
The Influence of Endogenous Derivatives on the Self-Assembly of Carbonized Polymer Dots

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Synthesis
2.2.1. Solvothermal Synthesis of CPDs (TM-EtOH)
2.2.2. Hydrothermal Synthesis of CPDs (TM-H2O)
2.2.3. High Pressure Reactor Synthesis of CPDs (TM-HP)
2.2.4. Airflow-Assisted Fusion Polymerization Synthesis of CPDs (TM)
2.3. Characterization
2.3.1. Ultra-High Performance Liquid Chromatography/Quadrupole Time-of-Flight Mass Spectrometer (UPLC-QTOF-MS)
2.3.2. Nuclear Magnetic Resonance (NMR)
2.3.3. Optical Spectroscopy
2.3.4. IR Spectroscopy
2.3.5. Transmission Electron Microscopy (TEM)
2.3.6. Photoluminescence Quantum Yield (QY)
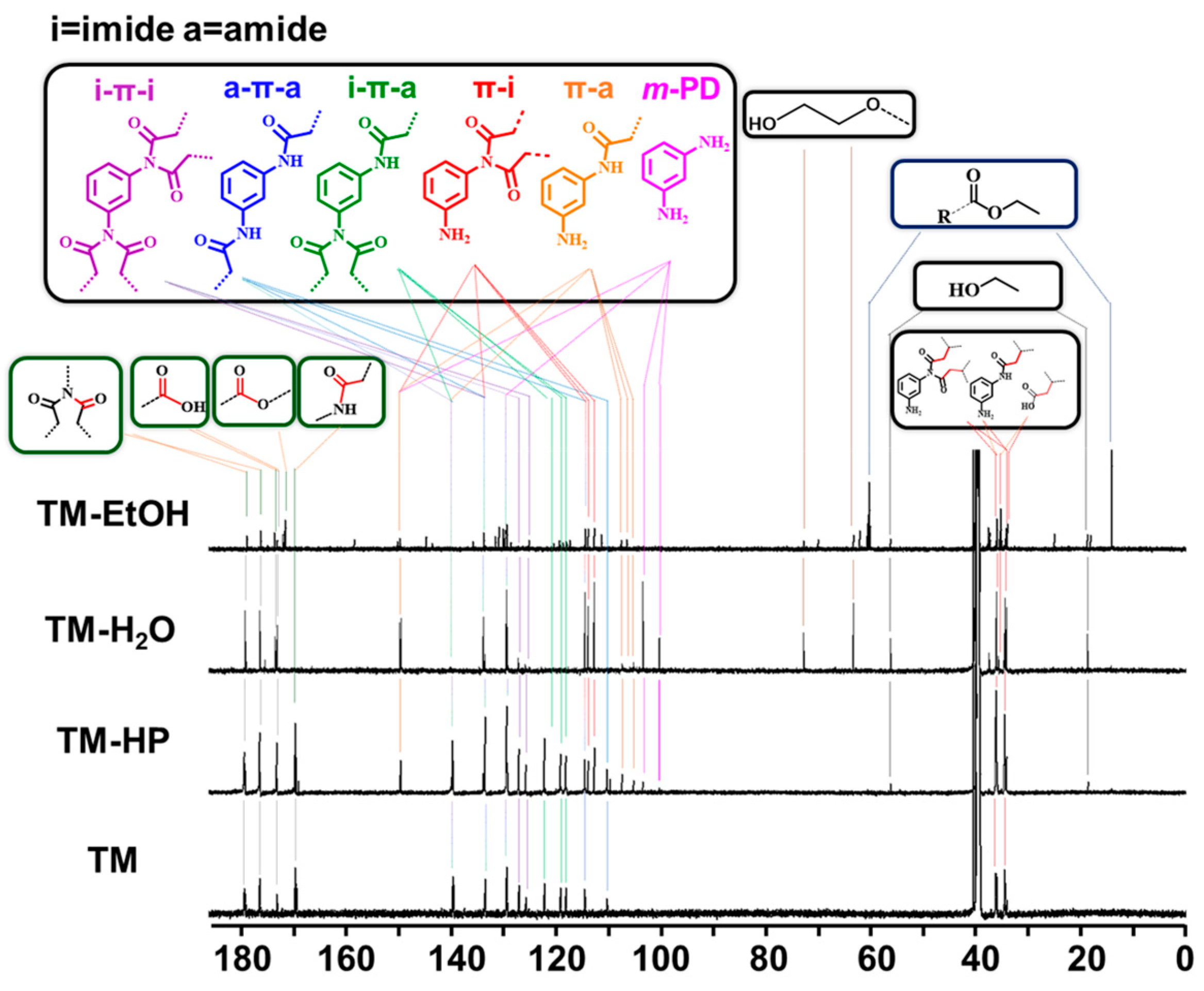
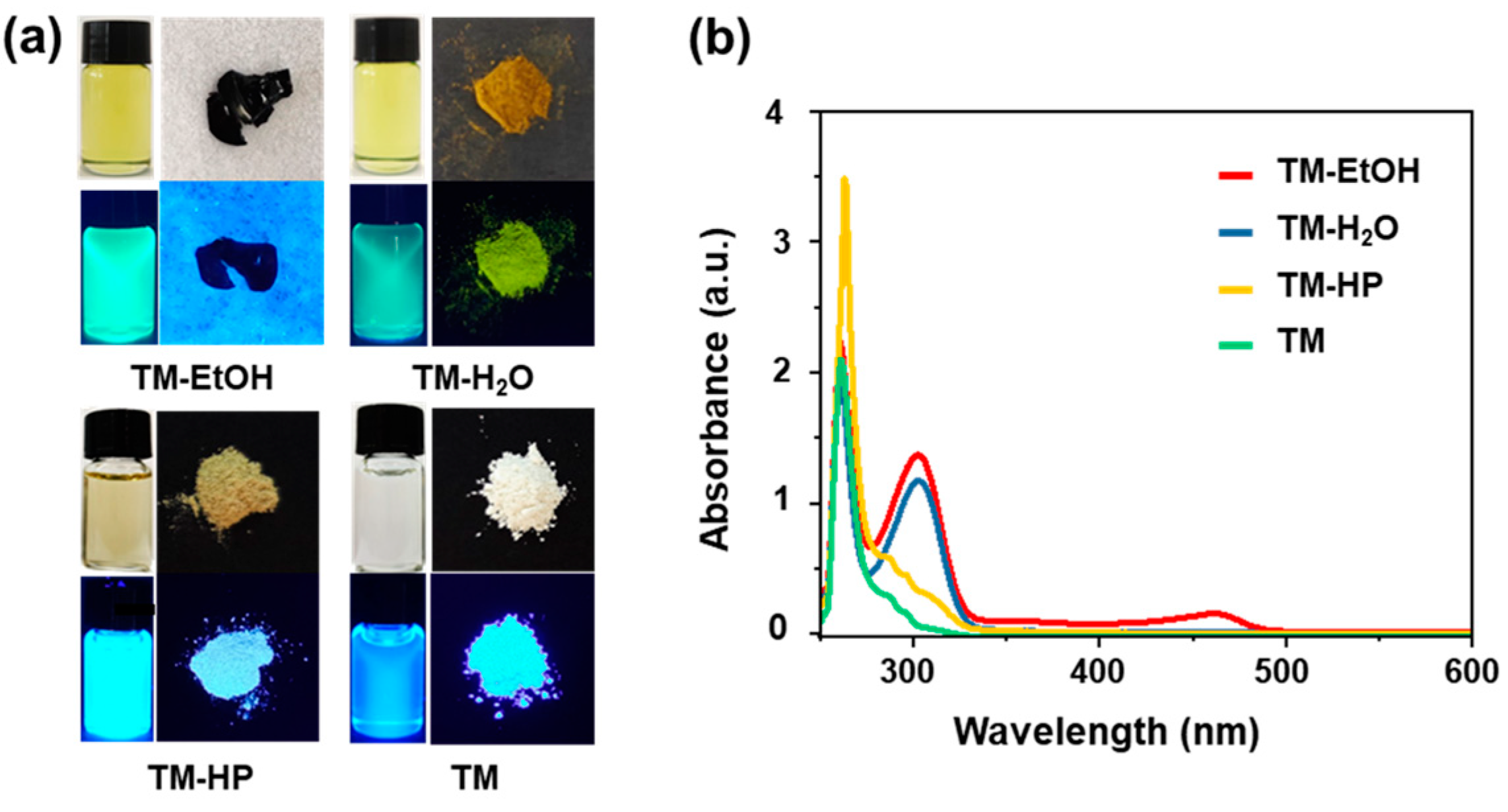
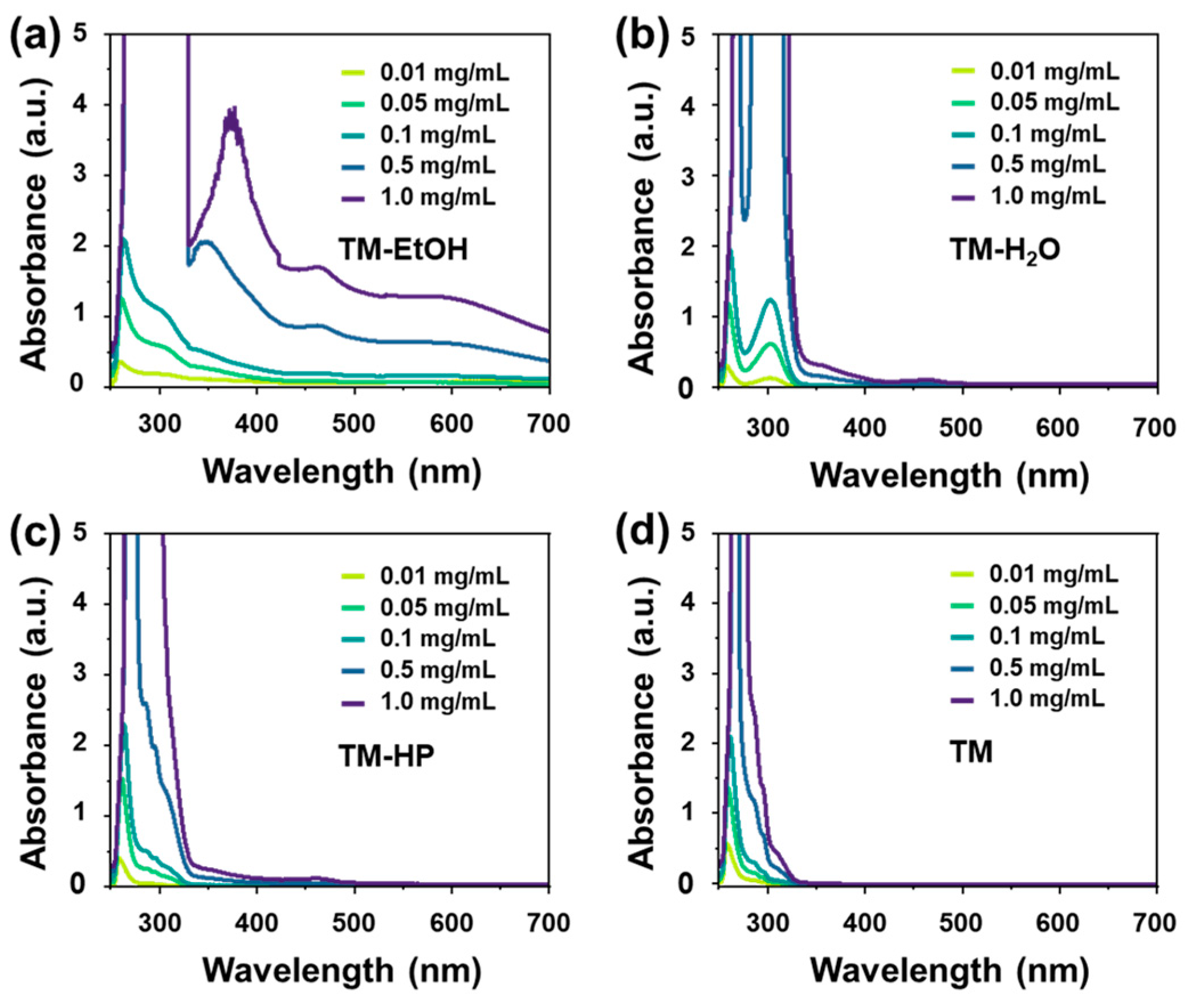
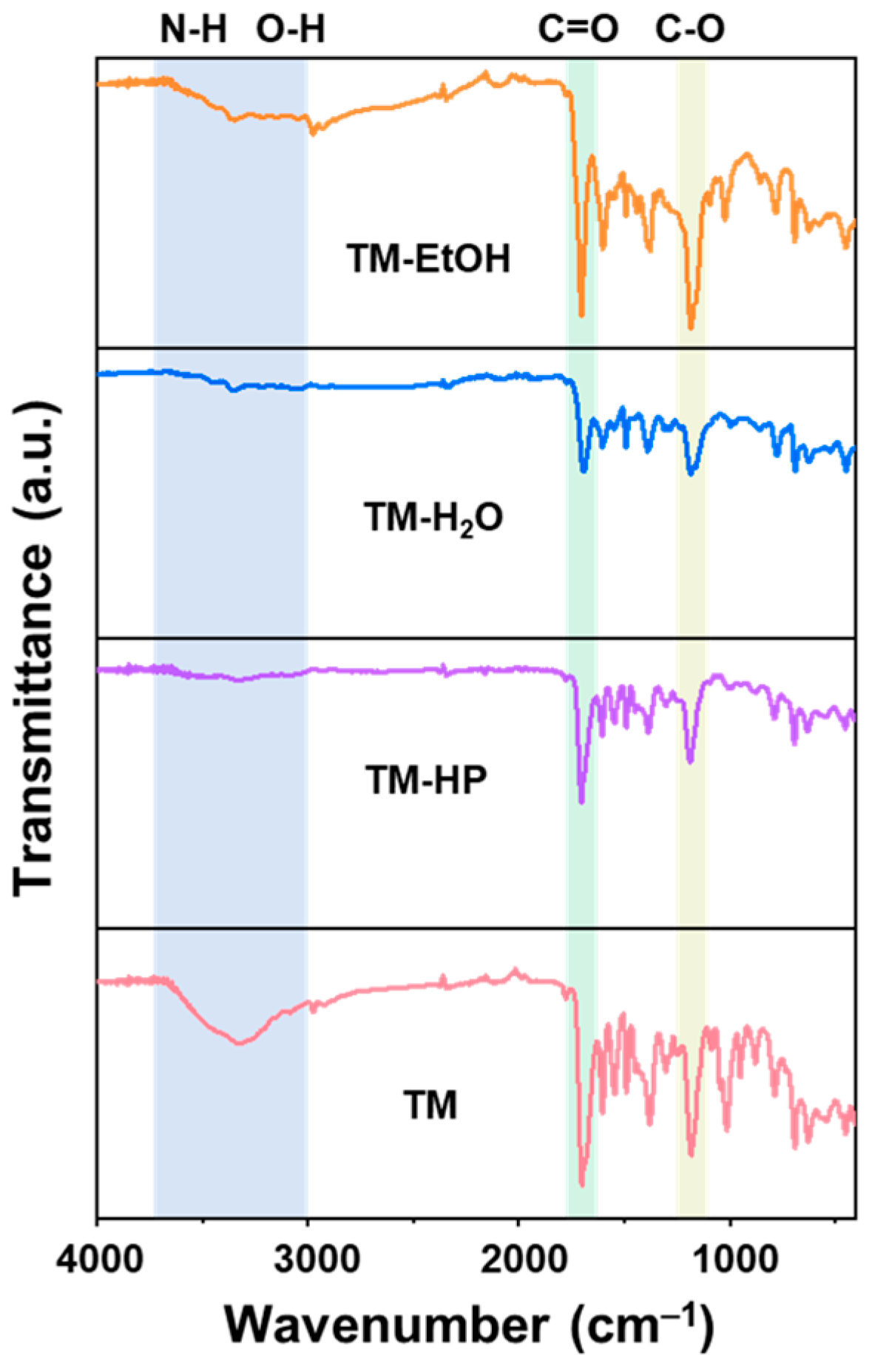
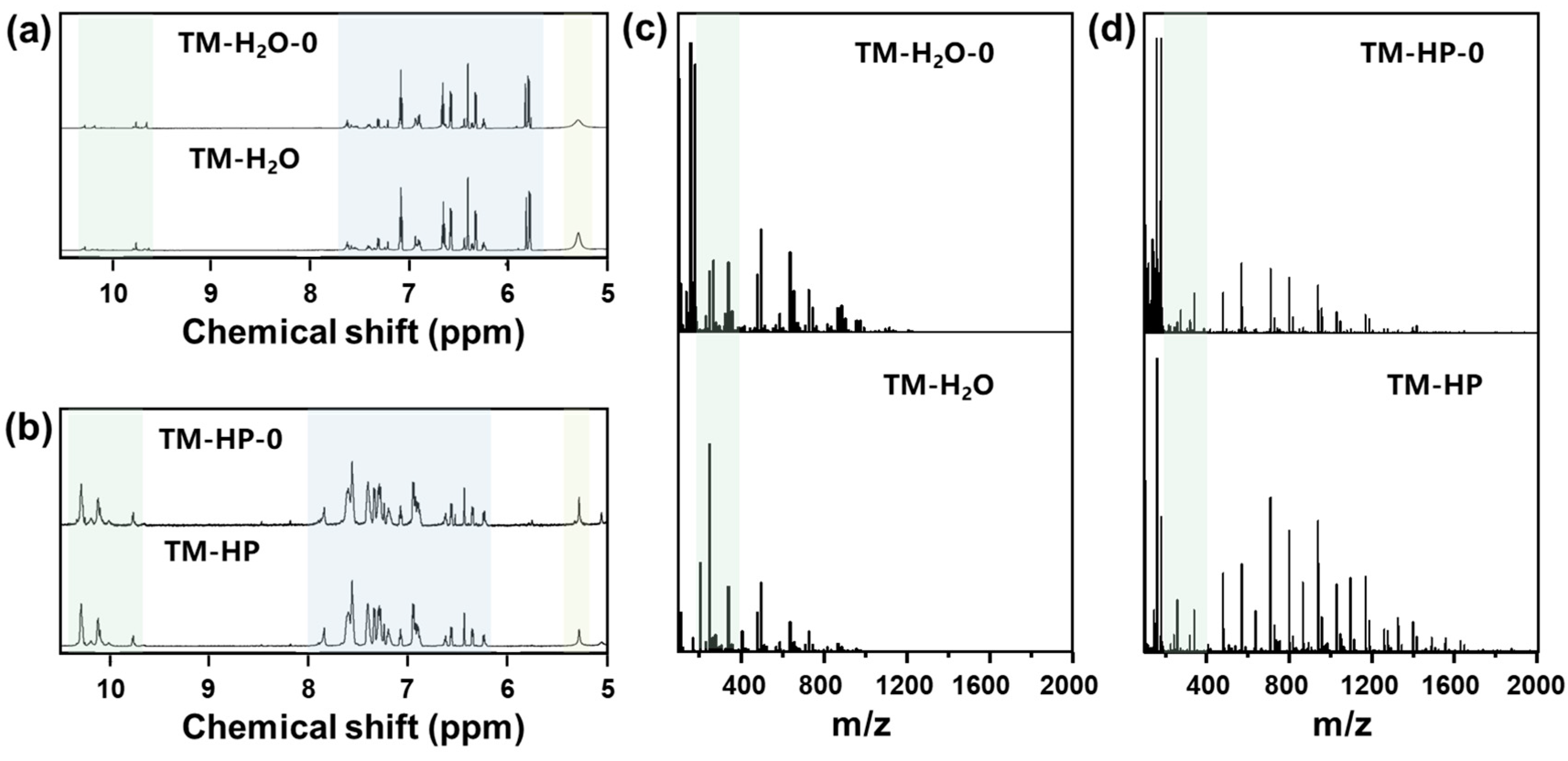
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, Y.; Qin, Y.; Wang, F.; Zhang, H.; Huangfu, C.; Shi, Y.; Chen, X.; Wang, Z.; Tian, W.; Feng, L. The Synthesis of Functionalized Carbonized Polymer Dots via Reversible Assembly of Oligomers for Anti-Counterfeiting, Catalysis, and Gas storage. Adv. Sci. 2024, 11, 2405043. [Google Scholar]
