
Experimental Studies on the Phase Separation Behavior of Molten Benzenesulfonate-Modified PET/PA6 Blends


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Polyester/Nylon Blends
2.3. Phase Separation Experiment
2.4. Characterization Method
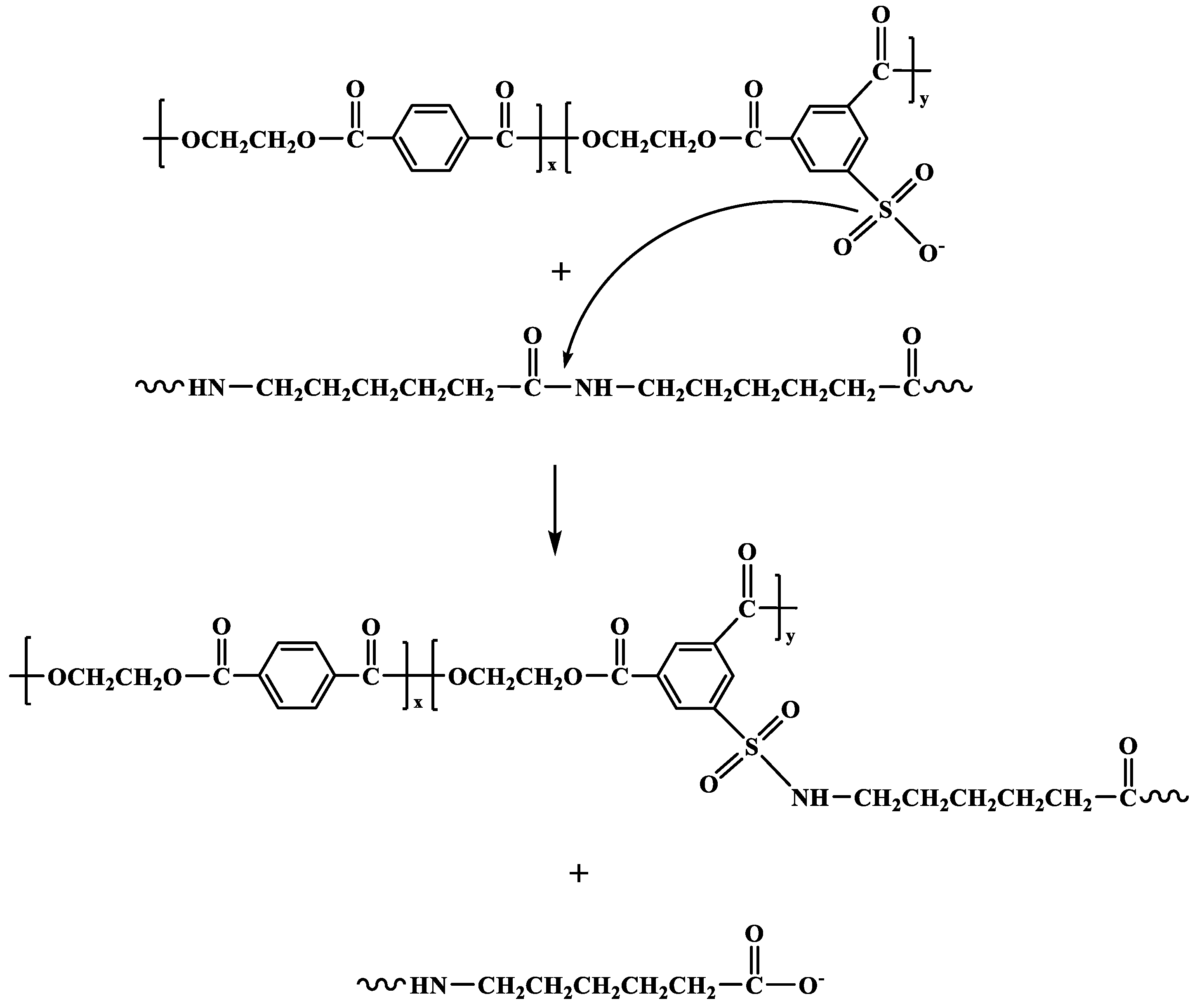
2.4.1. Nuclear Magnetic Resonance (13C NMR)
2.4.2. Scanning Electron Microscope (SEM)
2.4.3. Atomic Force Microscopy-Infrared (AFM-IR)
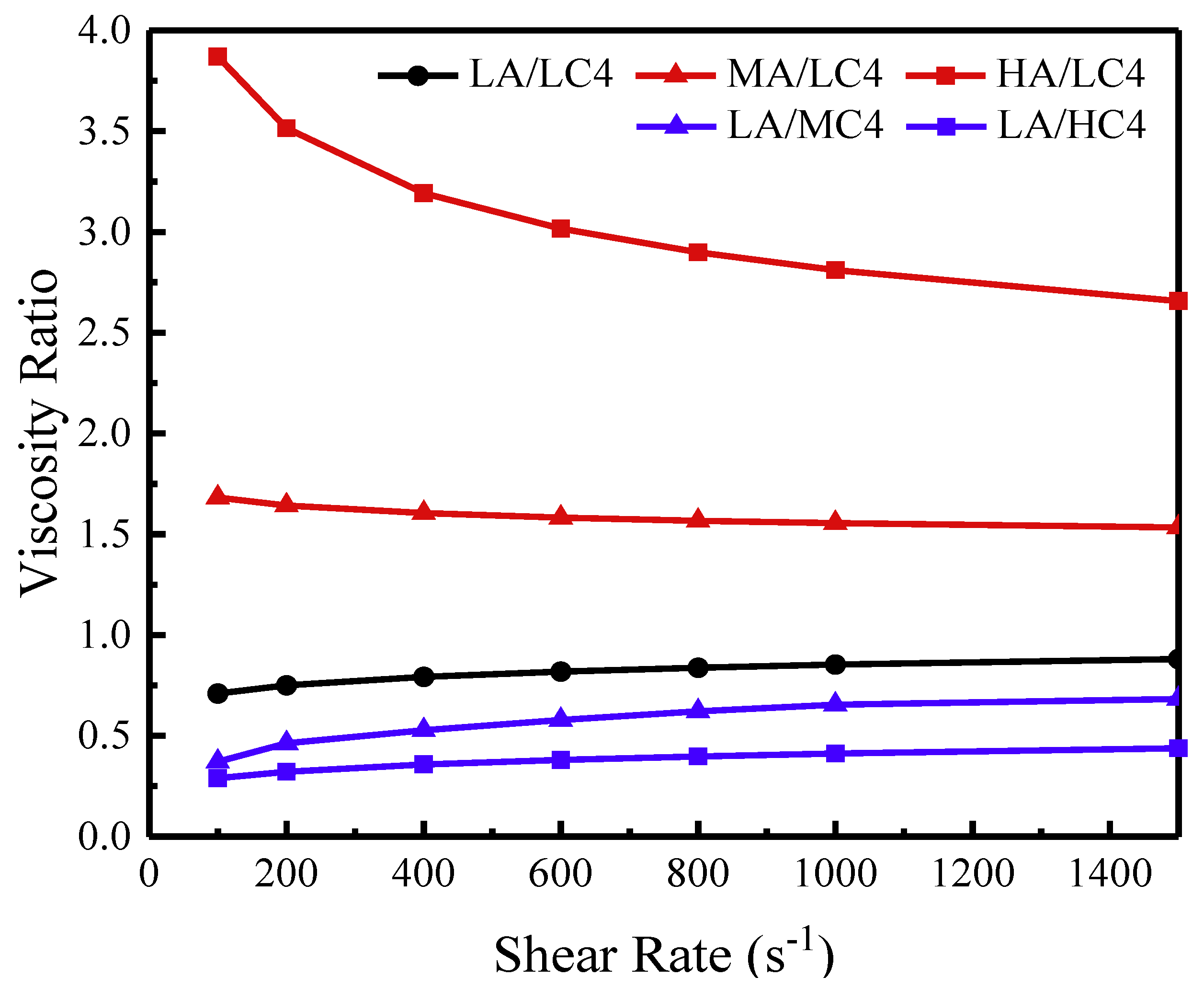
2.4.4. High-Pressure Capillary Rheology
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, C.; Zhao, R.; Lu, X.D. 2019 Review and 2020 outlook for world and china synthetic fiber industry. Pet. Petrochem. Today 2020, 28, 14–17. [Google Scholar]
- Nisticò, R. Polyethylene terephthalate (PET) in the packaging industry. Polym. Test. 2020, 90, 106707. [Google Scholar] [CrossRef]
- d’Ambrières, W. Plastics recycling worldwide: Current overview and desirable changes. Field Actions Sci. Rep. J. 2019, 19, 12–21. [Google Scholar]
- Anstey, A.; Codou, A.; Misra, M.; Mohanty, A.K. Novel compatibilized nylon-based ternary blends with polypropylene and poly (lactic acid): Fractionated crystallization phenomena and mechanical performance. ACS Omega 2018, 3, 2845–2854. [Google Scholar] [CrossRef] [PubMed]
