Impact of Oat Husk Extracts on Mid-Stage Cement Hydration and the Mechanical Strength of Mortar



Abstract
:1. Introduction
- Initial period, occurring immediately after contact with water;
- Induction or dormant period, a period of low reaction rate in which the matrix is still malleable;
- Acceleration period, which can take hours, characterised by the re-acceleration of reaction rates, consequently solidifying the matrix, with the beginning of this phase corresponding to the initial setting time measured by the Vicat method (this can take hours), but the Vicat’s final setting marks a point before its end;
2. Materials and Methods
2.1. Materials
2.1.1. Binder
2.1.2. Fine Aggregates
2.1.3. Oats Husks
2.2. Procedures
2.2.1. Binder Chemical Characterization
2.2.2. Husks’ Chemical Characterization
2.2.3. Extractives’ Removal
2.2.4. Leached Mixing Water
2.2.5. Mixing Procedure
2.2.6. Vicat Testing for Monitoring Cement Setting
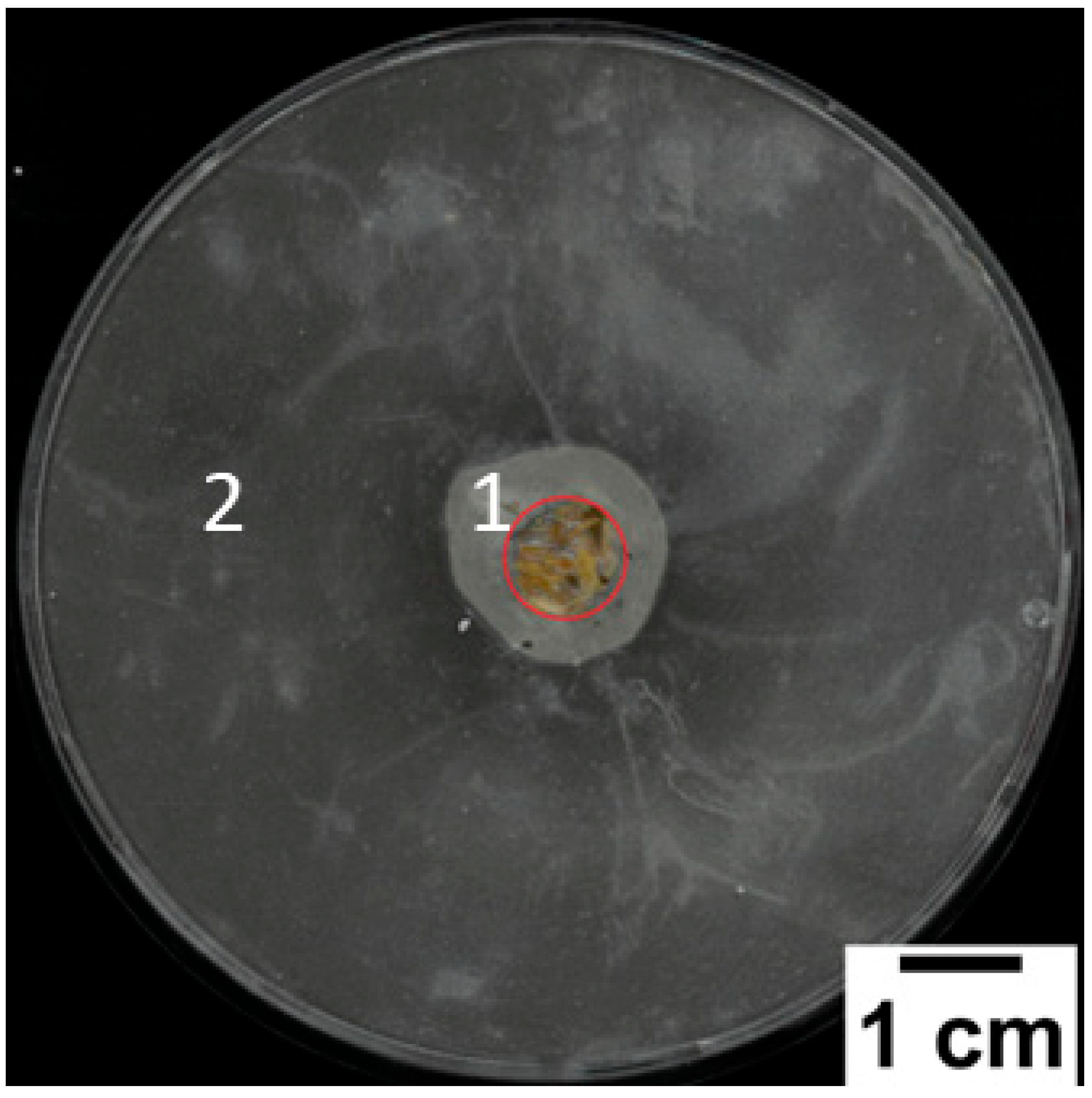
2.2.7. Analysis of the Direct Contact between the Sample and the Matrix
2.2.8. Hydration Analysis
2.2.9. Mechanical Tests
2.2.10. Statistical Analysis and Chart Production
3. Results and Discussions
3.1. Binder Mineral Phases Composition
3.2. Oat Husk Composition and Removal of Extractives
Oat Husks and the Removal of Extractives
3.3. Extractives’ Influence on Cement Setting Time and Solutions’ pH
3.4. Impact of Extractives on Hydration Product Formation
3.4.1. Identification of Functional Groups in the Presence of Extractive Solutions
3.4.2. Analysis of the Thermal Degradation of Cement Hydration Products
3.5. Influence of Extractives on Mechanical Properties
4. Conclusions
- The examined oat husks exhibited high lignocellulosic content, primarily holocellulosic cellulose content, greater than lignin;
- Hot water proved to be more effective than cold water in removing extractives;
- The duration of immersion is more relevant than the number of washing cycles for the effective removal of extractives;
- Cold water extractives resulted in a setting delay of 2.44% per gram of cement, while hot water extractives caused a delay of 4.03% per gram of cement;
- The presence of extractives significantly influences the duration to reaching the final setting of the matrix.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Horowitz, C.A. Paris Agreement. Int. Leg. Mater. 2016, 55, 740–755. [Google Scholar] [CrossRef]
- European Commission. Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the Regions. The European Green Deal. Brussels. 2019. Available online: https://cor.europa.eu/en/events/Documents/COTER/ec_communication_eu_missions.pdf (accessed on 30 October 2023).
