
Bioinformatic Analysis of Metabolomic Data: From Raw Spectra to Biological Insight



Abstract
1. Introduction
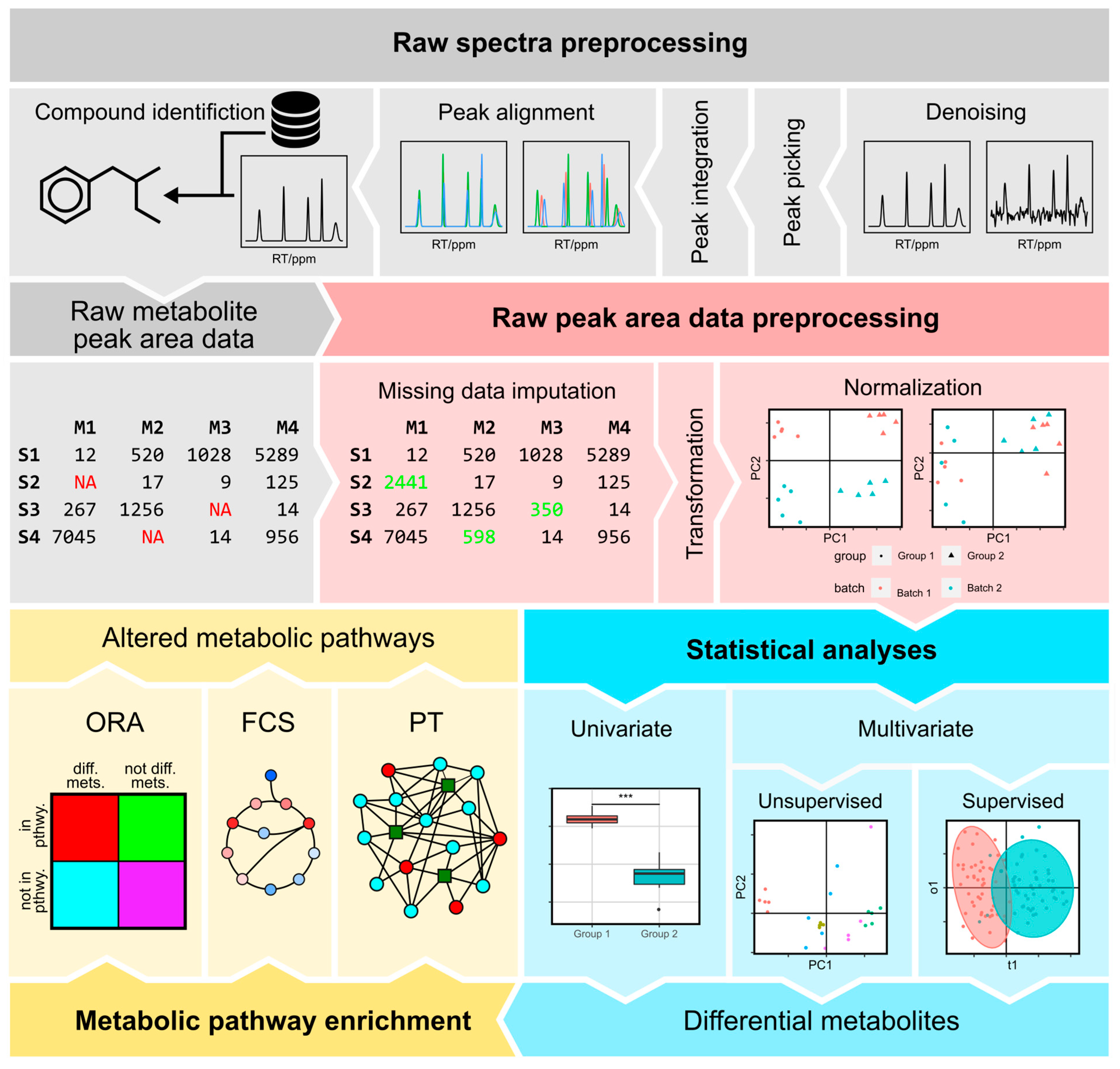
2. Raw Spectra Preprocessing
2.1. Denoising
2.2. Peak Picking
2.3. Peak Alignment
2.4. Compound Identification
3. Peak Area Preprocessing
3.1. Missing Value Imputation
3.2. Normalization
3.2.1. Normalization Methods
3.2.2. Assessment of Post-Acquisition Normalization Effectivity
4. Statistical Analysis of Metabolomic Data
4.1. Univariate Analyses
4.2. Multivariate Analyses
4.2.1. Non-Supervised Multivariate Analyses
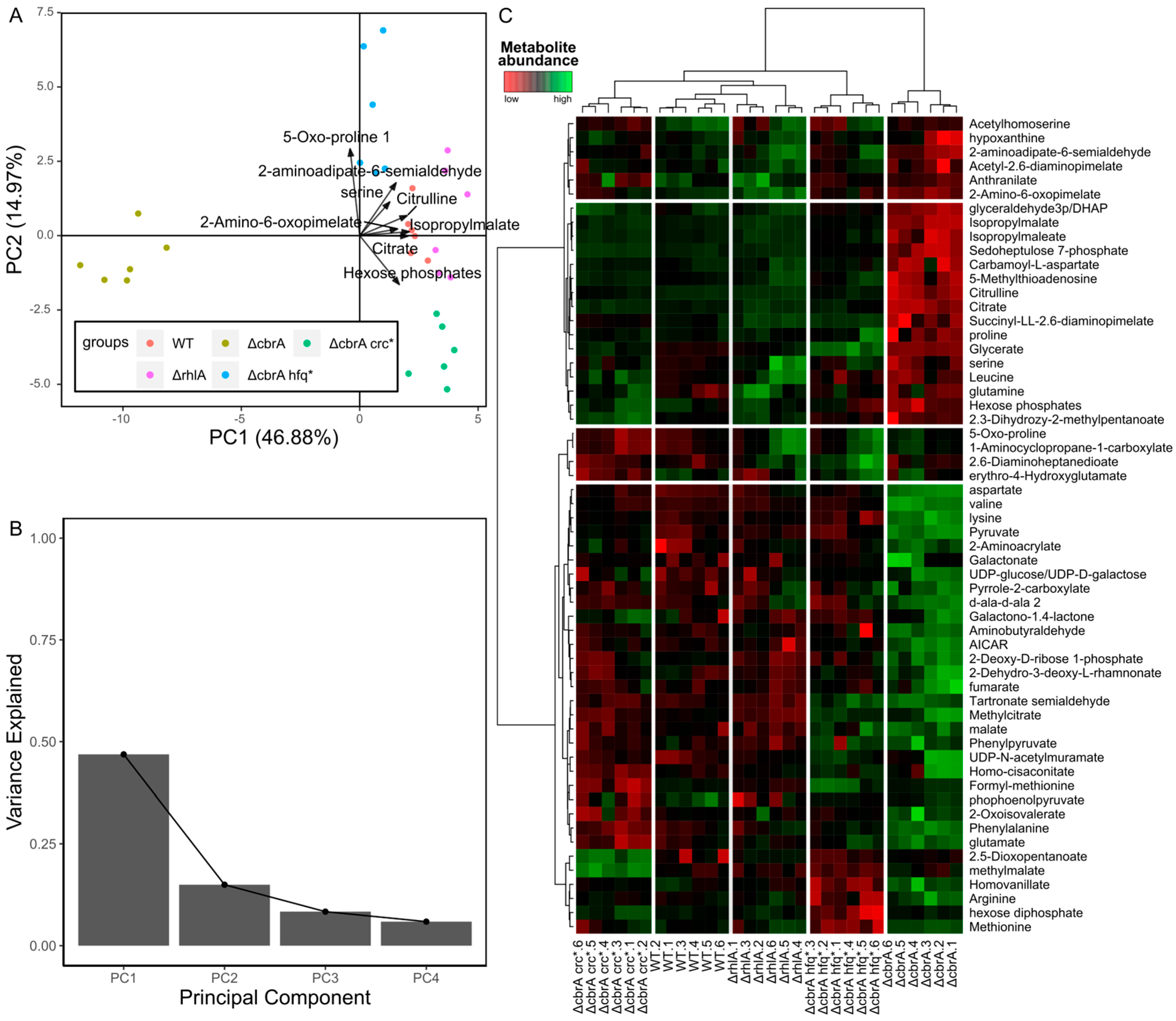
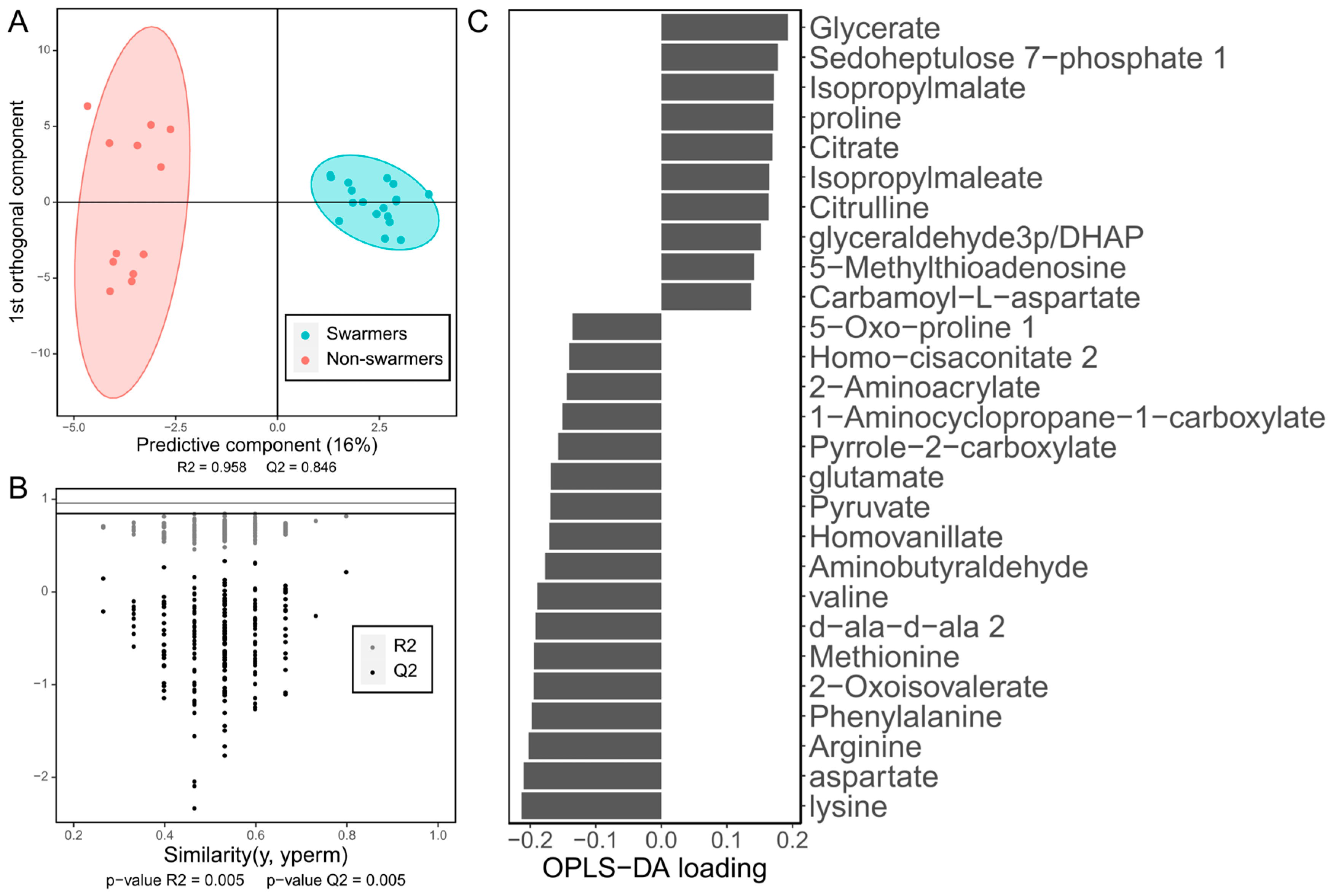
4.2.2. Supervised Multivariate Approaches
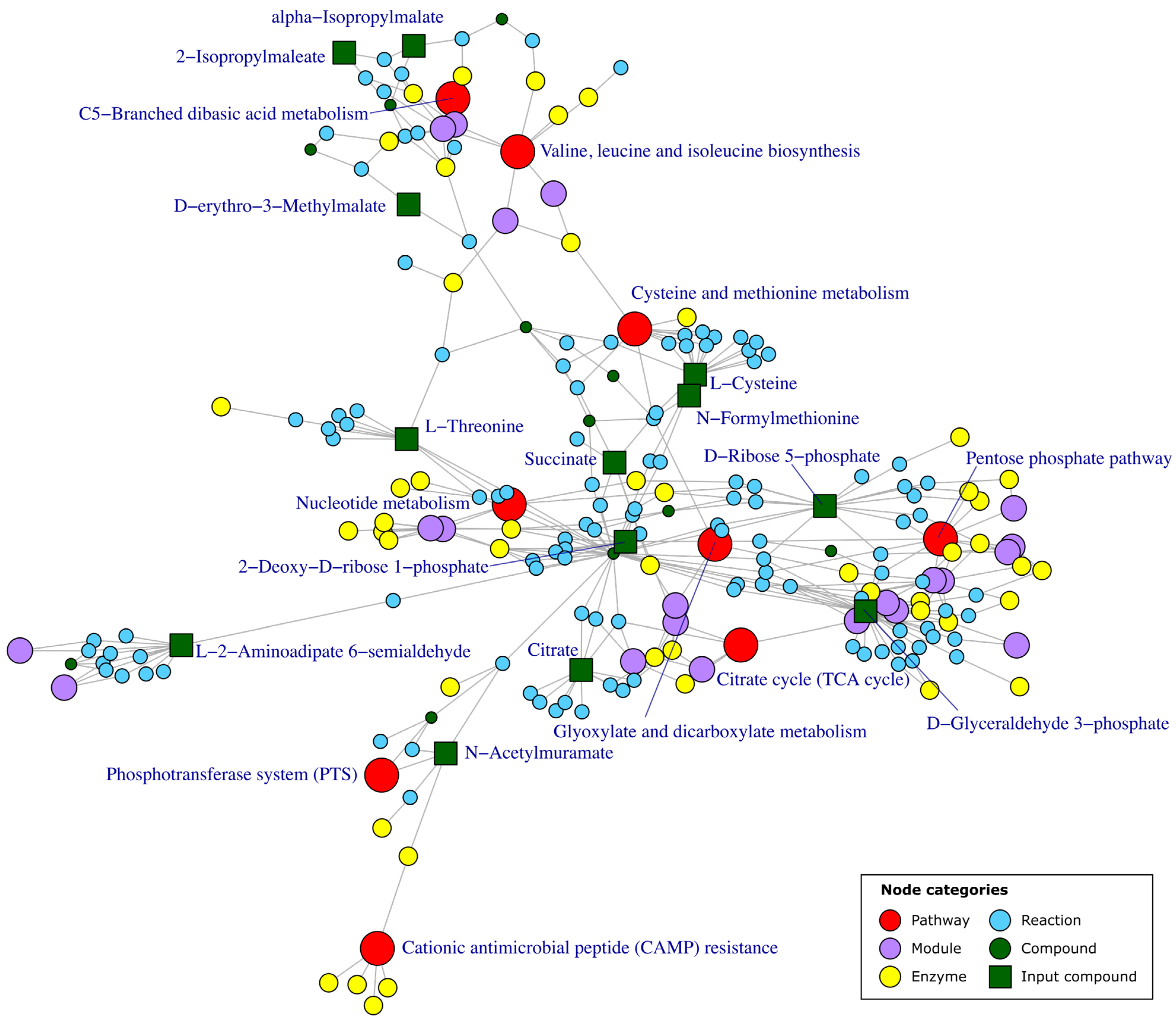
5. Metabolic Pathway Enrichment
5.1. Over-Representation Analysis (ORA)
5.2. Functional Class Scoring (FCS)
5.3. Pathway Topology (PT)
6. Generating Insight When Metabolomic Data Are Not Available: Genome-Scale Metabolic Models
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oliver, S.G.; Winson, M.K.; Kell, D.B.; Baganz, F. Systematic Functional Analysis of the Yeast Genome. Trends Biotechnol. 1998, 16, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolomics—The Link between Genotypes and Phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J. Molecular Genetic Studies of Complex Phenotypes. Transl. Res. 2012, 159, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Zulianello, L.; Canard, C.; Köhler, T.; Caille, D.; Lacroix, J.S.; Meda, P. Rhamnolipids Are Virulence Factors That Promote Early Infiltration of Primary Human Airway Epithelia by Pseudomonas aeruginosa. Infect. Immun. 2006, 74, 3134–3147. [Google Scholar] [CrossRef] [PubMed]
- Davey, M.E.; Caiazza, N.C.; O’Toole, G.A. Rhamnolipid Surfactant Production Affects Biofilm Architecture in Pseudomonas aeruginosa PAO1. J. Bacteriol. 2003, 185, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Caiazza, N.C.; Shanks, R.M.Q.; O’Toole, G.A. Rhamnolipids Modulate Swarming Motility Patterns of Pseudomonas aeruginosa. J. Bacteriol. 2005, 187, 7351–7361. [Google Scholar] [CrossRef] [PubMed]
- Sabra, W.; Kim, E.J.; Zeng, A.P. Physiological Responses of Pseudomonas aeruginosa PAO1 to Oxidative Stress in Controlled Microaerobic and Aerobic Cultures. Microbiology 2002, 148, 3195–3202. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Nair, S.; Ghosh, S. Pathogenesis in Tuberculosis: Transcriptomic Approaches to Unraveling Virulence Mechanisms and Finding New Drug Targets. FEMS Microbiol. Rev. 2012, 36, 463–485. [Google Scholar] [CrossRef] [PubMed]
- Galagan, J.E.; Minch, K.; Peterson, M.; Lyubetskaya, A.; Azizi, E.; Sweet, L.; Gomes, A.; Rustad, T.; Dolganov, G.; Glotova, I.; et al. The Mycobacterium tuberculosis Regulatory Network and Hypoxia. Nature 2013, 499, 178–183. [Google Scholar] [CrossRef]
- Raghunandanan, S.; Jose, L.; Gopinath, V.; Kumar, R.A. Comparative Label-Free Lipidomic Analysis of Mycobacterium tuberculosis during Dormancy and Reactivation. Sci. Rep. 2019, 9, 3660. [Google Scholar] [CrossRef]
- Ye, D.; Li, X.; Shen, J.; Xia, X. Microbial Metabolomics: From Novel Technologies to Diversified Applications. TrAC-Trends Anal. Chem. 2022, 148, 116540. [Google Scholar] [CrossRef]
- Emwas, A.H.; Roy, R.; McKay, R.T.; Tenori, L.; Saccenti, E.; Nagana Gowda, G.A.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. Nmr Spectroscopy for Metabolomics Research. Metabolites 2019, 9, 123. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Liang, Y.; Dunn, W.B.; Shen, H.; Kell, D.B. Comparative Evaluation of Software for Deconvolution of Metabolomics Data Based on GC-TOF-MS. TrAC-Trends Anal. Chem. 2008, 27, 215–227. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics by Gas Chromatography-Mass Spectrometry: The Combination of Targeted and Untargeted Profiling. Curr. Protoc. Mol. Biol. 2017, 114, 30.4.1–30.4.32. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.R.; Knapp, J.A.; Horn, C.K.; Stillman, S.L.; Evans, J.E.; Arfsten, D.P. Comparison of LC-MS-MS and GC-MS Analysis of Benzodiazepine Compounds Included in the Drug Demand Reduction Urinalysis Program. J. Anal. Toxicol. 2016, 40, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Gonzalez, F.J.; Idle, J.R. LC-MS-Based Metabolomics in Drug Metabolism. Drug Metab. Rev. 2007, 39, 581–597. [Google Scholar] [CrossRef]