- Ru, Y.; Sui, L.; Song, H.; Liu, X.; Tang, Z.; Zang, S.Q.; Yang, B.; Lu, S. Rational Design of Multicolor-Emitting Chiral Carbonized Polymer Dots for Full-Color and White Circularly Polarized Luminescence. Angew. Chem. Int. Ed. 2021, 60, 14091–14099. [Google Scholar]
- Shi, M.; Gao, Q.; Rao, J.; Lv, Z.; Chen, M.; Chen, G.; Bian, J.; Ren, J.; Lü, B.; Peng, F. Confinement-Modulated Clusterization-Triggered Time-Dependent Phosphorescence Color from Xylan-Carbonized Polymer Dots. J. Am. Chem. Soc. 2023, 146, 1294–1304. [Google Scholar] [PubMed]
- Tao, S.; Zhou, C.; Kang, C.; Zhu, S.; Feng, T.; Zhang, S.; Ding, Z.; Zheng, C.; Xia, C.; Yang, B. Confined-domain crosslink-enhanced emission effect in carbonized polymer dots. Light Sci. Appl. 2022, 11, 56. [Google Scholar]
- Vallan, L.; Urriolabeitia, E.P.; Ruipérez, F.; Matxain, J.M.; Canton-Vitoria, R.; Tagmatarchis, N.; Benito, A.M.; Maser, W.K. Supramolecular-Enhanced Charge Transfer within Entangled Polyamide Chains as the Origin of the Universal Blue Fluorescence of Polymer Carbon Dots. J. Am. Chem. Soc. 2018, 140, 12862–12869. [Google Scholar]
- Xia, C.; Zhong, J.; Han, X.; Zhu, S.; Li, Y.; Liu, H.; Yang, B. The Formation Mechanism of Carbonized Polymer Dots: Crosslinking-Induced Nucleation and Carbonization. Angew. Chem. Int. Ed. 2024, 63, e202410519. [Google Scholar]
