
Molecular Activation Mechanism and Structural Dynamics of Orange Carotenoid Protein


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
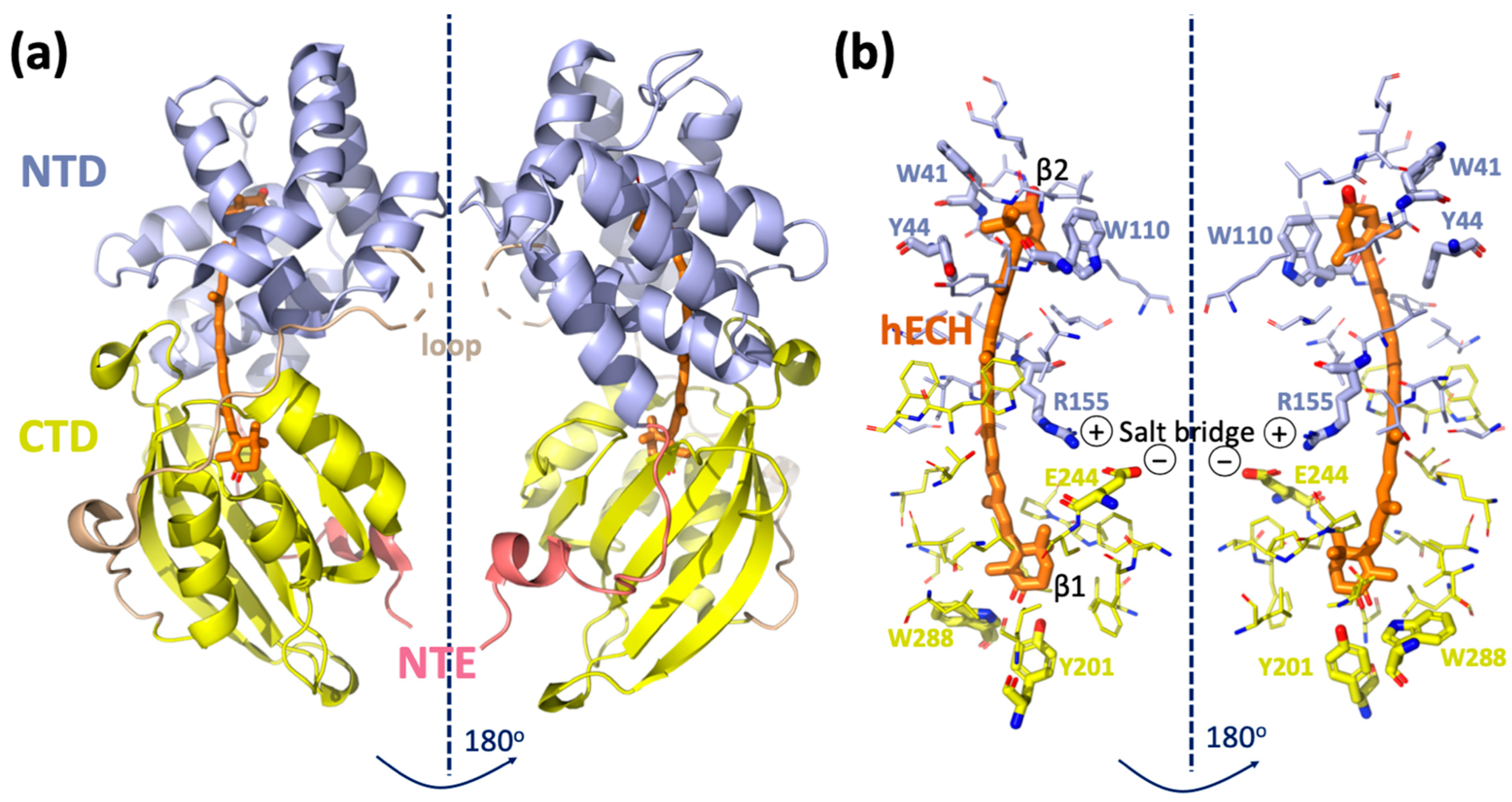
2. Structure and Function of OCP
3. Role of Individual Amino Acids in OCP Light Activation
4. OCPO Versus OCPR