- Ksouri, I.; De Almeida, O.; Haddar, N. Long term ageing of polyamide 6 and polyamide 6 reinforced with 30% of glass fibers: Physicochemical, mechanical and morphological characterization. J. Polym. Res. 2017, 24, 1–12. [Google Scholar] [CrossRef]
- Huang, S.; Toh, C.L.; Yang, L.; Phua, S.; Zhou, R.; Dasari, A.; Lu, X. Reinforcing nylon 6 via surface-initiated anionic ring-opening polymerization from stacked-cup carbon nanofibers. Compos. Sci. Technol. 2014, 93, 30–37. [Google Scholar] [CrossRef]
- Pillon, L.; Utracki, L. Compatibilization of polyester/polyamide blends via catalytic ester-amide interchange reaction. Polym. Eng. Sci. 1984, 24, 1300–1305. [Google Scholar] [CrossRef]
- Evstatiev, M.; Fakirov, S.; Schultz, J.; Friedrich, K. In situ fibrillar reinforced PET/PA-6/PA-66 blend. Polym. Eng. Sci. 2001, 41, 192–204. [Google Scholar] [CrossRef]
- Yao, Z.; Sun, J.-m.; Wang, Q.; Cao, K. Study on ester–amide exchange reaction between PBS and PA6IcoT. Ind. Eng. Chem. Res. 2012, 51, 751–757. [Google Scholar] [CrossRef]