- Johnson, C.H.; Ivanisevic, J.; Benton, H.P.; Siuzdak, G. Bioinformatics: The next Frontier of Metabolomics. Anal. Chem. 2015, 87, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Emwas, A.H.M. The Strengths and Weaknesses of NMR Spectroscopy and Mass Spectrometry with Particular Focus on Metabolomics Research. In Metabonomics: Methods and Protocols; Bjerrum, J.T., Ed.; Humana Press: New York, NY, USA, 2015; Volume 1277, pp. 161–194. [Google Scholar]
- Edison, A.S.; Colonna, M.; Gouveia, G.J.; Holderman, N.R.; Judge, M.T.; Shen, X.; Zhang, S. NMR: Unique Strengths That Enhance Modern Metabolomics Research. Anal. Chem. 2021, 93, 478–499. [Google Scholar] [CrossRef] [PubMed]
- Karaman, I.; Climaco Pinto, R.; Graça, G. Chapter 8—Metabolomics Data Preprocessing: From Raw Data to Features for Statistical Analysis. In Comprehensive Analytical Chemistry; Jaumot, J., Bedia, C., Tauler, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 82, ISBN 9780444640444. [Google Scholar]
- Alonso, A.; Marsal, S.; Julià, A. Analytical Methods in Untargeted Metabolomics: State of the Art in 2015. Front. Bioeng. Biotechnol. 2015, 3, 23. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Zhou, G.; De Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the Gap between Raw Spectra and Functional Insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A Multidimensional Spectral Processing System Based on UNIX Pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- van Beek, J.D. MatNMR: A Flexible Toolbox for Processing, Analyzing and Visualizing Magnetic Resonance Data in Matlab®. J. Magn. Reson. 2007, 187, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Yamamoto, M.; Xia, J. MetaboAnalystR 2.0: From Raw Spectra to Biological Insights. Metabolites 2019, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Altenhof, A.R.; Mason, H.; Schurko, R.W. DESPERATE: A Python Library for Processing and Denoising NMR Spectra. J. Magn. Reson. 2023, 346, 107320. [Google Scholar] [CrossRef]
- Qiu, T.; Wang, Z.; Liu, H.; Guo, D.; Qu, X. Review and Prospect: NMR Spectroscopy Denoising and Reconstruction with Low-Rank Hankel Matrices and Tensors. Magn. Reson. Chem. 2020, 59, 324–345. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.; Cramer, R.; Schuchhardt, J. Evaluation of Peak-Picking Algorithms for Protein Mass Spectrometry. In Data Mining in Proteomics: From Standards to Applications. Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2011; Volume 696, pp. 341–352. ISBN 9781607619871. [Google Scholar]
- Liu, Z.; Abbas, A.; Jing, B.Y.; Gao, X. WaVPeak: Picking NMR Peaks through Wavelet-Based Smoothing and Volume-Based Filtering. Bioinformatics 2012, 28, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A Web Server for Metabolomic Data Analysis and Interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Rocke, D.M. Baseline Correction for NMR Spectroscopic Metabolomics Data Analysis. BMC Bioinform. 2008, 9, 324. [Google Scholar] [CrossRef]
- Li, D.W.; Hansen, A.L.; Yuan, C.; Bruschweiler-Li, L.; Brüschweiler, R. DEEP Picker Is a Deep Neural Network for Accurate Deconvolution of Complex Two-Dimensional NMR Spectra. Nat. Commun. 2021, 12, 5229. [Google Scholar] [CrossRef]
- Bueschl, C.; Doppler, M.; Varga, E.; Seidl, B.; Flasch, M.; Warth, B.; Zanghellini, J. PeakBot: Machine-Learning-Based Chromatographic Peak Picking. Bioinformatics 2022, 38, 3422–3428. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, G.; Van Den Berg, F.; Andersson, C. Correlation Optimized Warping and Dynamic Time Warping as Preprocessing Methods for Chromatographic Data. J. Chemom. 2004, 18, 231–241. [Google Scholar] [CrossRef]