4.1. Global Structural Changes
4.2. Changes in the Secondary and Tertiary Structure
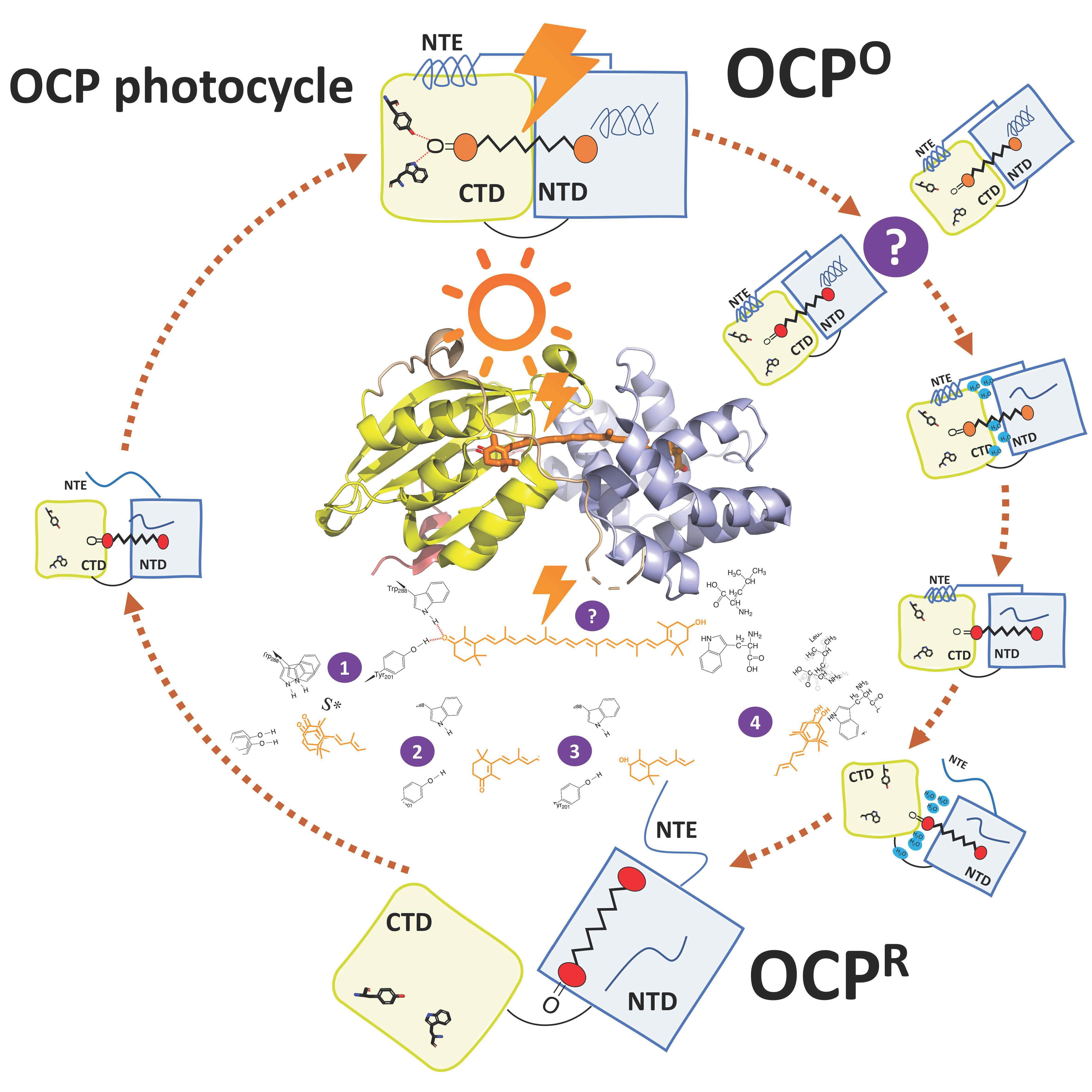
5. OCPO-OCPR-OCPO Photocycle
5.1. Flash Photolysis
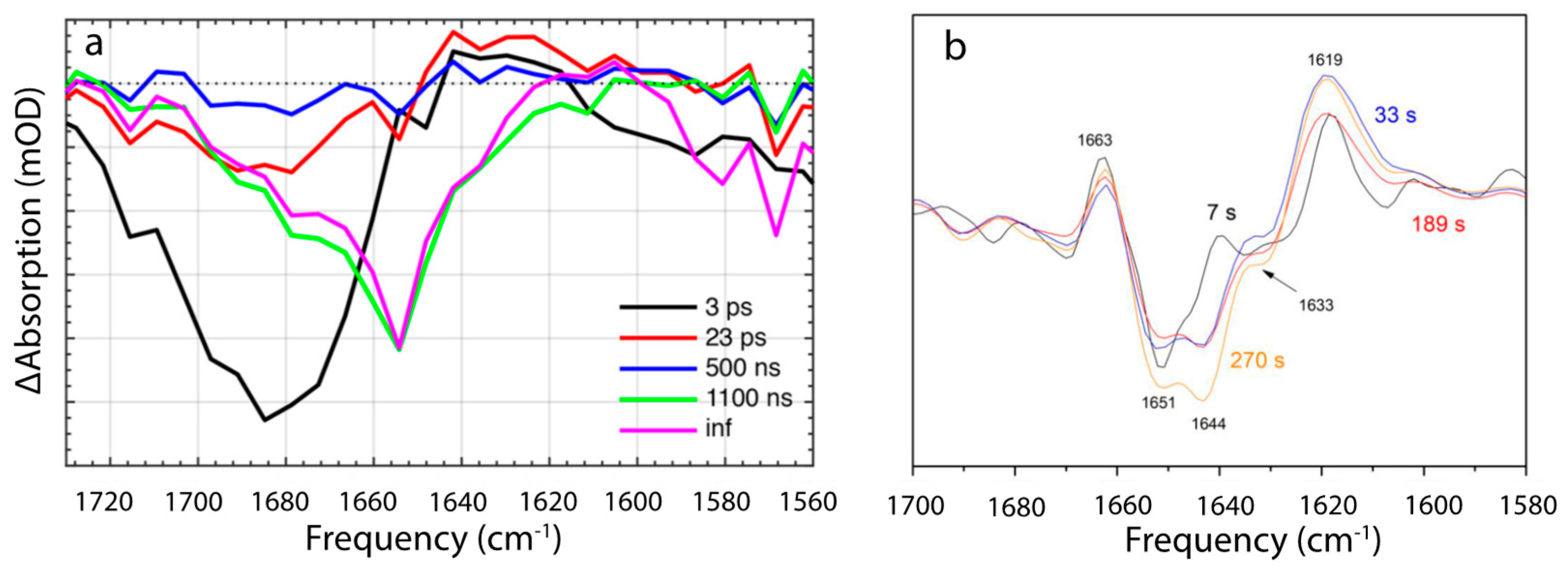
5.2. Intermediates Resolved by Transient Absorption Spectroscopy and FTIR