- Néry, L.; Lefebvre, H.; Fradet, A. Polyamide–polyester multiblock copolymers by chain-coupling reactions of carboxy-terminated polymers with phenylene and pyridylene bisoxazolines. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 1331–1341. [Google Scholar] [CrossRef]
- Ju, L.; Pretelt, J.; Chen, T.; Dennis, J.M.; Heifferon, K.V.; Baird, D.G.; Long, T.E.; Moore, R.B. Synthesis and characterization of phosphonated Poly (ethylene terephthalate) ionomers. Polymer 2018, 151, 154–163. [Google Scholar] [CrossRef]
- Ju, L.; Dennis, J.M.; Heifferon, K.V.; Long, T.E.; Moore, R.B. Compatibilization of polyester/polyamide blends with a phosphonated poly (ethylene terephthalate) ionomer: Comparison of monovalent and divalent pendant ions. ACS Appl. Polym. Mater. 2019, 1, 1071–1080. [Google Scholar] [CrossRef]
- Ju, L.; Mondschein, R.J.; Vandenbrande, J.A.; Arrington, C.B.; Long, T.E.; Moore, R.B. Phosphonated Poly (ethylene terephthalate) ionomers as compatibilizers in extruded Poly (ethylene terephthalate)/Poly (m-xylylene adipamide) blends and oriented films. Polymer 2020, 205, 122891. [Google Scholar] [CrossRef]