- Wang, B.; Waterhouse, G.I.N.; Yang, B.; Lu, S. Advances in Shell and Core Engineering of Carbonized Polymer Dots for Enhanced Applications. Acc. Chem. Res. 2024, 57, 2928–2939. [Google Scholar]
- Ru, Y.; Ai, L.; Jia, T.; Liu, X.; Lu, S.; Tang, Z.; Yang, B. Recent advances in chiral carbonized polymer dots: From synthesis and properties to applications. Nano Today 2020, 34, 100953. [Google Scholar]
- Guo, G.; Xia, Y. General Separation of Carbon Dots by Polyamide Chromatography. Anal. Chem. 2024, 96, 5095–5105. [Google Scholar]
- Wang, C.; He, Y.; Huang, J.; Sui, L.; Ran, G.; Zhu, H.; Song, Q. Intramolecular hydrogen bond-tuned thermal-responsive carbon dots and their application to abnormal body temperature imaging. J. Colloid Interface Sci. 2023, 634, 221–230. [Google Scholar]
- Fu, R.; Song, H.; Liu, X.; Zhang, Y.; Xiao, G.; Zou, B.; Waterhouse, G.I.N.; Lu, S. Disulfide Crosslinking-Induced Aggregation: Towards Solid-State Fluorescent Carbon Dots with Vastly Different Emission Colors. Chin. J. Chem. 2023, 41, 1007–1014. [Google Scholar] [CrossRef]
