
Vibrational-Excitation-Induced and Spontaneous Conformational Changes in Solid Para-H2—Diminished Matrix Effects



Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Determining the Column Densities and Quantum Efficiencies
3.2. Cis-Trans Tunneling Rates of the Carboxylic acids
3.3. Quantum Efficiency of the Processes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pettersson, M.; Lundell, J.; Khriachtchev, L.; Räsänen, M. IR Spectrum of the Other Rotamer of Formic Acid, cis -HCOOH. J. Am. Chem. Soc. 1997, 119, 11715–11716. [Google Scholar] [CrossRef]
- Pettersson, M.; Maçôas, E.M.S.; Khriachtchev, L.; Lundell, J.; Fausto, R.; Räsänen, M. Cis → trans conversion of formic acid by dissipative tunneling in solid rare gases: Influence of environment on the tunneling rate. J. Chem. Phys. 2002, 117, 9095–9098. [Google Scholar] [CrossRef] [Green Version]
- Maçôas, E.M.S.; Lundell, J.; Pettersson, M.; Khriachtchev, L.; Fausto, R.; Räsänen, M. Vibrational spectroscopy of cis- and trans-formic acid in solid argon. J. Mol. Spectrosc. 2003, 219, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Pettersson, M.; Maçôas, E.M.S.; Khriachtchev, L.; Fausto, R.; Räsänen, M. Conformational Isomerization of Formic Acid by Vibrational Excitation at Energies below the Torsional Barrier. J. Am. Chem. Soc. 2003, 125, 4058–4059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maçôas, E.M.S.S.; Khriachtchev, L.; Pettersson, M.; Juselius, J.; Fausto, R.; Räsänen, M. Reactive vibrational excitation spectroscopy of formic acid in solid argon: Quantum yield for infrared induced trans→cis isomerization and solid state effects on the vibrational spectrum. J. Chem. Phys. 2003, 119, 11765–11772. [Google Scholar] [CrossRef]
- Lopes, S.; Domanskaya, A.V.; Fausto, R.; Räsänen, M.; Khriachtchev, L. Formic and acetic acids in a nitrogen matrix: Enhanced stability of the higher-energy conformer. J. Chem. Phys. 2010, 133, 144507. [Google Scholar] [CrossRef] [Green Version]
- Maçôas, E.M.S.S.; Khriachtchev, L.; Pettersson, M.; Fausto, R.; Räsänen, M. Rotational Isomerism in Acetic Acid: The First Experimental Observation of the High-Energy Conformer. J. Am. Chem. Soc. 2003, 125, 16188–16189. [Google Scholar] [CrossRef] [Green Version]
- Maçôas, E.M.S.S.; Khriachtchev, L.; Fausto, R.; Räsänen, M. Photochemistry and Vibrational Spectroscopy of the Trans and Cis Conformers of Acetic Acid in Solid Ar. J. Phys. Chem. A 2004, 108, 3380–3389. [Google Scholar] [CrossRef] [Green Version]
- Maçôas, E.M.S.S.; Khriachtchev, L.; Pettersson, M.; Fausto, R.; Räsänen, M. Rotational isomerism of acetic acid isolated in rare-gas matrices: Effect of medium and isotopic substitution on IR-induced isomerization quantum yield and cis→ trans tunneling rate. J. Chem. Phys. 2004, 121, 1331–1338. [Google Scholar] [CrossRef] [Green Version]