- European Commission. A Renovation Wave for Europe—Greening Our Buildings, Creating Jobs, Improving Lives. Brussels. 2020. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A52020DC0662 (accessed on 30 October 2023).
- European Parliament. European Parliament Resolution of 14 September 2022 on the New European Bauhaus. 2022. Available online: https://policycommons.net/artifacts/11156998/european-parliament-resolution-of-14-september-2022-on-the-new-european-bauhaus-20212255ini/12035769/ (accessed on 30 October 2023).
- Bledzki, A. Composites Reinforced with Cellulose Based Fibres. Prog. Polym. Sci. 1999, 24, 221–274. [Google Scholar] [CrossRef]
- Elfaleh, I.; Abbassi, F.; Habibi, M.; Ahmad, F.; Guedri, M.; Nasri, M.; Garnier, C. A Comprehensive Review of Natural Fibers and Their Composites: An Eco-Friendly Alternative to Conventional Materials. Results Eng. 2023, 19, 101271. [Google Scholar] [CrossRef]
- Guo, A.; Sun, Z.; Feng, H.; Shang, H.; Sathitsuksanoh, N. State-of-the-Art Review on the Use of Lignocellulosic Biomass in Cementitious Materials. Sustain. Struct. 2023, 3, 000023. [Google Scholar] [CrossRef]
- Väisänen, T.; Haapala, A.; Lappalainen, R.; Tomppo, L. Utilization of Agricultural and Forest Industry Waste and Residues in Natural Fiber-Polymer Composites: A Review. Waste Manag. 2016, 54, 62–73. [Google Scholar] [CrossRef]
- Lisboa, F.J.N.; Scatolino, M.V.; de Paula Protásio, T.; Júnior, J.B.G.; Marconcini, J.M.; Mendes, L.M. Lignocellulosic Materials for Production of Cement Composites: Valorization of the Alkali Treated Soybean Pod and Eucalyptus Wood Particles to Obtain Higher Value-Added Products. Waste Biomass Valorization 2020, 11, 2235–2245. [Google Scholar] [CrossRef]
- Amziane, S.; Collet, F.; Lawrence, M.; Magniont, C.; Picandet, V.; Sonebi, M. Recommendation of the RILEM TC 236-BBM: Characterisation Testing of Hemp Shiv to Determine the Initial Water Content, Water Absorption, Dry Density, Particle Size Distribution and Thermal Conductivity. Mater. Struct./Mater. Constr. 2017, 50, 167. [Google Scholar] [CrossRef]
- Diquélou, Y.; Gourlay, E.; Arnaud, L.; Kurek, B. Impact of Hemp Shiv on Cement Setting and Hardening: Influence of the Extracted Components from the Aggregates and Study of the Interfaces with the Inorganic Matrix. Cem. Concr. Compos. 2015, 55, 112–121. [Google Scholar] [CrossRef]
- Liu, Z.; Han, C.; Li, Q.; Li, X.; Zhou, H.; Song, X.; Zu, F. Study on Wood Chips Modification and Its Application in Wood-Cement Composites. Case Stud. Constr. Mater. 2022, 17, e01350. [Google Scholar] [CrossRef]