- Xie, H.; Wang, J.; Lou, Z.; Hu, L.; Segawa, S.; Kang, X.; Wu, W.; Luo, Z.; Kwok, R.T.K.; Lam, J.W.Y.; et al. Mechanochemical Fabrication of Full-Color Luminescent Materials from Aggregation-Induced Emission Prefluorophores for Information Storage and Encryption. J. Am. Chem. Soc. 2024, 146, 18350–18359. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Tao, S.; Yang, F.; Yang, B. Aggregation and luminescence in carbonized polymer dots. Aggregate 2022, 3, 269. [Google Scholar] [CrossRef]
- Zheng, H.; Zhang, Z.; Cai, S.; An, Z.; Huang, W. Enhancing Purely Organic Room Temperature Phosphorescence via Supramolecular Self-Assembly. Adv. Mater. 2024, 36, 2311922. [Google Scholar] [CrossRef]
- Yang, J.; Fang, M.; Li, Z. Organic luminescent materials: The concentration on aggregates from aggregation-induced emission. Aggregate 2020, 1, 6–18. [Google Scholar] [CrossRef]
- Yang, H.; Liu, Y.; Guo, Z.; Lei, B.; Zhuang, J.; Zhang, X.; Liu, Z.; Hu, C. Hydrophobic carbon dots with blue dispersed emission and red aggregation-induced emission. Nat. Commun. 2019, 10, 1789. [Google Scholar] [CrossRef]
- He, Z.; Shan, L.; Mei, J.; Wang, H.; Lam, J.W.Y.; Sung, H.H.Y.; Williams, I.D.; Gu, X.; Miao, Q.; Tang, B.Z. Aggregation-induced emission and aggregation-promoted photochromism of bis(diphenylmethylene)dihydroacenes. Chem. Sci. 2015, 6, 3538–3543. [Google Scholar] [CrossRef]