- Apóstolo, R.F.G.F.G.; Bazsó, G.; Bento, R.R.F.R.F.; Tarczay, G.; Fausto, R. The first experimental observation of the higher-energy trans conformer of trifluoroacetic acid. J. Mol. Struct. 2016, 1125, 288–295. [Google Scholar] [CrossRef]
- Apóstolo, R.F.G.G.; Bazsó, G.; Ogruc-Ildiz, G.; Tarczay, G.; Fausto, R. Near-infrared in situ generation of the higher-energy trans conformer of tribromoacetic acid: Observation of a large-scale matrix-site changing mediated by conformational conversion. J. Chem. Phys. 2018, 148, 044303. [Google Scholar] [CrossRef] [PubMed]
- Lopes, S.; Nikitin, T.; Fausto, R. Propiolic Acid in Solid Nitrogen: NIR- and UV-Induced cis → trans Isomerization and Matrix-Site-Dependent trans → cis Tunneling. J. Phys. Chem. A 2019, 123, 1581–1593. [Google Scholar] [CrossRef]
- Maçôas, E.M.S.; Khriachtchev, L.; Pettersson, M.; Fausto, R.; Räsänen, M.; Maças, E.M.S.; Khriachtchev, L.; Pettersson, M.; Fausto, R.; Räsänen, M. Internal rotation in propionic acid: Near-infrared-induced isomerization in solid Argon. J. Phys. Chem. A 2005, 109, 3617–3625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazsó, G.; Góbi, S.; Tarczay, G. Near-Infrared Radiation Induced Conformational Change and Hydrogen Atom Tunneling of 2-Chloropropionic Acid in Low-Temperature Ar Matrix. J. Phys. Chem. A 2012, 116, 4823–4832. [Google Scholar] [CrossRef] [PubMed]
- Halasa, A.; Lapinski, L.; Reva, I.; Rostkowska, H.; Fausto, R.; Nowak, M.J. Near-Infrared Laser-Induced Generation of Three Rare Conformers of Glycolic Acid. J. Phys. Chem. A 2014, 118, 5626–5635. [Google Scholar] [CrossRef]
- Reva, I.; Nunes, C.M.; Biczysko, M.; Fausto, R.; Nunes, C.M.; Biczysko, M.; Fausto, R. Conformational switching in pyruvic acid isolated in Ar and N2 matrixes: Spectroscopic analysis, anharmonic simulation, and tunneling. J. Phys. Chem. A 2015, 119, 2614–2627. [Google Scholar] [CrossRef]
- Halasa, A.; Lapinski, L.; Rostkowska, H.; Reva, I.; Nowak, M.J. Tunable Diode Lasers as a Tool for Conformational Control: The Case of Matrix-Isolated Oxamic Acid. J. Phys. Chem. A 2015, 119, 2203–2210. [Google Scholar] [CrossRef]
- Maçôas, E.M.S.S.; Fausto, R.; Pettersson, M.; Khriachtchev, L.; Räsänen, M. Infrared-induced rotamerization of oxalic acid monomer in argon matrix. J. Phys. Chem. A 2000, 104, 6956–6961. [Google Scholar] [CrossRef] [Green Version]
- Maçôas, E.M.S.S.; Fausto, R.; Lundell, J.; Pettersson, M.; Khriachtchev, L.; Räsänen, M. Conformational Analysis and Near-Infrared-Induced Rotamerization of Malonic Acid in an Argon Matrix. J. Phys. Chem. A 2000, 104, 11725–11732. [Google Scholar] [CrossRef] [Green Version]
- Maçôas, E.M.S.S.; Fausto, R.; Lundell, J.; Pettersson, M.; Khriachtchev, L.; Räsänen, M. A matrix isolation spectroscopic and quantum chemical study of fumaric and maleic acid. J. Phys. Chem. A 2001, 105, 3922–3933. [Google Scholar] [CrossRef] [Green Version]