- Batool, F.; Islam, K.; Cakiroglu, C.; Shahriar, A. Effectiveness of Wood Waste Sawdust to Produce Medium- to Low-Strength Concrete Materials. J. Build. Eng. 2021, 44, 103237. [Google Scholar] [CrossRef]
- Magniont, C.; Escadeillas, G. Chemical Composition of Bio-Aggregates and Their Interactions with Mineral Binders. In Bio-Aggregates Based Building Materials; Springer: Dordrecht, The Netherlands, 2017; Volume 23, pp. 1–37. [Google Scholar]
- De Castro, V.G. Compósitos Madeira-Cimento: Um Produto Sustentável Para o Futuro; EdUFERSA: Mossoró, Brazil, 2021; ISBN 9786587108612. [Google Scholar]
- Prusty, J.K.; Patro, S.K.; Basarkar, S.S. Concrete Using Agro-Waste as Fine Aggregate for Sustainable Built Environment—A Review. Int. J. Sustain. Built Environ. 2016, 5, 312–333. [Google Scholar] [CrossRef]
- Jiang, D.; Jiang, D.; Lv, S.; Cui, S.; Sun, S.; Song, X.; He, S.; Zhang, J. Effect of Modified Wheat Straw Fiber on Properties of Fiber Cement-Based Composites at High Temperatures. J. Mater. Res. Technol. 2021, 14, 2039–2060. [Google Scholar] [CrossRef]
- Ataie, F. Influence of Rice Straw Fibers on Concrete Strength and Drying Shrinkage. Sustainability 2018, 10, 2445. [Google Scholar] [CrossRef]
- Danso, H. Effect of Rice Husk on the Mechanical Properties of Cement-Based Mortar. J. Inst. Eng. Ser. D 2020, 101, 205–213. [Google Scholar] [CrossRef]
- Demirbaş, A.; Aslan, A. Effects of Ground Hazelnut Shell, Wood, and Tea Waste on the Mechanical Properties of Cement2. Cem. Concr. Res. 1998, 28, 1101–1104. [Google Scholar] [CrossRef]
- Da Costa Santos, A.C.; Archbold, P. Suitability of Surface-Treated Flax and Hemp Fibers for Concrete Reinforcement. Fibers 2022, 10, 101. [Google Scholar] [CrossRef]
- Page, J.; Khadraoui, F.; Gomina, M.; Boutouil, M. Hydration of Flax Fibre-Reinforced Cementitious Composites: Influence of Fibre Surface Treatments. Eur. J. Environ. Civ. Eng. 2022, 26, 5798–5820. [Google Scholar] [CrossRef]
- Pickering, K.L.; Efendy, M.G.A.; Le, T.M. A Review of Recent Developments in Natural Fibre Composites and Their Mechanical Performance. Compos. Part A Appl. Sci. Manuf. 2016, 83, 98–112. [Google Scholar] [CrossRef]
- Burrows, V.D. Hulless Oat Development, Applications, and Opportunities. In Oats: Chemistry and Technology, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2011; pp. 31–50. ISBN 9780128104521. [Google Scholar]
- Food and Agriculture Organization of the United Nations. Available online: https://www.fao.org/faostat/en/#data/QCL (accessed on 8 January 2024).