- Samperi, F.; Montaudo, M.; Puglisi, C.; Alicata, R.; Montaudo, G. Essential role of chain ends in the Ny6/PBT exchange. A combined NMR and MALDI approach. Macromolecules 2003, 36, 7143–7154. [Google Scholar] [CrossRef]
- Xiao, L.; Wang, H.; Qian, Q.; Jiang, X.; Liu, X.; Huang, B.; Chen, Q. Molecular and structural analysis of epoxide-modified recycled poly (ethylene terephthalate) from rheological data. Polym. Eng. Sci. 2012, 52, 2127–2133. [Google Scholar] [CrossRef]
- Kammer, H.; Kummerloewe, C.; Kressler, J.; Melior, J. Shear-induced phase changes in polymer blends. Polymer 1991, 32, 1488–1492. [Google Scholar] [CrossRef]
- Sanviti, M.; Martinez-Tong, D.E.; Rebollar, E.; Ezquerra, T.A.; Garcia-Gutierrez, M.C. Crystallization and phase separation in PEDOT:PSS/PEO blend thin films: Influence on mechanical and electrical properties at the nanoscale. Polymer 2022, 262, 13. [Google Scholar] [CrossRef]
- Xiang, S.; Tang, X.X.; Rajabzadeh, S.; Zhang, P.F.; Cui, Z.Y.; Matsuyama, H. Fabrication of PVDF/EVOH blend hollow fiber membranes with hydrophilic property via thermally induced phase process. Sep. Purif. Technol. 2022, 301, 11. [Google Scholar] [CrossRef]