5.3. Open Questions
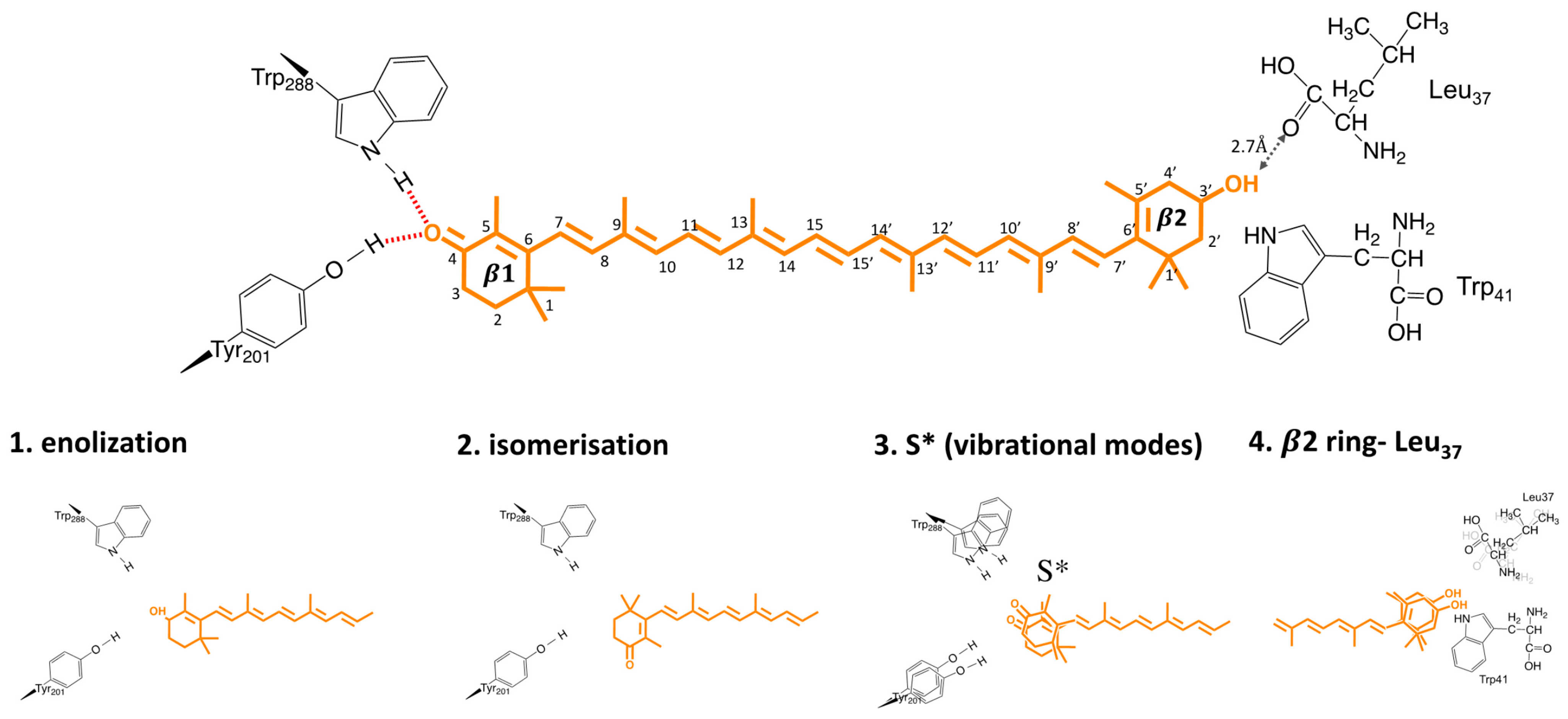
6. The Mechanism and Trigger of Light Activation
7. OCP Excited State Dynamics
7.1. Intramolecular Charge Transfer
7.2. Heterogeneity
7.3. RCP
7.4. Summary and Open Questions
8. General Conclusions and Future Prospects
- 1.
