lung Carcinoma | 2020 | CASPIAN (Phase 3) [115] | Combination with chemotherapy | |
| Biliary Tract Cancer | 2022 | TOPAZ-1 (Phase 3) [116] | Combination with chemotherapy | |
| Hepatocellular Carcinoma | 2020 | HIMALAYA (Phase 3) [117] | Combination with tremelimumab | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Younis, A.; Gribben, J. Immune Checkpoint Inhibitors: Fundamental Mechanisms, Current Status and Future Directions. Immuno 2024, 4, 186-210. https://doi.org/10.3390/immuno4030013
Younis A, Gribben J. Immune Checkpoint Inhibitors: Fundamental Mechanisms, Current Status and Future Directions. Immuno. 2024; 4(3):186-210. https://doi.org/10.3390/immuno4030013
Chicago/Turabian StyleYounis, Abdullah, and John Gribben. 2024. "Immune Checkpoint Inhibitors: Fundamental Mechanisms, Current Status and Future Directions" Immuno 4, no. 3: 186-210. https://doi.org/10.3390/immuno4030013
APA StyleYounis, A., & Gribben, J. (2024). Immune Checkpoint Inhibitors: Fundamental Mechanisms, Current Status and Future Directions. Immuno, 4(3), 186-210. https://doi.org/10.3390/immuno4030013