- Wu, X.X.; Song, T.T.; Wei, Z.Z.; Shen, L.; Jiang, H.Q.; Ke, Y.B.; He, C.Y.; Yang, H.; Shi, W.C. Promoted liquid-liquid phase separation of PEO/PS blends with very low LiTFSI fraction. Polymer 2022, 260, 6. [Google Scholar] [CrossRef]
- Kalourazi, S.F.; Wang, F.; Zhang, H.D.; Selzer, M.; Nestler, B. Phase-field simulation for the formation of porous microstructures due to phase separation in polymer solutions on substrates with different wettabilities. J. Phys. Condens. Matter 2022, 34, 16. [Google Scholar]
- Tseng, Y.H.; Fan, Y.C.; Chang, C.T.; Lin, Y.L.; Chang, C.W.; Liao, C.W.; Chen, J.T. Photoinduced alignment under solvent vapor annealing (PA-SVA): Enhanced ordering and patterning in block copolymer films. ACS Appl. Polym. Mater. 2022, 4, 8536–8542. [Google Scholar] [CrossRef]
- Pochivalov, K.V.; Basko, A.V. Formation of porous microspheres from semicrystalline polymer solutions: Diffusion-controlled and local phase separation. Polym. Plast. Technol. Mater. 2022, 61, 1279–1291. [Google Scholar]