- Hackett, R. A Comparison of Husked and Naked Oats under Irish Conditions. Ir. J. Agric. Food Res. 2018, 57, 1–8. [Google Scholar] [CrossRef]
- Bonifacio, A.L.; Archbold, P. The Effect of Calcination Conditions on Oat Husk Ash Pozzolanic Activity. Mater. Today Proc. 2022, 65, 622–628. [Google Scholar] [CrossRef]
- Ruviaro, A.S.; dos Santos Lima, G.T.; Silvestro, L.; Barraza, M.T.; Rocha, J.C.; de Brito, J.; Gleize, P.J.P.; Pelisser, F. Characterization and Investigation of the Use of Oat Husk Ash as Supplementary Cementitious Material as Partial Replacement of Portland Cement: Analysis of Fresh and Hardened Properties and Environmental Assessment. Constr. Build. Mater. 2023, 363, 129762. [Google Scholar] [CrossRef]
- Martínez-Toledo, C.; Valdés-Vidal, G.; Calabi-Floody, A.; González, M.E.; Reyes-Ortiz, O. Effect of Biochar from Oat Hulls on the Physical Properties of Asphalt Binder. Materials 2022, 15, 7000. [Google Scholar] [CrossRef]
- Gil Giraldo, G.A.; Mantovan, J.; Marim, B.M.; Kishima, J.O.F.; Mali, S. Surface Modification of Cellulose from Oat Hull with Citric Acid Using Ultrasonication and Reactive Extrusion Assisted Processes. Polysaccharides 2021, 2, 218–233. [Google Scholar] [CrossRef]
- Varanda, L.D.; do Nascimento, M.F.; Christoforo, A.L.; Silva, D.A.L.; Lahr, F.A.R. Oat Hulls as Addition to High Density Panels Production. Mater. Res. 2013, 16, 1355–1361. [Google Scholar] [CrossRef]
- Wang, L.; Lenorm, H.; Zmamou, H.; Leblanc, N. Effect of Soluble Components from Plant Aggregates on the Setting of the Lime-Based Binder. J. Renew. Mater. 2019, 7, 903–913. [Google Scholar] [CrossRef]
- Welch, R.W.; Hayward, M.V.; Jones, D.I.H. The Composition of Oat Husk and Its Variation Due to Genetic and Other Factors. J. Sci. Food Agric. 1983, 34, 417–426. [Google Scholar] [CrossRef]
- Schmitz, E.; Nordberg Karlsson, E.; Adlercreutz, P. Warming Weather Changes the Chemical Composition of Oat Hulls. Plant Biol. 2020, 22, 1086–1091. [Google Scholar] [CrossRef]
- Kochova, K.; Schollbach, K.; Gauvin, F.; Brouwers, H.J.H. Effect of Saccharides on the Hydration of Ordinary Portland Cement. Constr. Build. Mater. 2017, 150, 268–275. [Google Scholar] [CrossRef]
- Bullard, J.W.; Jennings, H.M.; Livingston, R.A.; Nonat, A.; Scherer, G.W.; Schweitzer, J.S.; Scrivener, K.L.; Thomas, J.J. Mechanisms of Cement Hydration. Cem. Concr. Res. 2011, 41, 1208–1223. [Google Scholar] [CrossRef]
- John, E.; Lothenbach, B. Cement Hydration Mechanisms through Time—A Review. J. Mater. Sci. 2023, 58, 9805–9833. [Google Scholar] [CrossRef]
- Aggarwal, L.K.; Singh, J. Effect of Plant Fibre Extractives on Properties of Cement. Cem. Concr. Compos. 1990, 12, 103–108. [Google Scholar] [CrossRef]
- Vaickelionis, G.; Vaickelioniene, R. Cement Hydration in the Presence of Wood Extractives and Pozzolan Mineral. Ceram. Silik. 2006, 50, 115–122. [Google Scholar]
- Young, J.F. A Review of the Mechanisms of Set-Retardation in Portland Cement Pastes Containing Organic Admixtures. Cem. Concr. Res. 1972, 2, 415–433. [Google Scholar] [CrossRef]
- BS EN 197-1; Cement Composition, Specifications and Conformity Criteria for Common Cements. British Standards Institution: Loughborough, UK, 2011.