- Nielsen, N.P.V.; Carstensen, J.M.; Smedsgaard, J. Aligning of Single and Multiple Wavelength Chromatographic Profiles for Chemometric Data Analysis Using Correlation Optimised Warping. J. Chromatogr. A 1998, 805, 17–35. [Google Scholar] [CrossRef]
- Vu, T.N.; Laukens, K. Getting Your Peaks in Line: A Review of Alignment Methods for NMR Spectral Data. Metabolites 2013, 3, 259–276. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Guo, A.C.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Guijas, C.; Benton, H.P.; Warth, B.; Siuzdak, G. METLIN MS2 Molecular Standards Database: A Broad Chemical and Biological Resource. Nat. Methods 2020, 17, 953–954. [Google Scholar] [CrossRef] [PubMed]
- Mamede, L.; Fall, F.; Schoumacher, M.; Ledoux, A.; De Tullio, P.; Govaerts, B.; Fr, M. Comparison of Extraction Methods in Vitro Plasmodium falciparum: A 1H NMR and LC-MS Joined Approach. Biochem. Biophys. Res. Commun. 2024, 703, 149684. [Google Scholar] [CrossRef] [PubMed]
- Hrydziuszko, O.; Viant, M.R. Missing Values in Mass Spectrometry Based Metabolomics: An Undervalued Step in the Data Processing Pipeline. Metabolomics 2012, 8, S161–S174. [Google Scholar] [CrossRef]
- Barnard, J.; Meng, X.L. Applications of Multiple Imputation in Medical Studies: From AIDS to NHANES. Stat. Methods Med. Res. 1999, 8, 17–36. [Google Scholar] [CrossRef]
- Bijlsma, S.; Bobeldijk, I.; Verheij, E.R.; Ramaker, R.; Kochhar, S.; Macdonald, I.A.; Van Ommen, B.; Smilde, A.K. Large-Scale Human Metabolomics Studies: A Strategy for Data (Pre-) Processing and Validation. Anal. Chem. 2006, 78, 567–574. [Google Scholar] [CrossRef]
- Kokla, M.; Virtanen, J.; Kolehmainen, M.; Paananen, J.; Hanhineva, K. Random Forest-Based Imputation Outperforms Other Methods for Imputing LC-MS Metabolomics Data: A Comparative Study. BMC Bioinform. 2019, 20, 492. [Google Scholar] [CrossRef]
- Hong, S.; Lynn, H.S. Accuracy of Random-Forest-Based Imputation of Missing Data in the Presence of Non-Normality, Non-Linearity, and Interaction. BMC Med. Res. Methodol. 2020, 20, 99. [Google Scholar] [CrossRef]
- Hu, L.Y.; Huang, M.W.; Ke, S.W.; Tsai, C.F. The Distance Function Effect on K-Nearest Neighbor Classification for Medical Datasets. Springerplus 2016, 5, 1304. [Google Scholar] [CrossRef]
- Kim, H.; Golub, G.H.; Park, H. Missing Value Estimation for DNA Microarray Gene Expression Data: Local Least Squares Imputation. Bioinformatics 2005, 21, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Troyanskaya, O.; Cantor, M.; Sherlock, G.; Brown, P.; Hastie, T.; Tibshirani, R.; Botstein, D.; Altman, R.B. Missing Value Estimation Methods for DNA Microarrays. Bioinformatics 2001, 17, 520–525. [Google Scholar] [CrossRef]
- Oba, S.; Sato, M.A.; Takemasa, I.; Monden, M.; Matsubara, K.I.; Ishii, S. A Bayesian Missing Value Estimation Method for Gene Expression Profile Data. Bioinformatics 2003, 19, 2088–2096. [Google Scholar] [CrossRef]
- Ilin, A.; Raiko, T. Practical Approaches to Principal Component Analysis in the Presence of Missing Values. J. Mach. Learn. Res. 2010, 11, 1957–2000. [Google Scholar]
- Leek, J.T.; Scharpf, R.B.; Bravo, H.C.; Simcha, D.; Langmead, B.; Johnson, W.E.; Geman, D.; Baggerly, K.; Irizarry, R.A. Tackling the Widespread and Critical Impact of Batch Effects in High-Throughput Data. Nat. Rev. Genet. 2010, 11, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-Seq: An Assessment of Technical Reproducibility and Comparison with Gene Expression Arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef]