- Trigger mechanism: Based on either structural or spectroscopic studies, proposed candidates for the trigger of OCP light activation are (1) β1-ring flip, due to photoisomerization; (2) enolization of β1-ring and protein moiety; (3) H-bond rupture due to a “hot” excited state; (4) out-of-plane motions in β2-ring. None of these mechanisms were exclusively supported in the literature, and there many results pointing in favor of different models. Unambiguously resolving the trigger and location of OCP light activation is essential to understand, and potentially controlling the OCP activation mechanism. The combination of ultrafast time- resolved spectroscopy, and structural time-resolved techniques, should be exploited to investigate this point.
- 2.
- Intermediates: To fully understand the process and potential gain control of OCP light activation, we need to resolve all OCP intermediate states, and their lifetimes. For example, there are spectroscopic results suggesting that OCP reaches its red state in less than 0.2 µs, before major structural reorganizations (which seem to be mainly localized in NTD), while other results suggest that this process is 50 times slower, and happens after NTD reorganizations. It is also not clear when H-bond rupture occurs, what its spectroscopic signature is, and its role within light activation. Other intermediates and lifetimes of OCPO-OCPR-OCPO photocycle still need to be either resolved or confirmed, based on spectroscopic characters and more advanced structural insights.
- 3.
- Carotenoid/OCP specificity: Spectroscopic studies suggest keto-carotenoid specificity, affecting energetic landscape and excited state kinetics of OCP and RCP. Furthermore, genomic data are beginning to hint of a wider role for OCP homologs beyond photoprotection, possibly within other types of adaptation to changing environment. Therefore, it is important to elucidate whether the trigger and the mechanism of OCP light activation are universal, or if there is some carotenoid/OCP-specificity.
- 4.
- NPQ mechanism: The biological function of OCP is established as a fluorescence quenching of the phycobilisome light harvesting complexes (NPQ); however, the transfer mechanism is unknown. Neither Förster theory or Dexter theory support a mechanism of excitation energy transfer from allophycocyanin (APC), to the S1 (21 Ag−) state of OCPR; therefore, a charge transfer mechanism has been proposed [11]. Potential mechanisms include a dipole-quadrupole interaction, a breakdown of C2h symmetry, or intensity borrowing from the S2 (11 Bu+) state through higher vibrational levels of the S1 (21 Ag−) state. An evaluation of the possible mechanism requires a measurement of the energy transfer rate, which is proportional to the square of the coupling strength, KET = 1.18|V|2J (V is the coupling constant, cm−1, and J is the spectral overlap integral). In principle, the rate can be measured from an equilibration experiment of OCPR with APC660 phycobilisome antenna pigment, that can be prepared by illumination of OCPO to form OCPR, which will bind APC [60]. The rate constant could be determined by ultrafast spectroscopy of APC660-OCPR complex, in order to measure the overlap integral from high-sensitivity fluorescence measurements. Additionally, direct population of the S1 (21 Ag−) state could be achieved by two-photon absorption. While the S0 (11 Ag−) → S1 (21 Ag−) state is one-photon disallowed, the transition is two-photon allowed using ~1.2–1.3 µm wavelength excitation. Similar experiments have been successfully demonstrated for the Car (S1)-BChl (S1) transfer in B800–B850 complexes [61].