- Lapinski, L.; Reva, I.; Rostkowska, H.; Halasa, A.; Fausto, R.; Nowak, M.J. Conformational transformation in squaric acid induced by near-IR laser light. J. Phys. Chem. A 2013, 117, 5251–5259. [Google Scholar] [CrossRef]
- Halasa, A.; Lapinski, L.; Reva, I.; Rostkowska, H.; Fausto, R.; Nowak, M.J. Three Conformers of 2-Furoic Acid: Structure Changes Induced with Near-IR Laser Light. J. Phys. Chem. A 2015, 119, 1037–1047. [Google Scholar] [CrossRef]
- Kuş, N.; Fausto, R. Effects of the matrix and intramolecular interactions on the stability of the higher-energy conformers of 2-fluorobenzoic acid. J. Chem. Phys. 2017, 146, 124305. [Google Scholar] [CrossRef]
- Bazsó, G.; Magyarfalvi, G.; Tarczay, G. Near-infrared laser induced conformational change and UV laser photolysis of glycine in low-temperature matrices: Observation of a short-lived conformer. J. Mol. Struct. 2012, 1025, 33–42. [Google Scholar] [CrossRef]
- Bazsó, G.; Magyarfalvi, G.; Tarczay, G. Tunneling Lifetime of the ttc /VIp Conformer of Glycine in Low-Temperature Matrices. J. Phys. Chem. A 2012, 116, 10539–10547. [Google Scholar] [CrossRef]
- Bazsó, G.; Najbauer, E.E.; Magyarfalvi, G.; Tarczay, G. Near-Infrared Laser Induced Conformational Change of Alanine in Low-Temperature Matrixes and the Tunneling Lifetime of Its Conformer VI. J. Phys. Chem. A 2013, 117, 1952–1962. [Google Scholar] [CrossRef]
- Nunes, C.M.; Lapinski, L.; Fausto, R.; Reva, I. Near-IR laser generation of a high-energy conformer of L-alanine and the mechanism of its decay in a low-temperature nitrogen matrix. J. Chem. Phys. 2013, 138, 125101. [Google Scholar] [CrossRef]
- Najbauer, E.E.; Bazsó, G.; Góbi, S.; Magyarfalvi, G.; Tarczay, G. Exploring the Conformational Space of Cysteine by Matrix Isolation Spectroscopy Combined with Near-Infrared Laser Induced Conformational Change. J. Phys. Chem. B 2014, 118, 2093–2103. [Google Scholar] [CrossRef]
- Najbauer, E.E.; Bazsó, G.; Apóstolo, R.; Fausto, R.; Biczysko, M.; Barone, V.; Tarczay, G. Identification of Serine Conformers by Matrix-Isolation IR Spectroscopy Aided by Near-Infrared Laser-Induced Conformational Change, 2D Correlation Analysis, and Quantum Mechanical Anharmonic Computations. J. Phys. Chem. B 2015, 119, 10496–10510. [Google Scholar] [CrossRef] [Green Version]
- Lapinski, L.; Nowak, M.J.; Reva, I.; Rostkowska, H.; Fausto, R. NIR-laser-induced selective rotamerization of hydroxy conformers of cytosine. Phys. Chem. Chem. Phys. 2010, 12, 9615. [Google Scholar] [CrossRef] [Green Version]
- Reva, I.; Nowak, M.J.; Lapinski, L.; Fausto, R. Spontaneous tunneling and near-infrared-induced interconversion between the amino-hydroxy conformers of cytosine. J. Chem. Phys. 2012, 136, 064511. [Google Scholar] [CrossRef] [Green Version]