- Yang, Q.; Zhu, J.; Li, Z.; Chen, X.; Jiang, Y.; Luo, Z.; Wang, P.; Xie, H. Luminescent Liquid Crystals Based on Carbonized Polymer Dots and Their Polarized Luminescence Application. ACS Appl. Mater. Interfaces 2021, 13, 26522–26532. [Google Scholar] [CrossRef]
- Xu, X.; Mo, L.; Li, Y.; Pan, X.; Hu, G.; Lei, B.; Zhang, X.; Zheng, M.; Zhuang, J.; Liu, Y.; et al. Construction of Carbon Dots with Color-Tunable Aggregation-Induced Emission by Nitrogen-Induced Intramolecular Charge Transfer. Adv. Mater. 2021, 33, 2104872. [Google Scholar] [CrossRef]
- Wu, S.; Shi, H.; Lu, W.; Wei, S.; Shang, H.; Liu, H.; Si, M.; Le, X.; Yin, G.; Theato, P.; et al. Aggregation-Induced Emissive Carbon Dots Gels for Octopus-Inspired Shape/Color Synergistically Adjustable Actuators. Angew. Chem. Int. Ed. 2021, 60, 21890–21898. [Google Scholar] [CrossRef]
- Ji, C.; Zeng, F.; Xu, W.; Zhu, M.; Yu, H.; Yang, H.; Peng, Z. Hydrogen Bond-Mediated Self-Assembly of Carbon Dots Enabling Precise Tuning of Particle and Cluster Luminescence for Advanced Optoelectronic Applications. Adv. Mater. 2024, 37, 2414450. [Google Scholar]
- Wang, Y.; Li, Y.; Feng, L. Exploring Solvent-Related Reactions and Corresponding Band Gap Tuning Strategies for Carbon Nanodots Based on Solvothermal Synthesis. J. Phys. Chem. Lett. 2020, 11, 10439–10445. [Google Scholar] [PubMed]