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Ernst, O.P.; Lodowski, D.T.; Elstner, M.; Hegemann, P.; Brown, L.S.; Kandori, H. Microbial and animal rhodopsins: Structures, functions, and molecular mechanisms. Chem. Rev. 2014, 114, 126–163. [Google Scholar] [CrossRef] [PubMed]
- Nagy, F.; Schäfer, E. Phytochromes control photomorphogenesis by differentially regulated, interacting signaling pathways in higher plants. Annu. Rev. Plant Biol. 2002, 53, 329–355. [Google Scholar] [CrossRef] [PubMed]
- Christie, J.M.; Blackwood, L.; Petersen, J.; Sullivan, S. Plant flavoprotein photoreceptors. Plant Cell Physiol. 2015, 56, 401–413. [Google Scholar] [CrossRef] [Green Version]
- Horton, P.; Hague, A. Studies on the induction of chlorophyll fluorescence in isolated barley protoplasts. IV. Resolution of non-photochemical quenching. Biochim. Biophys. Acta-Bioenerg. 1988, 932, 107–115. [Google Scholar] [CrossRef]
- Adams, W.W.; Demmig-Adams, B. Operation of the xanthophyll cycle in higher plants in response to diurnal changes in incident sunlight. Planta 1992, 186, 390–398. [Google Scholar] [CrossRef]
- Kirilovsky, D.; Kerfeld, C.A. The Orange Carotenoid Protein: A blue-green light photoactive protein. Photochem. Photobiol. Sci. 2013, 2, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Kerfeld, C.A.; Sawaya, M.R.; Brahmandam, V.; Cascio, D.; Ho, K.K.; Trevithick-Sutton, C.C.; Krogmann, D.W.; Yeates, T.O. The crystal structure of a cyanobacterial water-soluble carotenoid binding protein. Structure 2003, 11, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.; Punginelli, C.; Gall, A.; Bonetti, C.; Alexandre, M.; Routaboul, J.M.; Kerfeld, C.A.; van Grondelle, R.; Robert, B.; Kennis, J.T.; et al. A photoactive carotenoid protein acting as light intensity sensor. Proc. Natl. Acad. Sci. USA 2008, 105, 12075–12080. [Google Scholar] [CrossRef] [Green Version]
- Gwizdala, M.; Wilson, A.; Kirilovsky, D. In vitro reconstitution of the cyanobacterial photoprotective mechanism mediated by the Orange Carotenoid Protein in Synechocystis PCC 6803. Plant Cell 2011, 23, 2631–2643. [Google Scholar] [CrossRef] [Green Version]
- Maksimov, E.G.; Schmitt, F.J.; Shirshin, E.A.; Svirin, M.D.; Elanskaya, I.V.; Friedrich, T.; Fadeev, V.V.; Paschenko, V.Z.; Rubin, A.B. The time course of non-photochemical quenching in phycobilisomes of Synechocystis sp. PCC6803 as revealed by picosecond time-resolved fluorimetry. Biochim. Biophys. Acta-Bioenerg. 2014, 1837, 1540–1547. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; van Stokkum, I.H.M.; Koehorst, R.B.M.; Jongerius, A.; Kirilovsky, D.; van Amerongen, H. Site, rate, and mechanism of photoprotective quenching in cyanobacteria. J. Am. Chem. Soc. 2011, 133, 18304–18311. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.; Tal, O.; Jallet, D.; Wilson, A.; Kirilovsky, D.; Adir, N. Orange carotenoid protein burrows into the phycobilisome to provide photoprotection. Proc. Natl. Acad. Sci. USA 2016, 113, E1655–E1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muzzopappa, F.; Kirilovsky, D. Changing Color for Photoprotection: The Orange Carotenoid Protein. Trends Plant Sci. 2020, 25, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Punginelli, C.; Wilson, A.; Routaboul, J.M.; Kirilovsky, D. Influence of zeaxanthin and echinenone binding on the activity of the orange carotenoid protein. Biochim. Biophys. Acta-Bioenerg. 2009, 1787, 280–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leverenz, R.L.; Sutter, M.; Wilson, A.; Gupta, S.; Thurotte, A.; de Carbon, C.B.; Petzold, C.J.; Ralston, C.; Perreau, F.; Kirilovsky, D.; et al. A 12 Å carotenoid translocation in a photoswitch associated with cyanobacterial photoprotection. Science 2015, 348, 1463–1466. [Google Scholar] [CrossRef] [Green Version]
- Bandara, S.; Ren, Z.; Lu, L.; Zeng, X.; Shin, H.; Zhao, K.-H.H.; Yang, X. Photoactivation mechanism of a carotenoid-based photoreceptor. Proc. Natl. Acad. Sci. USA 2017, 114, 6286–6291. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.; Punginelli, C.; Couturier, M.; Perreau, F.; Kirilovsky, D. Essential role of two tyrosines and two tryptophans on the photoprotection activity of the Orange Carotenoid Protein. Biochim. Biophys. Acta-Bioenerg. 2011, 1807, 293–301. [Google Scholar] [CrossRef]