- BS EN 13139; Aggregates for Mortar. British Standards Institute: Loughborough, UK, 2013.
- Rietveld, H.M. A Profile Refinement Method for Nuclear and Magnetic Structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Santos Nobre, T.R.; González Pérez, M.; Carlos Bloise, A., Jr.; Cirelli Ângulo, S.; Ota, S.; Angelo Quarcioni, V. Caracterização de Fases de Cimento Portland Por Meio Das Técnicas de Difratometria de Raios X e Spectroscopia de Ressonância Magnética Nuclear de 29Si No Estado Sólido. Rev. Ipt. Tecnol. Inov. 2022, 6, 67–88. [Google Scholar] [CrossRef]
- TAPPI T 222 om-02; Acid-Insoluble Lignin in Wood and Pulp, Test Method T 222 Om-02. Technical Association of the Pulp and Paper Industry: Peachtree Corners, GA, USA, 2002.
- ASTM D1110-21; Standard Test Methods for Water Solubility of Wood. American Society for Testing and Materials: West Conshohocken, PA, USA, 2021.
- David, S. Etudes Des Interactions Physico-Chimiques Aux Interfaces Fibres de Chanvre/Ciment: Influence Sur Les Propriétés Mécaniques Du Composite. Ph.D. Thesis, Universite de Limoges, Limoges, France, 2007. [Google Scholar]
- BS EN 196-3; Methods of Testing Cement—Part 3: Determination of Setting Times and Soundness. British Standards Institution: Loughborough, UK, 2016.
- Deboucha, W.; Leklou, N.; Khelidj, A.; Oudjit, M.N. Hydration Development of Mineral Additives Blended Cement Using Thermogravimetric Analysis (TGA): Methodology of Calculating the Degree of Hydration. Constr. Build. Mater. 2017, 146, 687–701. [Google Scholar] [CrossRef]
- Pane, I.; Hansen, W. Investigation of Blended Cement Hydration by Isothermal Calorimetry and Thermal Analysis. Cem. Concr. Res. 2005, 35, 1155–1164. [Google Scholar] [CrossRef]
- BS EN 196-1; Methods of Testing Cement Part 1: Determination of Strength. British Standards Institution: Loughborough, UK, 2016.