- Zhong, Z.X.; Peng, L.; Zhang, N.; Su, J.X.; Ye, N.B.; Luo, Z.F.; Han, C.C.; Huang, X.B.; Su, Z.H. Miscibility of isotactic polypropylene with random and block ethylene-octene copolymers studied by atomic force microscopy-infrared. Polymer 2022, 259, 10. [Google Scholar] [CrossRef]
- Moonprasith, N.; Tatsumichi, M.; Nakamura, K.; Kida, T.; Tsubouchi, K.; Hiraoka, T.; Yamaguchi, M. Preparation of graded materials for miscible polycarbonate/poly(methyl methacrylate) blends by segregation under shear flow. J. Appl. Polym. Sci. 2023, 140, 8. [Google Scholar] [CrossRef]
- Chuang, P.Y.; Liao, S.Y.; Wu, K.H.; Hu, Y.R.; Lo, C.T. Competitive Effects of Hydrogen Bonds and Molecular Weights on the Phase and Crystallization Behaviors of Binary Block Copolymers. Macromolecules 2022, 55, 7411–7424. [Google Scholar] [CrossRef]
- Ma, X.J. Studies on the Preparation of PET/PA6 Alloy by Reactive Extrusion and Its Performance. Master’s Thesis, Zhejiang University, Hangzhou, China, 2021. [Google Scholar]




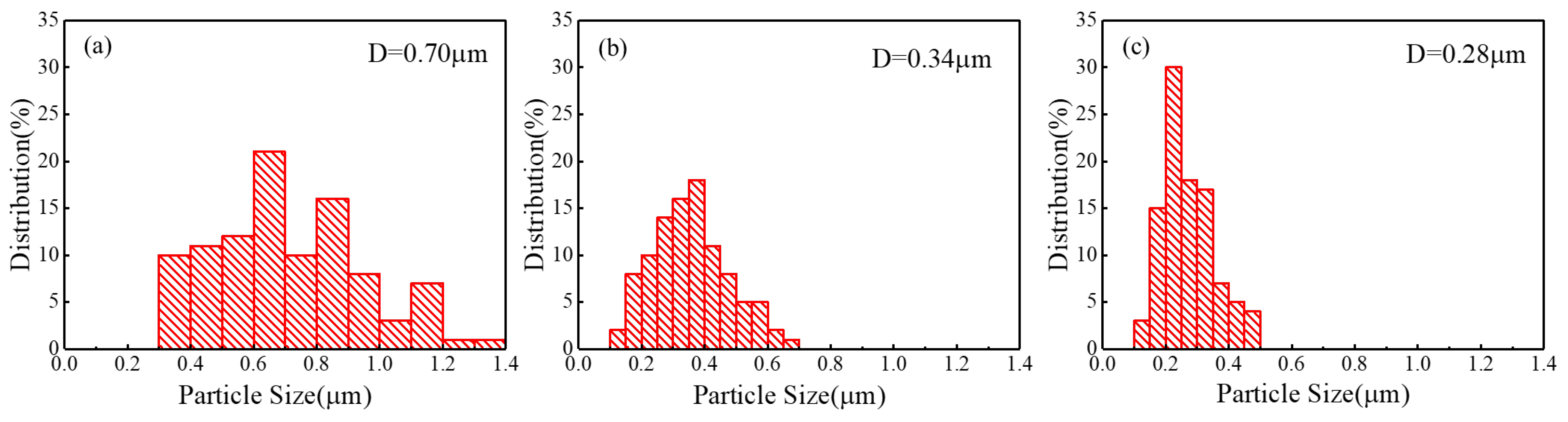


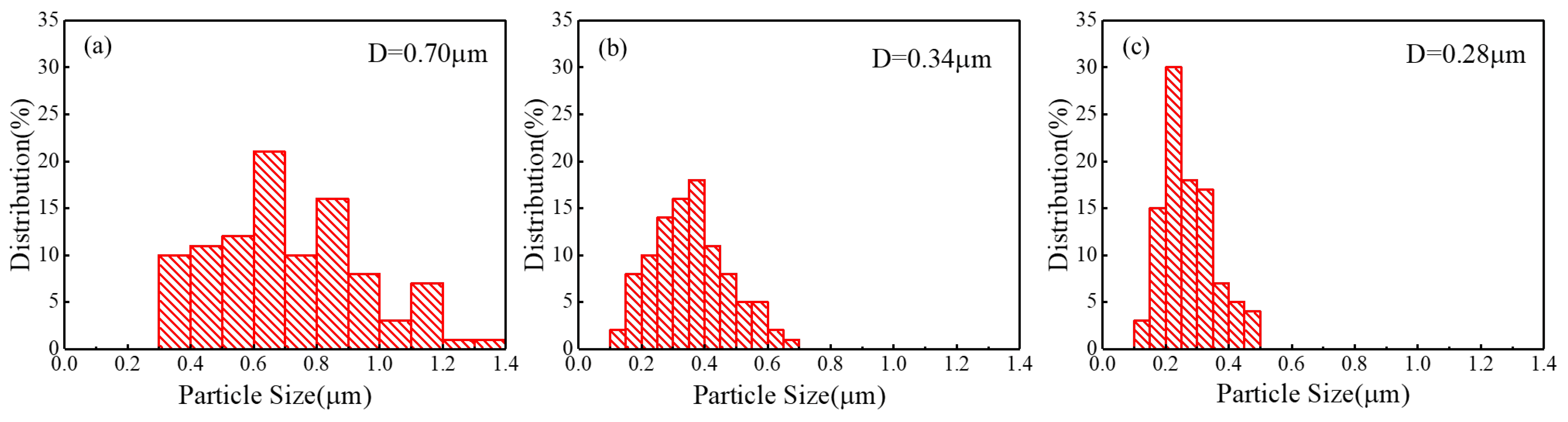
{kind=link}
{kind=link}
{kind=link}
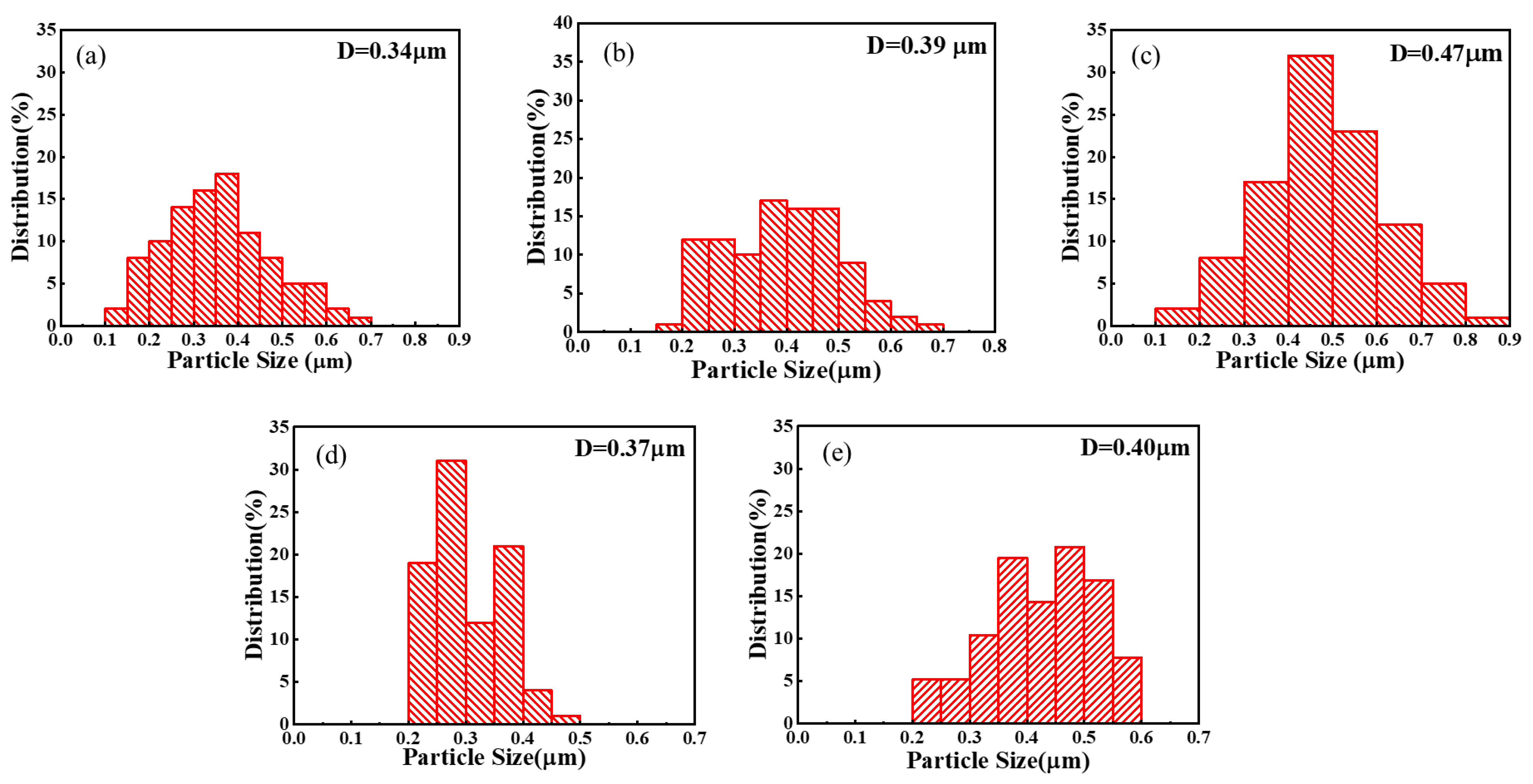