- Wilson, A.; Gwizdala, M.; Mezzetti, A.; Alexandre, M.; Kerfeld, C.A.; Kirilovsky, D. The essential role of the N-terminal domain of the orange carotenoid protein in cyanobacterial photoprotection: Importance of a positive charge for phycobilisome binding. Plant Cell 2012, 24, 1972–1983. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.; Kinney, J.N.; Zwart, P.H.; Punginelli, C.; D’Haene, S.; Perreau, F.; Klein, M.G.; Kirilovsky, D.; Kerfeld, C.A. Structural determinants underlying photoprotection in the photoactive orange carotenoid protein of cyanobacteria. J. Biol. Chem. 2010, 285, 18364–18375. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.P.; Krogmann, D.W. The orange carotenoid protein of Synechocystis PCC 6803. Biochim. Biophys. Acta-Bioenerg. 1997, 1322, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Guttman, M.; Leverenz, R.L.; Zhumadilova, K.; Pawlowski, E.G.; Petzold, C.J.; Lee, K.K.; Ralston, C.Y.; Kerfeld, C.A. Local and global structural drivers for the photoactivation of the orange carotenoid protein. Proc. Natl. Acad. Sci. USA 2015, 112, E5567–E5574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leverenz, R.L.; Jallet, D.; de Li, M.; Mathies, R.A.; Kirilovsky, D.; Kerfeld, C.A. Structural and functionalmodularity of the orange carotenoid protein: Distinct roles for the N- and C-terminal domains in cyanobacterial photoprotection. Plant Cell 2014, 26, 426–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, Y.G.; Song, P.S.; Cordonnier, M.M.; Pratt, L.H. A Photoreversible Circular Dichroism Spectral Change in Oat Phytochrome Is Suppressed by a Monoclonal Antibody That Binds near Its N-Terminus and by Chromophore Modification. Biochemistry 1987, 26, 4947–4952. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, N.; Imamoto, Y.; Harigai, M.; Kamikubo, H.; Yamazaki, Y.; Kataoka, M. pH-dependent equilibrium between long lived near-UV intermediates of photoactive yellow protein. J. Biol. Chem. 2006, 281, 4318–4325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zhang, H.; King, J.D.; Wolf, N.R.; Prado, M.; Gross, M.L.; Blankenship, R.E. Mass spectrometry footprinting reveals the structural rearrangements of cyanobacterial orange carotenoid protein upon light activation. Biochim. Biophys. Acta-Bioenerg. 2014, 1837, 1955–1963. [Google Scholar] [CrossRef] [Green Version]
- Chábera, P.; Durchan, M.; Shih, P.M.; Kerfeld, C.A.; Polívka, T. Excited-state properties of the 16kDa red carotenoid protein from Arthrospira maxima. Biochim. Biophys. Acta-Bioenerg. 2011, 1807, 30–35. [Google Scholar] [CrossRef] [Green Version]
- King, J.D.; Liu, H.; He, G.; Orf, G.S.; Blankenship, R.E. Chemical activation of the cyanobacterial orange carotenoid protein. FEBS Lett. 2014, 588, 4561–4565. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.J.; Zhang, H.; Niedzwiedzki, D.M.; Prado, M.; He, G.N.; Gross, M.L.; Blankenship, R.E. Phycobilisomes Supply Excitations to Both Photosystems in a Megacomplex in Cyanobacteria. Science 2013, 342, 1104–1107. [Google Scholar] [CrossRef] [Green Version]
- Kennis, J.T.M.; Crosson, S.; Gauden, M.; van Stokkum, I.H.M.; Moffat, K.; van Grondelle, R. Primary reactions of the LOV2 domain of phototropin, a plant blue-light photoreceptor. Biochemistry 2003, 42, 3385–3392. [Google Scholar] [CrossRef]
- Gauden, M.; Yeremenko, S.; Laan, W.; van Stokkum, I.H.M.; Ihalainen, J.A.; van Grondelle, R.; Hellingwerf, K.J.; Kennis, J.T.M. Photocycle of the flavin-binding photoreceptor AppA, a bacterial transcriptional antirepressor of photosynthesis genes. Biochemistry 2005, 44, 3653–3662. [Google Scholar] [CrossRef]
- Hutchison, C.D.M.; Kaucikas, M.; Tenboer, J.; Kupitz, C.; Moffat, K.; Schmidt, M.; van Thor, J.J. Photocycle populations with femtosecond excitation of crystalline photoactive yellow protein. Chem. Phys. Lett. 2016, 654, 63–71. [Google Scholar] [CrossRef] [Green Version]
- van Thor, J.J.; Gensch, T.; Hellingwerf, K.J.; Johnson, L.N. Phototransformation of green fluorescent protein with UV and visible light leads to decarboxylation of glutamate 222. Nat. Struct. Biol. 2002, 9, 37–41. [Google Scholar] [CrossRef]