- ACI Committee 214. Guide to Evaluation of Strength Test Results of Concrete; American Concrete Institute: Farmington Hills, MI, USA, 2011. [Google Scholar]
- Toby, B.H. R Factors in Rietveld Analysis: How Good Is Good Enough? Powder Diffr. 2006, 21, 67–70. [Google Scholar] [CrossRef]
- Neitzel, N.; Eder, M.; Hosseinpourpia, R.; Walther, T.; Adamopoulos, S. Chemical Composition, Particle Geometry, and Micro-Mechanical Strength of Barley Husks, Oat Husks, and Wheat Bran as Alternative Raw Materials for Particleboards. Mater. Today Commun. 2023, 36, 106602. [Google Scholar] [CrossRef]
- Krishnarao, R.V.; Subrahmanyam, J.; Jagadishkumar, T. Preparation of Black Amorphous Silica from Rice Husks. Trans. Indian Ceram. Soc. 2001, 60, 95–99. [Google Scholar] [CrossRef]
- Chauhan, K.; Sharma, K.; Dutt, B. Variation in Hot and Cold Water Soluble Extractive Content in Gymnosperms from Western Himalayas. J. Pharmacogn. Phytochem. 2020, 9, 1151–1154. [Google Scholar] [CrossRef]
- Utepov, Y.; Tulebekova, A.; Aldungarova, A.; Mkilima, T.; Zharassov, S.; Shakhmov, Z.; Bazarbayev, D.; Tolkynbayev, T.; Kaliyeva, Z. Investigating the Influence of Initial Water PH on Concrete Strength Gain Using a Sensors and Sclerometric Test Combination. Infrastructures 2022, 7, 159. [Google Scholar] [CrossRef]
- Bishop, M.; Barron, A.R. Cement Hydration Inhibition with Sucrose, Tartaric Acid, and Lignosulfonate: Analytical and Spectroscopic Study. Ind. Eng. Chem. Res. 2006, 45, 7042–7049. [Google Scholar] [CrossRef]
- Mollah, M.Y.A.; Yu, W.; Schennach, R.; Cocke, D.L. A Fourier Transform Infrared Spectroscopic Investigation of the Early Hydration of Portland Cement and the Influence of Sodium Lignosulfonate. Cem. Concr. Res. 2000, 30, 267–273. [Google Scholar] [CrossRef]
- Ylmén, R.; Jäglid, U. Carbonation of Portland Cement Studied by Diffuse Reflection Fourier Transform Infrared Spectroscopy. Int. J. Concr. Struct. Mater. 2013, 7, 119–125. [Google Scholar] [CrossRef]
- Delgado, A.H.; Paroli, R.M.; Beaudoin, J.J. Comparison of IR Techniques for the Characterization of Construction Cement Minerals and Hydrated Products. Appl. Spectrosc. 1996, 50, 970–976. [Google Scholar] [CrossRef]
- Collier, N.C. Transition and Decomposition Temperatures of Cement Phases—A Collection of Thermal Analysis Data. Ceram.-Silik. 2016, 60, 338–343. [Google Scholar] [CrossRef]
- Aprianti, E.; Shafigh, P.; Bahri, S.; Farahani, J.N. Supplementary Cementitious Materials Origin from Agricultural Wastes—A Review. Constr. Build. Mater. 2015, 74, 176–187. [Google Scholar] [CrossRef]
- ASTM C511-03; Standard Specification for Mixing Rooms, Moist Cabinets, Moist Rooms, and Water Storage Tanks Used in the Testing of Hydraulic Cements and Concretes. American Society for Testing and Materials: West Conshohocken, PA, USA, 2017.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mineral Phase | Cement Chemist Notation | (%) | Chemical Formula | ICSD Code |
|---|---|---|---|---|
| Alite | C3S(monoclinic) | 58.0 | Ca3SiO5-Mg, Al | 94742 |
| Belite | C2S(monoclinic) | 17.2 | Ca2SiO4 | 81096 |
| Aluminate | C3A(cubic) | 4.6 | Ca3Al2O6 | 1841 |
| Aluminate | C3A(orthorhombic) | 0.1 | Ca8.5NaAl6O18 | 100220 |
| Ferrite | C4AF | 9.2 | Ca2AlFeO5 | 9197 |
| Gypsum | CŠH2 | 2.4 | CaSO4.2H2O | 151692 |
| Anhydrite | CŠ | 2.3 | CaSO4 | 24473 |