{kind=link}
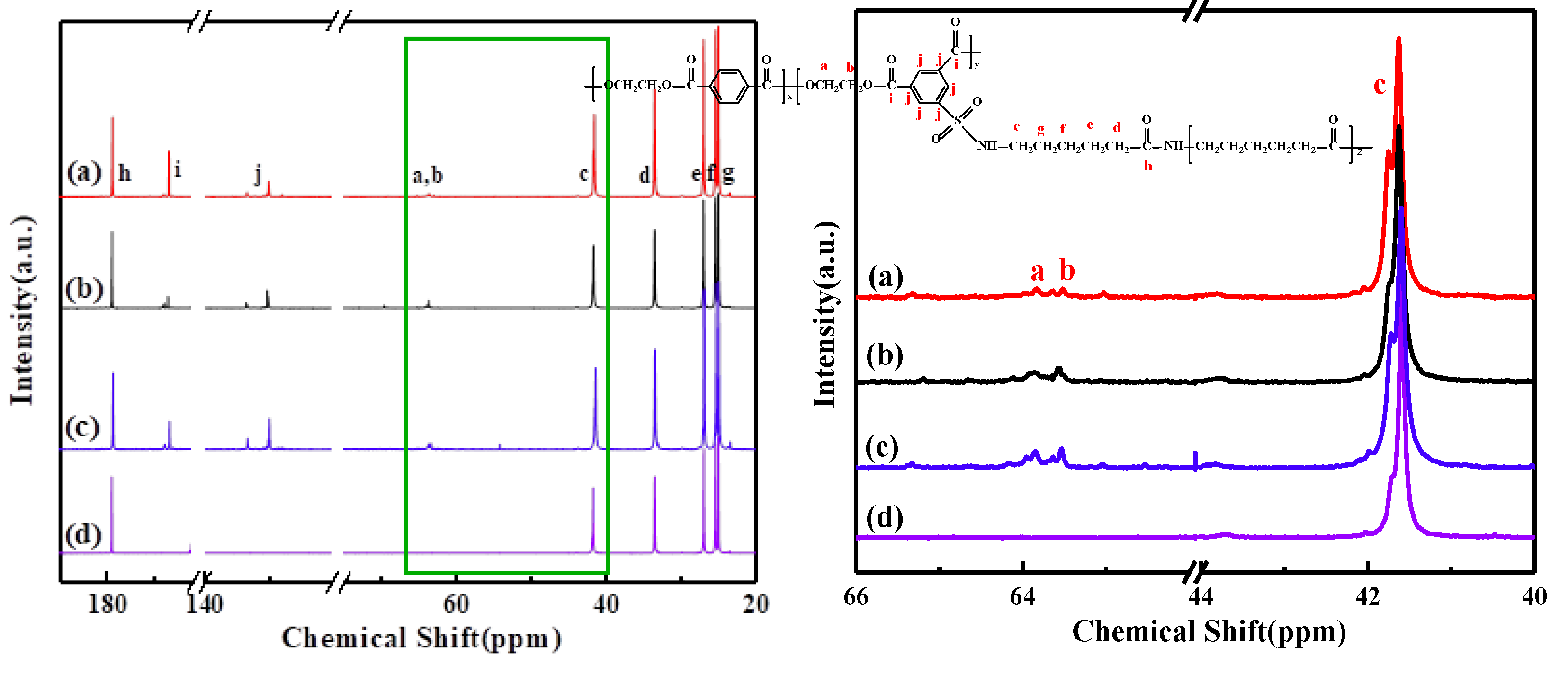{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CDP | Molecular Weight (g/mol) | Sulfonic Acid Content (mol %) | Supplier |
|---|---|---|---|
| PET | 18,400 | 0 | Zhejiang Henglan Technology Co., Ltd. |
| LC2 | 14,500 | 2.0 | Zhejiang Henglan Technology Co., Ltd. |
| LC4 | 11,700 | 4.0 | Zhejiang Henglan Technology Co., Ltd. |
| LC8 | 11,400 | 8.0 | Self-made |
| MC4 | 23,000 | 4.0 | Self-made |
| HC4 | 25,400 | 4.0 | Self-made |
| PA6 | Molecular Weight (g/mol) | Supplier |
|---|---|---|
| LA | 19,600 | Zhejiang Henglan Technology Co., Ltd. |
| MA | 31,600 | Ube Co., Ltd., Thailand |
| HA | 39,700 | Ube Co., Ltd., Thailand |
| Sample | Temperature (°C) | CDP | PA6 | CDP Content (wt%) | PA6 Content (wt%) |
|---|---|---|---|---|---|
| LC2/LA(70/30) | 250 | LC2 | LA | 70 | 30 |
| LC4/LA(70/30) | 250 | LC4 | LA | 70 | 30 |
| LC8/LA(70/30) | 250 | LC8 | LA | 70 | 30 |
| LC4/LA(50/50) | 250 | LC4 | LA | 50 | 50 |
| LC4/LA(30/70) | 250 | LC4 | LA | 30 | 70 |
| LC4/MA(70/30) | 250 | LC4 | MA | 70 | 30 |
| LC4/HA(70/30) | 250 | LC4 | HA | 70 | 30 |