- Wang, Y.; Qin, Y.; Tian, W.; Zhang, H.; Wang, F.; Yan, X.; Rong, S.; Huangfu, C.; Shi, Y.; Wang, Z.; et al. Dye-Incorporated Carbonized Polymer Dots with Tunable Solid-State Emission Based on Intraparticle Förster Resonance Energy Transfer. Adv. Funct. Mater. 2024, 34, 242825. [Google Scholar] [CrossRef]
- Xue, S.; Li, P.; Sun, L.; An, L.; Qu, D.; Wang, X.; Sun, Z. The Formation Process and Mechanism of Carbon Dots Prepared from Aromatic Compounds as Precursors: A Review. Small 2023, 19, 2206180. [Google Scholar]
- Liu, H.; Zhong, X.; Pan, Q.; Zhang, Y.; Deng, W.; Zou, G.; Hou, H.; Ji, X. A review of carbon dots in synthesis strategy. Coord. Chem. Rev. 2024, 498, 215468. [Google Scholar] [CrossRef]
- Zheng, G.; Shen, C.; Deng, Y.; Liu, K.; Zang, J.; Dong, L.; Lou, Q.; Shan, C. Rational design multi-color-emissive chemiluminescent carbon nanodots in a single solvothermal reaction. Nano Res. 2024, 17, 4651–4660. [Google Scholar]
- Li, P.; Xue, S.; Sun, L.; Zong, X.; An, L.; Qu, D.; Wang, X.; Sun, Z. Formation and fluorescent mechanism of red emissive carbon dots from o-phenylenediamine and catechol system. Light Sci. Appl. 2022, 11, 298. [Google Scholar] [CrossRef]