- Karpievitch, Y.V.; Dabney, A.R.; Smith, R.D. Normalization and Missing Value Imputation for Label-Free LC-MS Analysis. BMC Bioinform. 2012, 13, S5. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate Normalization of Real-Time Quantitative RT-PCR Data by Geometric Averaging of Multiple Internal Control Genes. Rock. Mech. Rock. Eng. 2002, 3, research0034.1. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Mann, M. A Proteomics Approach to the Protein Normalization Problem: Selection of Unvarying Proteins for MS-Based Proteomics and Western Blotting. J. Proteome Res. 2016, 15, 2321–2326. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, L. Sample Normalization Methods in Quantitative Metabolomics. J. Chromatogr. A 2016, 1430, 80–95. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, P.; Lv, M.; Guo, H.; Huang, Y.; Zhang, Z.; Xu, F. Influences of Normalization Method on Biomarker Discovery in Gas Chromatography-Mass Spectrometry-Based Untargeted Metabolomics: What Should Be Considered? Anal. Chem. 2017, 89, 5342–5348. [Google Scholar] [CrossRef]
- Temmerman, L.; De Livera, A.M.; Browne, J.B.; Sheedy, J.R.; Callahan, D.L.; Nahid, A.; De Souza, D.P.; Schoofs, L.; Tull, D.L.; McConville, M.J.; et al. Cross-Platform Urine Metabolomics of Experimental Hyperglycemia in Type 2 Diabetes. J. Diabetes Metab. 2012, 6, 1–7. [Google Scholar] [CrossRef]
- De Livera, A.M.; Sysi-Aho, M.; Jacob, L.; Gagnon-Bartsch, J.A.; Castillo, S.; Simpson, J.A.; Speed, T.P. Statistical Methods for Handling Unwanted Variation in Metabolomics Data. Anal. Chem. 2015, 87, 3606–3615. [Google Scholar] [CrossRef] [PubMed]
- Edmands, W.M.B.; Ferrari, P.; Scalbert, A. Normalization to Specific Gravity Prior to Analysis Improves Information Recovery from High Resolution Mass Spectrometry Metabolomic Profiles of Human Urine. Anal. Chem. 2014, 86, 10925–10931. [Google Scholar] [CrossRef]
- Marcinowska, R.; Trygg, J.; Wolf-Watz, H.; Mortiz, T.; Surowiec, I. Optimization of a Sample Preparation Method for the Metabolomic Analysis of Clinically Relevant Bacteria. J. Microbiol. Methods 2011, 87, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shen, G.; Zhang, R.; He, J.; Zhang, Y.; Xu, J.; Yang, W.; Chen, X.; Song, Y.; Abliz, Z. Combination of Injection Volume Calibration by Creatinine and MS Signals’ Normalization to Overcome Urine Variability in LC-MS-Based Metabolomics Studies. Anal. Chem. 2013, 85, 7659–7665. [Google Scholar] [CrossRef]
- De Livera, A.M.; Dias, D.A.; De Souza, D.; Rupasinghe, T.; Pyke, J.; Tull, D.; Roessner, U.; McConville, M.; Speed, T.P. Normalizing and Integrating Metabolomics Data. Anal. Chem. 2012, 84, 10768–10776. [Google Scholar] [CrossRef]
- Antonelli, J.; Claggett, B.L.; Henglin, M.; Kim, A.; Ovsak, G.; Kim, N.; Deng, K.; Rao, K.; Tyagi, O.; Watrous, J.D.; et al. Statistical Workflow for Feature Selection in Human Metabolomics Data. Metabolites 2019, 9, 143. [Google Scholar] [CrossRef]
- van den Berg, R.A.; Hoefsloot, H.C.J.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, Scaling, and Transformations: Improving the Biological Information Content of Metabolomics Data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef]
- De Livera, A.M.; Olshansky, G.; Simpson, J.A.; Creek, D.J. NormalizeMets: Assessing, Selecting and Implementing Statistical Methods for Normalizing Metabolomics Data. Metabolomics 2018, 14, 54. [Google Scholar] [CrossRef]
- Sysi-Aho, M.; Katajamaa, M.; Yetukuri, L.; Orešič, M. Normalization Method for Metabolomics Data Using Optimal Selection of Multiple Internal Standards. BMC Bioinform. 2007, 8, 93. [Google Scholar] [CrossRef] [PubMed]
- Grocholska, P.; Bachor, R. Trends in the Hydrogen−deuterium Exchange at the Carbon Centers. Preparation of Internal Standards for Quantitative Analysis by Lc-Ms. Molecules 2021, 26, 2989. [Google Scholar] [CrossRef] [PubMed]
- Gullberg, J.; Jonsson, P.; Nordström, A.; Sjöström, M.; Moritz, T. Design of Experiments: An Efficient Strategy to Identify Factors Influencing Extraction and Derivatization of Arabidopsis thaliana Samples in Metabolomic Studies with Gas Chromatography/Mass Spectrometry. Anal. Biochem. 2004, 331, 283–295. [Google Scholar] [CrossRef]