- Lapinski, L.; Reva, I.; Rostkowska, H.; Fausto, R.; Nowak, M.J. Near-IR-Induced, UV-Induced, and Spontaneous Isomerizations in 5-Methylcytosine and 5-Fluorocytosine. J. Phys. Chem. B 2014, 118, 2831–2841. [Google Scholar] [CrossRef]
- Rostkowska, H.; Lapinski, L.; Kozankiewicz, B.; Nowak, M.J. Photochemical isomerizations of thiosemicarbazide, a matrix isolation study. J. Phys. Chem. A 2012, 116, 9863–9871. [Google Scholar] [CrossRef]
- Halasa, A.; Reva, I.; Lapinski, L.; Nowak, M.J.; Fausto, R. Conformational Changes in Thiazole-2-carboxylic Acid Selectively Induced by Excitation with Narrowband Near-IR and UV Light. J. Phys. Chem. A 2016, 120, 2078–2088. [Google Scholar] [CrossRef]
- Halasa, A.; Lapinski, L.; Rostkowska, H.; Nowak, M.J. Intramolecular Vibrational Energy Redistribution in 2-Thiocytosine: SH Rotamerization Induced by Near-IR Selective Excitation of NH 2 Stretching Overtone. J. Phys. Chem. A 2015, 119, 9262–9271. [Google Scholar] [CrossRef]
- Halasa, A.; Reva, I.; Lapinski, L.; Rostkowska, H.; Fausto, R.; Nowak, M.J. Conformers of Kojic Acid and Their Near-IR-Induced Conversions: Long-Range Intramolecular Vibrational Energy Transfer. J. Phys. Chem. A 2016, 120, 2647–2656. [Google Scholar] [CrossRef]
- Lopes Jesus, A.J.; Reva, I.; Araujo-Andrade, C.; Fausto, R. Conformational Switching by Vibrational Excitation of a Remote NH Bond. J. Am. Chem. Soc. 2015, 137, 14240–14243. [Google Scholar] [CrossRef]
- Lapinski, L.; Reva, I.; Rostkowska, H.; Lopes Jesus, A.J.; Vieira Pinto, S.M.; Fausto, R.; Nowak, M.J. Conformational Isomerizations by Rotation around C–C or C–N Bonds: A Comparative Study on Matrix-Isolated Glycolamide and N-Hydroxyurea Excited with Near-IR Laser Light. J. Phys. Chem. A 2019, 123, 3831–3839. [Google Scholar] [CrossRef]
- Kovács, B.; Kuş, N.; Tarczay, G.; Fausto, R. Experimental Evidence of Long-Range Intramolecular Vibrational Energy Redistribution through Eight Covalent Bonds: NIR Irradiation Induced Conformational Transformation of E-Glutaconic Acid. J. Phys. Chem. A 2017, 121, 3392–3400. [Google Scholar] [CrossRef]
- Góbi, S.; Reva, I.; Csonka, I.P.; Nunes, C.M.; Tarczay, G.; Fausto, R. Selective conformational control by excitation of NH imino vibrational antennas. Phys. Chem. Chem. Phys. 2019, 21, 24935–24949. [Google Scholar] [CrossRef]
- Góbi, S.; Balbisi, M.; Tarczay, G. Local and Remote Conformational Switching in 2-Fluoro-4-Hydroxy Benzoic Acid. Photochem 2022, 2, 102–121. [Google Scholar] [CrossRef]
- Marushkevich, K.; Khriachtchev, L.; Räsänen, M. High-energy conformer of formic acid in solid neon: Giant difference between the proton tunneling rates of cis monomer and trans-cis dimer. J. Chem. Phys. 2007, 126, 241102. [Google Scholar] [CrossRef]