- Qin, Y.; Chi, J.; Zhang, W.; Pan, X.; Liu, Z.; Chen, X.; Shi, Y.; Yang, L.; Zhang, H.; Qin, A.; et al. Influence of Endogenous Derivatives on the Chemical Sensing Performance of Carbonized Polymer Dots. J. Phys. Chem. Lett. 2025, 16, 2151–2159. [Google Scholar] [CrossRef]
- Sun, J.; Liu, Y.; Han, Y.; Li, W.; Wang, N.; Zhang, L.; Zhang, Y.; Deng, F.; Wang, D.; Zhang, X. Enabling controllable time-dependent phosphorescence in carbonized polymer dots based on chromophore excited triplet energy level modulation by ionic bonding. Angew. Chem. Int. Ed. 2024, 64, 202415042. [Google Scholar]
- Xia, C.; Zhu, S.; Feng, T.; Yang, M.; Yang, B. Evolution and Synthesis of Carbon Dots: From Carbon Dots to Carbonized Polymer Dots. Adv. Sci. 2019, 6, 19013616. [Google Scholar] [CrossRef]
- Tao, S.; Zhu, S.; Feng, T.; Xia, C.; Song, Y.; Yang, B. The polymeric characteristics and photoluminescence mechanism in polymer carbon dots: A review. Mater. Today Chem. 2017, 6, 13–25. [Google Scholar]
- Li, P.; Xue, S.; Sun, L.; Ma, X.; Liu, W.; An, L.; Liu, Y.; Qu, D.; Sun, Z. Formation and Fluorescent Mechanism of Multiple Color Emissive Carbon Dots from o-Phenylenediamine. Small 2024, 20, 2310563. [Google Scholar]