- Liu, R.H.; Lin, D.L.; Chang, W.-T.; Liu, C.; Tsay, W.-I.; Li, J.-H.; Kuo, T.-L. Isotopically Labeled Analogues for Drug Quantitation. Anal. Chem. 2002, 74, 618A–626A. [Google Scholar] [CrossRef] [PubMed]
- Redestig, H.; Fukushima, A.; Stenlund, H.; Moritz, T.; Arita, M.; Saito, K.; Kusano, M. Compensation for Systematic Cross-Contribution Improves Normalization of Mass Spectrometry Based Metabolomics Data. Anal. Chem. 2009, 81, 7974–7980. [Google Scholar] [CrossRef]
- Gagnon-Bartsch, J.A.; Speed, T.P. Using Control Genes to Correct for Unwanted Variation in Microarray Data. Biostatistics 2012, 13, 539–552. [Google Scholar] [CrossRef]
- Santamaria, G.; Liao, C.; Lindberg, C.; Chen, Y.; Wang, Z.; Rhee, K.; Pinto, F.; Yan, J.; Xavier, J.B. Evolution and Regulation of Microbial Secondary Metabolism. eLife 2022, 11, e76119. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-Mcintyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for Large-Scale Metabolic Profiling of Serum and Plasma Using Gas Chromatography and Liquid Chromatography Coupled to Mass Spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef] [PubMed]
- Sangster, T.; Major, H.; Plumb, R.; Wilson, A.J.; Wilson, I.D. A Pragmatic and Readily Implemented Quality Control Strategy for HPLC-MS and GC-MS-Based Metabonomic Analysis. Analyst 2006, 131, 1075–1078. [Google Scholar] [CrossRef] [PubMed]
- Gika, H.G.; Theodoridis, G.A.; Wingate, J.E.; Wilson, I.D. Within-Day Reproducibility of an HPLC-MS-Based Method for Metabonomic Analysis: Application to Human Urine. J. Proteome Res. 2007, 6, 3291–3303. [Google Scholar] [CrossRef] [PubMed]
- Broadhurst, D.; Goodacre, R.; Reinke, S.N.; Kuligowski, J.; Wilson, I.D.; Lewis, M.R.; Dunn, W.B. Guidelines and Considerations for the Use of System Suitability and Quality Control Samples in Mass Spectrometry Assays Applied in Untargeted Clinical Metabolomic Studies. Metabolomics 2018, 14, 72. [Google Scholar] [CrossRef]
- Schiffman, C.; Petrick, L.; Perttula, K.; Yano, Y.; Carlsson, H.; Whitehead, T.; Metayer, C.; Hayes, J.; Rappaport, S.; Dudoit, S. Filtering Procedures for Untargeted Lc-Ms Metabolomics Data. BMC Bioinform. 2019, 20, 334. [Google Scholar] [CrossRef] [PubMed]
- Begley, P.; Francis-McIntyre, S.; Dunn, W.B.; Broadhurst, D.I.; Halsall, A.; Tseng, A.; Knowles, J.; HUSERMET Consortium; Goodacre, R.; Kell, D.B. Development and Performance of a Gas Chromatography-Time-of-Flight Mass Spectrometry Analysis for Large-Scale Nontargeted Metabolomic Studies of Human Serum. Anal. Chem. 2009, 81, 7038–7046. [Google Scholar] [CrossRef]
- Zelena, E.; Dunn, W.B.; Broadhurst, D.; Francis-McIntyre, S.; Carroll, K.M.; Begley, P.; O’Hagan, S.; Knowles, J.D.; Halsall, A.; HUSERMET Consortium; et al. Development of a Robust and Repeatable UPLC-MS Method for the Long-Term Metabolomic Study of Human Serum. Anal. Chem. 2009, 81, 1357–1364. [Google Scholar] [CrossRef]
- Rousseeuw, P.J. Silhouettes: A Graphical Aid to the Interpretation and Validation of Cluster Analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- De Livera, A.M.; Olshansky, M.; Speed, T.P. Statistical Analysis of Metabolomics Data. In Metabolomics Tools for Natural Product Discovery; Humana Press: Totowa, NJ, USA, 2013; pp. 291–307. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. Author Correction: SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 352. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Sokal, R.R.; Rohlf, F.J. Biometry. The Principles and Practice of Statistics in Biological Research, 3rd ed.; W. H. Freeman and Company: New York, NY, USA, 1995. [Google Scholar]
- Bewick, V.; Cheek, L.; Ball, J. Statistics Review 14: Logistic Regression. Crit. Care 2005, 9, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Broadhurst, D.I.; Kell, D.B. Statistical Strategies for Avoiding False Discoveries in Metabolomics and Related Experiments. Metabolomics 2006, 2, 171–196. [Google Scholar] [CrossRef]