| Hemihydrate | CŠH0.5 | 0.5 | CaSO4.0.5H2O | 79528 |
| Portlandite | CH | 2.8 | Ca(OH)2 | 15471 |
| Calcite | CČ | 2.9 | CaCO3 | 80869 |
| Components | (% of Dry Weight Mass) | C.I (95%) | Adapted from Neitzel et al. [54] | Adapted from Schmitz et al. [34] |
|---|---|---|---|---|
| (%) | (%) | |||
| Lignin (Acid Soluble) | 20.28 | ±0.47 | 25.44 | 25.40 |
| Holocellulose | 70.90 | ±1.96 | 66.19 | 50.30 |
| Cellulose | 33.65 | ±2.07 | 29.80 | 17.20 |
| Hemicellulose | 37.25 | 36.39 | 33.10 | |
| Soxhlet extractives (Water as solvent) | 5.01 | ±0.26 | 16.98 b | |
| Ash | 1.99 c | ±0.01 | 6.27 b | 6.00 b |
| Others | 1.81 | ±0.00 | 2.10 a | 18.30 a |
| Component | C | O | Al | Si | K | Ca |
|---|---|---|---|---|---|---|
| Weight husk (%) | 35.21 | 59.65 | 0.59 | 3.47 | 0.97 | 0.11 |
| Weight ash (%) | 0 | 46.94 | 0.64 | 23.84 | 15.17 | 4.79 |
| Washing Cycles | Duration | Extraction Method | Extractives Removed (%) | C.I (95%) | Number of Samples | S.D |
|---|---|---|---|---|---|---|
| 1 | 2 h | Hot Water | 8.702 | ±0.349 | 5 | 0.281 |
| 1 | 6 h | Hot Water | 16.266 | ±0.353 | 5 | 0.285 |
| 1 | 12 h | Cold Water | 13.385 | ±0.179 | 5 | 0.144 |
| 1 | 24 h | Cold Water | 15.441 | ±0.107 | 5 | 0.086 |
| 1 | 6 h | Ca(OH)2 | 9.517 | ±0.961 | 4 | 0.774 |
| 1 | 24 h | Ca(OH)2 | 27.345 | ±0.519 | 5 | 0.326 |
| 1 | 1 h | Cold Water | 1.362 | ±0.537 | 4 | 0.338 |
| 2 | 1 h | Cold Water | 0.275 | ±0.080 | 4 | 0.050 |
| 3 | 1 h | Cold Water | 0.162 | ±0.076 | 4 | 0.048 |
| 4 | 1 h | Cold Water | 0.150 | ±0.113 | 4 | 0.071 |
| 5 | 1 h | Cold Water | 0.083 | ±0.313 | 3 | 0.126 |
| Σ_{i = 1}^{5} i | Cold Water | 2.033 |
| Measurement | Sample CW | Sample CCH | Sample LW | Sample LCH |
|---|---|---|---|---|
| Initial pH | 6.532 | 12.855 | 6.954 | 12.855 |
| Final pH | 6.421 | 12.741 | 5.652 | 10.245 |
| Formulation | Initial Setting Time (Hour:Minute:Second) | C.I (95%) | Final Setting Time (Hour:Minute:Second) | C.I (95%) |
|---|---|---|---|---|
| Distilled water (Sample CW) | 02:19:02 | ±00:14:21 | 02:36:59 | ±00:18:18 |
| Cold-water leached (Sample LW) | 02:39:21 | ±00:19:03 | 03:26:48 | ±00:10:32 |
| Hot-water leached (Sample LBCW) | 02:50:55 | ±00:16:53 | 03:59:14 | ±00:22:54 |
| Ca(OH)2 solution (Sample CCH) | 02:24:04 | ±00:01:00 | 03:19:04 | ±00:08:04 |
| Ca(OH)2 leached (Sample LCH) | 03:02:03 | ±00:05:26 | 04:32:03 | ±00:03:02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonifacio, A.L.; Archbold, P. Impact of Oat Husk Extracts on Mid-Stage Cement Hydration and the Mechanical Strength of Mortar. Constr. Mater. 2024, 4, 91-109. https://doi.org/10.3390/constrmater4010006
Bonifacio AL, Archbold P. Impact of Oat Husk Extracts on Mid-Stage Cement Hydration and the Mechanical Strength of Mortar. Construction Materials. 2024; 4(1):91-109. https://doi.org/10.3390/constrmater4010006
Chicago/Turabian StyleBonifacio, Alysson Larsen, and Paul Archbold. 2024. "Impact of Oat Husk Extracts on Mid-Stage Cement Hydration and the Mechanical Strength of Mortar" Construction Materials 4, no. 1: 91-109. https://doi.org/10.3390/constrmater4010006
APA StyleBonifacio, A. L., & Archbold, P. (2024). Impact of Oat Husk Extracts on Mid-Stage Cement Hydration and the Mechanical Strength of Mortar. Construction Materials, 4(1), 91-109. https://doi.org/10.3390/constrmater4010006