- Ai, L.; Wang, H.; Wang, B.; Liu, S.; Song, H.; Lu, S. Concentration-Switchable Assembly of Carbon Dots for Circularly Polarized Luminescent Amplification in Chiral Logic Gates and Deep-Red Light-Emitting Diodes. Adv. Mater. 2024, 36, 210094. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, Y.; Zhang, W.; Liu, Z.; Jia, M.; Chi, J.; Liu, Y.; Wang, Y.; Qin, A.; Wang, Y.; Feng, L. The Influence of Endogenous Derivatives on the Self-Assembly of Carbonized Polymer Dots. Solids 2025, 6, 14. https://doi.org/10.3390/solids6010014
Qin Y, Zhang W, Liu Z, Jia M, Chi J, Liu Y, Wang Y, Qin A, Wang Y, Feng L. The Influence of Endogenous Derivatives on the Self-Assembly of Carbonized Polymer Dots. Solids. 2025; 6(1):14. https://doi.org/10.3390/solids6010014
Chicago/Turabian StyleQin, Yingxi, Wenkai Zhang, Ziwei Liu, Mingyan Jia, Jie Chi, Yujia Liu, Yue Wang, Aimiao Qin, Yu Wang, and Liang Feng. 2025. "The Influence of Endogenous Derivatives on the Self-Assembly of Carbonized Polymer Dots" Solids 6, no. 1: 14. https://doi.org/10.3390/solids6010014
APA StyleQin, Y., Zhang, W., Liu, Z., Jia, M., Chi, J., Liu, Y., Wang, Y., Qin, A., Wang, Y., & Feng, L. (2025). The Influence of Endogenous Derivatives on the Self-Assembly of Carbonized Polymer Dots. Solids, 6(1), 14. https://doi.org/10.3390/solids6010014