- Góbi, S.; Nunes, C.M.; Reva, I.; Tarczay, G.; Fausto, R. S–H rotamerization via tunneling in a thiol form of thioacetamide. Phys. Chem. Chem. Phys. 2019, 21, 17063–17071. [Google Scholar] [CrossRef]
- Rostkowska, H.; Lapinski, L.; Khvorostov, A.; Nowak, M.J. Proton-Transfer Processes in Thiourea: UV Induced Thione → Thiol Reaction and Ground State Thiol → Thione Tunneling. J. Phys. Chem. A 2003, 107, 6373–6380. [Google Scholar] [CrossRef]
- Rostkowska, H.; Lapinski, L.; Nowak, M.J. Hydrogen-atom tunneling through a very high barrier; Spontaneous thiol → thione conversion in thiourea isolated in low-temperature Ar, Ne, H2 and D2 matrices. Phys. Chem. Chem. Phys. 2018, 20, 13994–14002. [Google Scholar] [CrossRef]
- Rostkowska, H.; Lapinski, L.; Khvorostov, A.; Nowak, M.J. Proton transfer processes in selenourea: UV-induced selenone→selenol photoreaction and ground state selenol→selenone proton tunneling. Chem. Phys. 2004, 298, 223–232. [Google Scholar] [CrossRef]
- Lapinski, L.; Rostkowska, H.; Khvorostov, A.; Yaman, M.; Fausto, R.; Nowak, M.J. Double-Proton-Transfer Processes in Dithiooxamide: UV-Induced Dithione → Dithiol Reaction and Ground-State Dithiol → Dithione Tunneling. J. Phys. Chem. A 2004, 108, 5551–5558. [Google Scholar] [CrossRef] [Green Version]
- Marushkevich, K.; Khriachtchev, L.; Räsänen, M. High-energy conformer of formic acid in solid hydrogen: Conformational change promoted by host excitation. Phys. Chem. Chem. Phys. 2007, 9, 5748–5751. [Google Scholar] [CrossRef]
- Ryazantsev, S.V.; Feldman, V.I.; Khriachtchev, L. Conformational Switching of HOCO Radical: Selective Vibrational Excitation and Hydrogen-Atom Tunneling. J. Am. Chem. Soc. 2017, 139, 9551–9557. [Google Scholar] [CrossRef]
- Räsänen, M.; Kunttu, H.; Murto, J. Infrared Induced Conformer Interconversion Processes in Low-Temperature Matrices. Laser Chem. 1988, 9, 123–145. [Google Scholar] [CrossRef] [Green Version]
- Bondybey, V.E. Relaxation and Vibrational Energy Redistribution Processes in Polyatomic Molecules. Annu. Rev. Phys. Chem. 1984, 35, 591–612. [Google Scholar] [CrossRef]
- Domanskaya, A.; Marushkevich, K.; Khriachtchev, L.; Räsänen, M. Spectroscopic study of cis-to-trans tunneling reaction of HCOOD in rare gas matrices. J. Chem. Phys. 2009, 130, 154509. [Google Scholar] [CrossRef]
- Tsuge, M.; Khriachtchev, L. Tunneling Isomerization of Small Carboxylic Acids and Their Complexes in Solid Matrixes: A Computational Insight. J. Phys. Chem. A 2015, 119, 2628–2635. [Google Scholar] [CrossRef] [PubMed]
- Trakhtenberg, L.I.; Fokeyev, A.A.; Zyubin, A.S.; Mebel, A.M.; Lin, S.H. Matrix reorganization with intramolecular tunneling of H atom: Formic acid in Ar matrix. J. Chem. Phys. 2009, 130, 144502. [Google Scholar] [CrossRef]