| MC4/LA(70/30) | 250 | MC4 | LA | 70 | 30 |
| HC4/LA(70/30) | 250 | HC4 | LA | 70 | 30 |
| PET/LA(70/30) | 250 | PET | LA | 70 (PET content) | 30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, X.-J.; Ye, Q.-Y.; Zheng, S.-J.; Hu, J.-J.; Yao, Z. Experimental Studies on the Phase Separation Behavior of Molten Benzenesulfonate-Modified PET/PA6 Blends. Macromol 2023, 3, 54-64. https://doi.org/10.3390/macromol3010005
Ma X-J, Ye Q-Y, Zheng S-J, Hu J-J, Yao Z. Experimental Studies on the Phase Separation Behavior of Molten Benzenesulfonate-Modified PET/PA6 Blends. Macromol. 2023; 3(1):54-64. https://doi.org/10.3390/macromol3010005
Chicago/Turabian StyleMa, Xiao-Jun, Qi-Yu Ye, Shao-Jie Zheng, Ji-Jiang Hu, and Zhen Yao. 2023. "Experimental Studies on the Phase Separation Behavior of Molten Benzenesulfonate-Modified PET/PA6 Blends" Macromol 3, no. 1: 54-64. https://doi.org/10.3390/macromol3010005
APA StyleMa, X.-J., Ye, Q.-Y., Zheng, S.-J., Hu, J.-J., & Yao, Z. (2023). Experimental Studies on the Phase Separation Behavior of Molten Benzenesulfonate-Modified PET/PA6 Blends. Macromol, 3(1), 54-64. https://doi.org/10.3390/macromol3010005