- Maksimov, E.G.; Shirshin, E.A.; Sluchanko, N.N.; Zlenko, D.V.; Parshina, E.Y.; Tsoraev, G.V.; Klementiev, K.E.; Budylin, G.S.; Schmitt, F.-J.; Friedrich, T.; et al. The Signaling State of Orange Carotenoid Protein. Biophys. J. 2015, 109, 595–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maksimov, E.G.; Sluchanko, N.N.; Slonimskiy, Y.B.; Slutskaya, E.A.; Stepanov, A.V.; Argentova-Stevens, A.M.; Shirshin, E.A.; Tsoraev, G.V.; Klementiev, K.E.; Slatinskaya, O.V.; et al. The photocycle of orange carotenoid protein conceals distinct intermediates and asynchronous changes in the carotenoid and protein components. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konold, P.E.; van Stokkum, I.H.M.; Muzzopappa, F.; Wilson, A.; Groot, M.L.; Kirilovsky, D.; Kennis, J.T.M. Photoactivation Mechanism, Timing of Protein Secondary Structure Dynamics and Carotenoid Translocation in the Orange Carotenoid Protein. J. Am. Chem. Soc. 2019, 141, 520–530. [Google Scholar] [CrossRef] [Green Version]
- Mezzetti, A.; Alexandre, M.; Thurotte, A.; Wilson, A.; Gwizdala, M.; Kirilovsky, D. Two-Step Structural Changes in Orange Carotenoid Protein Photoactivation Revealed by Time-Resolved Fourier Transform Infrared Spectroscopy. J. Phys. Chem. B 2019, 123, 3259–3266. [Google Scholar] [CrossRef]
- Warren, M.M.; Kaucikas, M.; Fitzpatrick, A.; Champion, P.; Sage, J.T.; van Thor, J.J. Ground-state proton transfer in the photoswitching reactions of the fluorescent protein Dronpa. Nat. Commun. 2013, 4, 1461. [Google Scholar] [CrossRef] [Green Version]
- Warren, C.K.; Weedon, B.C.L. 804. Carotenoids and related compounds. Part VII. Synthesis of canthaxanthin and echinenone. J. Chem. Soc. 1958, 3986–3993. [Google Scholar] [CrossRef]
- Kish, E.; Pinto, M.M.M.; Kirilovsky, D.; Spezia, R.; Robert, B. Echinenone vibrational properties: From solvents to the orange carotenoid protein. Biochim. Biophys. Acta-Bioenerg. 2015, 1847, 1044–1054. [Google Scholar] [CrossRef]
- Gurchiek, J.K.; Bao, H.; Domínguez-Martín, M.A.; McGovern, S.E.; Marquardt, C.E.; Roscioli, J.D.; Ghosh, S.; Kerfeld, C.A.; Beck, W.F. Fluorescence and Excited-State Conformational Dynamics of the Orange Carotenoid Protein. J. Phys. Chem. B 2018, 122, 1792–1800. [Google Scholar] [CrossRef]
- Bondanza, M.; Cupellini, L.; Lipparini, F.; Mennucci, B. The Multiple Roles of the Protein in the Photoactivation of Orange Carotenoid Protein. Chem 2020, 6, 187–203. [Google Scholar] [CrossRef]
- Pande, K.; Hutchison, C.D.M.; Groenhof, G.; Aquila, A.; Robinson, J.S.; Tenboer, J.; Basu, S.; Boutet, S.; DePonte, D.P.; Liang, M.N.; et al. Femtosecond structural dynamics drives the trans/cis isomerization in photoactive yellow protein. Science 2016, 352, 725–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.; Krasselt, A.; Reuter, W. Local protein flexibility as a prerequisite for reversible chromophore isomerization in α-phycoerythrocyanin. Biochim. Biophys. Acta-Proteins Proteom. 2006, 1764, 55–62. [Google Scholar] [CrossRef]
- Schoenlein, R.W.; Peteanu, L.A.; Mathies, R.A.; Shank, C.V. The first step in vision: Femtosecond isomerization of rhodopsin. Science 1991, 254, 412–415. [Google Scholar] [CrossRef]
- Liguori, N.; Xu, P.; van Stokkum, I.H.M.; van Oort, B.; Lu, Y.; Karcher, D.; Bock, R.; Croce, R. Different carotenoid conformations have distinct functions in light-harvesting regulation in plants. Nat. Commun. 2017, 8, 1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balevičius, V.; Wei, T.; di Tommaso, D.; Abramavicius, D.; Hauer, J.; Polívka, T.; Duffy, C.D.P. The full dynamics of energy relaxation in large organic molecules: From photo-excitation to solvent heating. Chem. Sci. 2019, 10, 4792–4804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, T.; Kuznetsova, V.; Dominguez-Martin, M.A.; Kerfeld, C.A.; Polívka, T. UV Excitation of Carotenoid Binding Proteins OCP and HCP: Excited-State Dynamics and Product Formation. ChemPhotoChem 2022, 6, e202100194. [Google Scholar] [CrossRef]
- Niziński, S.; Wilson, A.; Uriarte, L.M.; Ruckebusch, C.; Andreeva, E.A.; Schlichting, I.; Colletier, J.-P.; Kirilovsky, D.; Burdzinski, G.; Sliwa, M. Unifying Perspective of the Ultrafast Photodynamics of Orange Carotenoid Proteins from Synechocystis: Peril of High-Power Excitation, Existence of Different S* States, and Influence of Tagging. JACS Au 2022, 2, 1084–1095. [Google Scholar] [CrossRef]
- Berera, R.; van Stokkum, I.H.M.; Gwizdala, M.; Wilson, A.; Kirilovsky, D.; van Grondelle, R. The photophysics of the orange carotenoid protein, a light-powered molecular switch. J. Phys. Chem. B 2012, 116, 2568–2574. [Google Scholar] [CrossRef]