- Saccenti, E.; Hoefsloot, H.C.J.; Smilde, A.K.; Westerhuis, J.A.; Hendriks, M.M.W.B. Reflections on Univariate and Multivariate Analysis of Metabolomics Data. Metabolomics 2014, 10, 361–374. [Google Scholar] [CrossRef]
- Worley, B.; Powers, R. Multivariate Analysis in Metabolomics. Curr. Metabolomics 2013, 1, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Şenbabaoğlu, Y.; Michailidis, G.; Li, J.Z. Critical Limitations of Consensus Clustering in Class Discovery. Sci. Rep. 2014, 4, 6207. [Google Scholar] [CrossRef]
- John, C.R.; David, W.; Russ, D.; Goldmann, K.; Ehrenstein, M.; Pitzalis, C.; Lewis, M.; Barnes, M. M3C: Monte Carlo Reference-Based Consensus Clustering. Sci. Rep. 2020, 10, 1816. [Google Scholar] [CrossRef] [PubMed]
- Boyle, K.E.; Monaco, H.T.; Deforet, M.; Yan, J.; Wang, Z.; Rhee, K.; Xavier, J.B. Metabolism and the Evolution of Social Behavior. Mol. Biol. Evol. 2017, 34, 2367–2379. [Google Scholar] [CrossRef]
- van der Maaten, L.; Hinton, G. Visualizing Data Using T-SNE. J. Mach. Learn. Res. 2008, 219, 2579–2605. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A Class Discovery Tool with Confidence Assessments and Item Tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Wold, S.; Sjöström, M.; Eriksson, L. PLS-Regression: A Basic Tool of Chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar] [CrossRef]
- Trygg, J.; Wold, S. Orthogonal Projections to Latent Structures (O-PLS). J. Chemom. 2002, 16, 119–128. [Google Scholar] [CrossRef]
- Mehmood, T.; Liland, K.H.; Snipen, L.; Sæbø, S. A Review of Variable Selection Methods in Partial Least Squares Regression. Chemom. Intell. Lab. Syst. 2012, 118, 62–69. [Google Scholar] [CrossRef]
- Rizvi, A.; Shankar, A.; Chatterjee, A.; More, T.H.; Bose, T.; Dutta, A.; Balakrishnan, K.; Madugulla, L.; Rapole, S.; Mande, S.S.; et al. Rewiring of Metabolic Network in Mycobacterium tuberculosis during Adaptation to Different Stresses. Front. Microbiol. 2019, 10, 2417. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Liu, Z.; Zhong, S.; Li, R.; Xia, H.; Jie, Z.; Wen, B.; Chen, X.; Yan, W.; Fan, Y.; et al. Integrated Metabolomics and Metagenomics Analysis of Plasma and Urine Identified Microbial Metabolites Associated with Coronary Heart Disease. Sci. Rep. 2016, 6, 22525. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chi, Y.H.; Niu, M.; Zhu, Y.; Zhao, Y.L.; Chen, Z.; Wang, J.B.; Zhang, C.E.; Li, J.Y.; Wang, L.F.; et al. Metabolomics Coupled with Multivariate Data and Pathway Analysis on Potential Biomarkers in Cholestasis and Intervention Effect of Paeonia lactiflora Pall. Front. Pharmacol. 2016, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Palmer, D.S.; Nigsch, F.; Mitchell, J.B. Simultaneous Feature Selection and Parameter Optimisation Using an Artificial Ant Colony: Case Study of Melting Point Prediction. Chem. Cent. J. 2008, 2, 21. [Google Scholar] [CrossRef] [PubMed]
- Szymańska, E.; Saccenti, E.; Smilde, A.K.; Westerhuis, J.A. Double-Check: Validation of Diagnostic Statistics for PLS-DA Models in Metabolomics Studies. Metabolomics 2012, 8, 3–16. [Google Scholar] [CrossRef]
- Breiman, L.; Friedman, J.H.; Olshen, R.A.; Stone, C.J. Classification and Regression Trees; Chapman & Hall: London, UK; CRC: Boca Raton, FL, USA, 1984; ISBN 0412048418. [Google Scholar]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Breiman, L. Bagging Predictors. Mach. Learn. 1996, 24, 123–140. [Google Scholar] [CrossRef]
- Amit, Y.; Geman, D. Shape Quantization and Recognition with Randomized Trees. Neural Comput. 1997, 9, 1545–1588. [Google Scholar] [CrossRef]
- Devroye, L.; Lugosi, G. Consistency of Random Forests and Other Averaging Classifiers. J. Mach. Learn. Res. 2008, 9, 2015–2033. [Google Scholar]
- Venables, W.N.; Ripley, B.D. Modern Applied Statistics with S, 4th ed.; Springer: New York, NY, USA, 2002. [Google Scholar]
- Thévenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the Human Adult Urinary Metabolome Variations with Age, Body Mass Index, and Gender by Implementing a Comprehensive Workflow for Univariate and OPLS Statistical Analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef] [PubMed]
- Mevik, B.-H.; Wehrens, R. The Pls Package: Principal Component and Partial Least Squares Regression in R. J. Stat. Softw. 2007, 18. [Google Scholar] [CrossRef]
- BiRG—Wright State University Pyopls. Available online: https://pypi.org/project/pyopls/ (accessed on 15 April 2024).
- Kuhn, M. Building Predictive Models in R Using the Caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and Regression by RandomForest. R News 2002, 2, 18–22. [Google Scholar] [CrossRef]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten Years of Pathway Analysis: Current Approaches and Outstanding Challenges. PLoS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef]
- Goeman, J.J.; Bühlmann, P. Analyzing Gene Expression Data in Terms of Gene Sets: Methodological Issues. Bioinformatics 2007, 23, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Maleki, F.; Ovens, K.; Hogan, D.J.; Kusalik, A.J. Gene Set Analysis: Challenges, Opportunities, and Future Research. Front. Genet. 2020, 11, 654. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-Responsive Genes Involved in Oxidative Phosphorylation Are Coordinately Downregulated in Human Diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Li, S.; Xia, J. MetaboAnalystR 3.0: Toward an Optimized Workflow for Global Metabolomics. Metabolites 2020, 10, 186. [Google Scholar] [CrossRef] [PubMed]
- Tomfohr, J.; Lu, J.; Kepler, T.B. Pathway Level Analysis of Gene Expression Using Singular Value Decomposition. BMC Bioinform. 2005, 6, 225. [Google Scholar] [CrossRef] [PubMed]
- McLuskey, K.; Wandy, J.; Vincent, I.; van der Hooft, J.J.J.; Rogers, S.; Burgess, K.; Daly, R. Ranking Metabolite Sets by Their Activity Levels. Metabolites 2021, 11, 103. [Google Scholar] [CrossRef] [PubMed]
- Shojaie, A.; Michailidis, G. Analysis of Gene Sets Based on the Underlying Regulatory Network. J. Comput. Biol. 2009, 16, 407–426. [Google Scholar] [CrossRef] [PubMed]
- Hellstern, M.; Ma, J.; Yue, K.; Shojaie, A. Netgsa: Fast Computation and Interactive Visualization for Topology-Based Pathway Enrichment Analysis. PLoS Comput. Biol. 2021, 17, e1008979. [Google Scholar] [CrossRef] [PubMed]
- Picart-Armada, S.; Fernández-Albert, F.; Vinaixa, M.; Yanes, O.; Perera-Lluna, A. FELLA: An R Package to Enrich Metabolomics Data. BMC Bioinform. 2018, 19, 538–546. [Google Scholar] [CrossRef]
- Jacob, L.; Neuvial, P.; Dudoit, S. More Power via Graph-Structured Tests for Differential Expression of Gene Networks. Ann. Appl. Stat. 2012, 6, 561–600. [Google Scholar] [CrossRef]
- Santamaria, G.; Ruiz-Rodríguez, P.; Renau-Mínguez, C.; Pinto, F.R.; Coscollá, M. In Silico Exploration of Mycobacterium tuberculosis Metabolic Networks Shows Host-Associated Convergent Fluxomic Phenotypes. Biomolecules 2022, 12, 376. [Google Scholar] [CrossRef]
- Baart, G.J.; Martens, D.E. Genome-Scale Metabolic Models: Reconstruction and Analysis. In Neisseria meningiditis: Advanced Methods and Protocols; Christodoulides, M., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 107–126. [Google Scholar]
- Bartell, J.A.; Blazier, A.S.; Yen, P.; Thøgersen, J.C.; Jelsbak, L.; Goldberg, J.B.; Papin, J.A. Reconstruction of the Metabolic Network of Pseudomonas aeruginosa to Interrogate Virulence Factor Synthesis. Nat. Commun. 2017, 8, 14631. [Google Scholar] [CrossRef]
- Edwards, J.S.; Palsson, B.O. Systems Properties of the Haemophilus Influenzae Rd Metabolic Genbotype. Mol. Biol. 1999, 274, 17410–17416. [Google Scholar]
- Karp, P.D.; Weaver, D.; Latendresse, M. How Accurate Is Automated Gap Filling of Metabolic Models? BMC Syst. Biol. 2018, 12, 73. [Google Scholar] [CrossRef] [PubMed]