- Šlouf, V.; Kuznetsova, V.; Fuciman, M.; de Carbon, C.B.; Wilson, A.; Kirilovsky, D.; Polívka, T. Ultrafast spectroscopy tracks carotenoid configurations in the orange and red carotenoid proteins from cyanobacteria. Photosynth. Res. 2017, 131, 105–117. [Google Scholar] [CrossRef]
- Berera, R.; Gwizdala, M.; van Stokkum, I.H.; Kirilovsky, D.; van Grondelle, R. Excited states of the inactive and active forms of the orange carotenoid protein. J. Phys. Chem. B 2013, 117, 9121–9128. [Google Scholar] [CrossRef] [PubMed]
- Polívka, T.; Kerfeld, C.A.; Pascher, T.; Sundström, V. Spectroscopic properties of the carotenoid 3′-hydroxyechinenone in the orange carotenoid protein from the cyanobacterium Arthrospira maxima. Biochemistry 2005, 44, 3994–4003. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiedzki, D.M.; Liu, H.; Blankenship, R.E. Excited state properties of 3′-hydroxyechinenone in solvents and in the orange carotenoid protein from synechocystis sp. PCC 6803. J. Phys. Chem. B 2014, 118, 6141–6149. [Google Scholar] [CrossRef] [PubMed]
- Frank, H.A.; Bautista, J.A.; Josue, J.; Pendon, Z.; Hiller, R.G.; Sharples, F.P.; Gosztola, D.; Wasielewski, M.R. Effect of the Solvent Environment on the Spectroscopic Properties and Dynamics of the Lowest Excited States of Carotenoids. J. Phys. Chem. B 2000, 104, 4569–4577. [Google Scholar] [CrossRef]
- Zigmantas, D.; Hiller, R.G.; Sharples, F.P.; Frank, H.A.; Sundström, V.; Polívka, T. Effect of a conjugated carbonyl group on the photophysical properties of carotenoids. Phys. Chem. Chem. Phys. 2004, 6, 3009–3016. [Google Scholar] [CrossRef]
- Chábera, P.; Fuciman, M.; Híbek, P.; Polívka, T. Effect of carotenoid structure on excited-state dynamics of carbonyl carotenoids. Phys. Chem. Chem. Phys. 2009, 11, 8795–8803. [Google Scholar] [CrossRef]
- Fujisawa, T.; Leverenz, R.L.; Nagamine, M.; Kerfeld, C.A.; Unno, M. Raman Optical Activity Reveals Carotenoid Photoactivation Events in the Orange Carotenoid Protein in Solution. J. Am. Chem. Soc. 2017, 139, 10456–10460. [Google Scholar] [CrossRef]
- de Re, E.; Schlau-Cohen, G.S.; Leverenz, R.L.; Huxter, V.M.; Oliver, T.A.A.; Mathies, R.A.; Fleming, G.R. Insights into the structural changes occurring upon photoconversion in thse orange carotenoid protein from broadband two-dimensional electronic spectroscopy. J. Phys. Chem. B 2014, 118, 5382–5389. [Google Scholar] [CrossRef]
- Polivka, T.; Chabera, P.; Kerfeld, C.A. Carotenoid-protein interaction alters the S(1) energy of hydroxyechinenone in the Orange Carotenoid Protein. Biochim. Biophys. Acta-Bioenerg. 2013, 1827, 248–254. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Gwizdala, M.; van Stokkum, I.H.; Koehorst, R.B.; Kirilovsky, D.; van Amerongen, H. Picosecond kinetics of light harvesting and photoprotective quenching in wild-type and mutant phycobilisomes isolated from the cyanobacterium Synechocystis PCC 6803. Biophys. J. 2012, 102, 1692–1700. [Google Scholar] [CrossRef] [Green Version]
- Walla, P.J.; Linden, P.A.; Hsu, C.-P.; Scholes, G.D.; Fleming, G.R. Femtosecond dynamics of the forbidden carotenoid S1 state in light-harvesting complexes of purple bacteria observed after two-photon excitation. Proc. Natl. Acad. Sci. USA 2000, 97, 10808–10813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngamwonglumlert, L.; Devahastin, S.; Chiewchan, N. Natural colorants: Pigment stability and extraction yield enhancement via utilization of appropriate pretreatment and extraction methods. Crit. Rev. Food Sci. Nutr. 2017, 57, 3243–3259. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chukhutsina, V.U.; van Thor, J.J. Molecular Activation Mechanism and Structural Dynamics of Orange Carotenoid Protein. Physchem 2022, 2, 235-252. https://doi.org/10.3390/physchem2030017
Chukhutsina VU, van Thor JJ. Molecular Activation Mechanism and Structural Dynamics of Orange Carotenoid Protein. Physchem. 2022; 2(3):235-252. https://doi.org/10.3390/physchem2030017
Chicago/Turabian StyleChukhutsina, Volha U., and Jasper J. van Thor. 2022. "Molecular Activation Mechanism and Structural Dynamics of Orange Carotenoid Protein" Physchem 2, no. 3: 235-252. https://doi.org/10.3390/physchem2030017
APA StyleChukhutsina, V. U., & van Thor, J. J. (2022). Molecular Activation Mechanism and Structural Dynamics of Orange Carotenoid Protein. Physchem, 2(3), 235-252. https://doi.org/10.3390/physchem2030017