- Palsson, B.Ø. Systems Biology: Properties of Reconstructed Networks; Cambridge University Press: Cambridge, UK, 2006; ISBN 9780521859035. [Google Scholar]
- Varma, A.; Palsson, B.Ø. Metabolic Flux Balancing: Basic Concepts, Scientific and Practical Use. Nat. Biotechnol. 1994, 12, 994–998. [Google Scholar] [CrossRef]
- Feist, A.M.; Palsson, B.Ø. The Biomass Objective Function. Curr. Opin. Microbiol. 2010, 13, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, R.; Kuepfer, L.; Sauer, U. Systematic Evaluation of Objective Functions for Predicting Intracellular Fluxes in Escherichia Coli. Mol. Syst. Biol. 2007, 3, 119. [Google Scholar] [CrossRef] [PubMed]
- Piddington, D.L.; Kashkouli, A.; Buchmeier, N.A. Growth of Mycobacterium tuberculosis in a Defined Medium Is Very Restricted by Acid pH and Mg2+ Levels Mycobacterium tuberculosis Grows within the Phagocytic Vacuoles of Macrophages, Where It Encounters a Moderately Acidic and Possibly Nutrient-Restricted. Infect. Immun. 2000, 68, 4518–4522. [Google Scholar] [CrossRef] [PubMed]
- Boyle, K.E.; Monaco, H.; van Ditmarsch, D.; Deforet, M.; Xavier, J.B. Integration of Metabolic and Quorum Sensing Signals Governing the Decision to Cooperate in a Bacterial Social Trait. PLoS Comput. Biol. 2015, 11, e1004279. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.A.; Dyson, B.C.; Vass, L.; Johnson, G.N.; Schwartz, J.M. Flux Sampling Is a Powerful Tool to Study Metabolism under Changing Environmental Conditions. npj Syst. Biol. Appl. 2019, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Wiback, S.J.; Famili, I.; Greenberg, H.J.; Palsson, B. Monte Carlo Sampling Can Be Used to Determine the Size and Shape of the Steady-State Flux Space. J. Theor. Biol. 2004, 228, 437–447. [Google Scholar] [CrossRef]
- Wishart, D.S. Emerging Applications of Metabolomics in Drug Discovery and Precision Medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Øyås, O.; Borrell, S.; Trauner, A.; Zimmermann, M.; Feldmann, J.; Liphardt, T.; Gagneux, S.; Stelling, J.; Sauer, U.; Zampieri, M. Model-Based Integration of Genomics and Metabolomics Reveals SNP Functionality in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2020, 117, 8494–8502. [Google Scholar] [CrossRef]
- Gustafsson, J.; Anton, M.; Roshanzamir, F.; Jörnsten, R.; Kerkhiven, E.J.; Robinson, J.; Nielsen, J. Generation and Analysis of Context-Specific Genome-Scale Metabolic Models Derived from Single-Cell RNA-Seq Data. Proc. Natl. Acad. Sci. USA 2023, 120, e2217868120. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scaling Methods | Internal Standard/Nonchanging Metabolite-Based | QC Sample-Based | |
|---|---|---|---|
| Scaling | Statistical Modeling | ||
| mean median sum | IS RI | NOMIS CCMN RUV-2 | QC-RLSC |
| Quantitative Factors | Categorical Factors | |||
|---|---|---|---|---|
| Parametric | Non-Parametric | |||
| 2 Classes | >2 Classes | 2 Classes | >2 Classes | |
| Simple linear regression | Student’s t test Logistic regression (Wald test) | ANOVA | Mann–Whitney U test | Kruskal–Wallis test |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santamaria, G.; Pinto, F.R. Bioinformatic Analysis of Metabolomic Data: From Raw Spectra to Biological Insight. BioChem 2024, 4, 90-114. https://doi.org/10.3390/biochem4020005
Santamaria G, Pinto FR. Bioinformatic Analysis of Metabolomic Data: From Raw Spectra to Biological Insight. BioChem. 2024; 4(2):90-114. https://doi.org/10.3390/biochem4020005
Chicago/Turabian StyleSantamaria, Guillem, and Francisco R. Pinto. 2024. "Bioinformatic Analysis of Metabolomic Data: From Raw Spectra to Biological Insight" BioChem 4, no. 2: 90-114. https://doi.org/10.3390/biochem4020005
APA StyleSantamaria, G., & Pinto, F. R. (2024). Bioinformatic Analysis of Metabolomic Data: From Raw Spectra to Biological Insight. BioChem, 4(2), 90-114. https://doi.org/10.3390/biochem4020005