- Trakhtenberg, L.I.; Fokeyev, A.A.; Zyubin, A.S.; Mebel, A.M.; Lin, S.H. Effect of the Medium on Intramolecular H-Atom Tunneling: Cis−Trans Conversion of Formic Acid in Solid Matrixes of Noble Gases. J. Phys. Chem. B 2010, 114, 17102–17112. [Google Scholar] [CrossRef] [PubMed]
- Marushkevich, K.; Räsänen, M.; Khriachtchev, L. Interaction of Formic Acid with Nitrogen: Stabilization of the Higher-Energy Conformer. J. Phys. Chem. A 2010, 114, 10584–10589. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, P.R.; Wagner, J.P.; Reisenauer, H.P.; Gerbig, D.; Ley, D.; Sarka, J.; Császár, A.G.; Vaughn, A.; Allen, W.D. Domino Tunneling. J. Am. Chem. Soc. 2015, 137, 7828–7834. [Google Scholar] [CrossRef]
- Tsuge, M.; Lee, Y.-P. Spectroscopy of molecules confined in solid para-hydrogen. In Molecular and Laser Spectroscopy; Gupta, V.P., Ozaki, Y., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 167–215. ISBN 978-0-12-818870-5. [Google Scholar]
- Bazsó, G.; Csonka, I.P.; Góbi, S.; Tarczay, G. VIZSLA—Versatile Ice Zigzag Sublimation Setup for Laboratory Astrochemistry. Rev. Sci. Instrum. 2021, 92, 124104. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Maçôas, E.M.S.S.; Khriachtchev, L.; Pettersson, M.; Fausto, R.; Räsänen, M.M. Rotational isomerization of small carboxylic acids isolated in argon matrices: Tunnelling and quantum yields for the photoinduced processes. Phys. Chem. Chem. Phys. 2005, 7, 743–749. [Google Scholar] [CrossRef] [Green Version]
- Hocking, W.H. The Other Rotamer of Formic Acid, cis-HCOOH. Zeitschrift fur Naturforsch. Sect. A J. Phys. Sci. 1976, 31, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- CRC Handbook of Chemistry and Physics; Lide, D.R. (Ed.) CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Jodl, H.J. Solid state aspects of matrices. In Chemistry and Physics of Matrix-Isolated Species; Andrews, L., Moskovits, M., Eds.; North-Holland: Amsterdam, The Netherlands, 1989; p. 430. ISBN 9780444705495. [Google Scholar]
- Cao, Q.; Melavuori, M.; Lundell, J.; Räsänen, M.; Khriachtchev, L. Matrix-isolation and ab initio study of the complex between formic acid and xenon. J. Mol. Struct. 2012, 1025, 132–139. [Google Scholar] [CrossRef]
- Ceponkus, J.; Nelander, B. A simple model for the water o-H 2 complex. J. Chem. Phys. 2006, 124, 2–6. [Google Scholar] [CrossRef]
- Nucara, A.; Calvani, P.; Cunsolo, S.; Lupi, S.; Ruzicka, B. Translational and rotational spectra in the fundamental infrared band of liquid and solid parahydrogen. Phys. Rev. B 1993, 47, 2590–2595. [Google Scholar] [CrossRef]
- Fausto, R.; Khriachtchev, L.; Hamm, P. Conformational changes in cryogenic matrices. In Physics and Chemistry at Low Temperatures; Jenny Stanford Publishing: New York, NY, USA, 2011; pp. 51–84. ISBN 9789814267519. [Google Scholar]

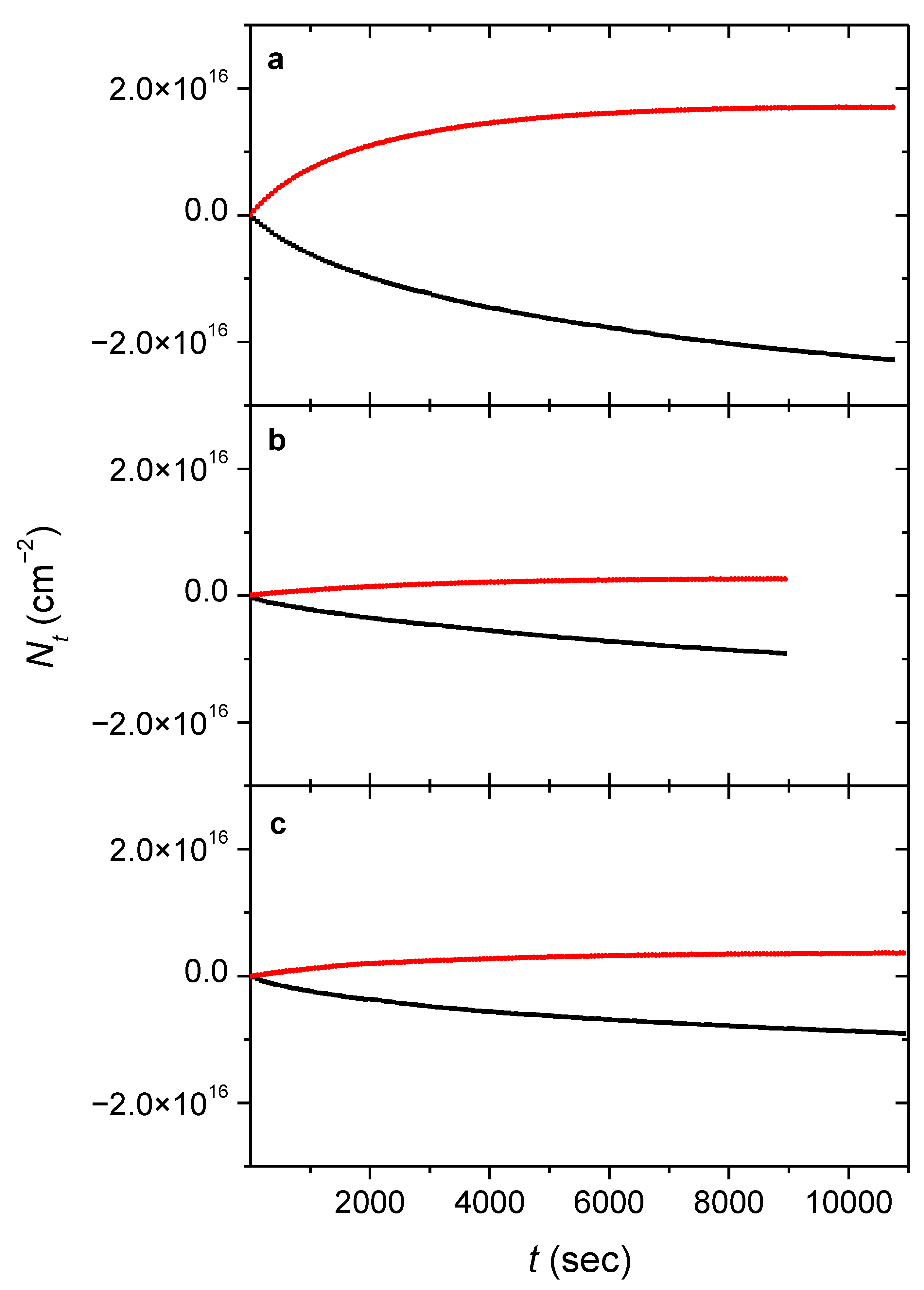
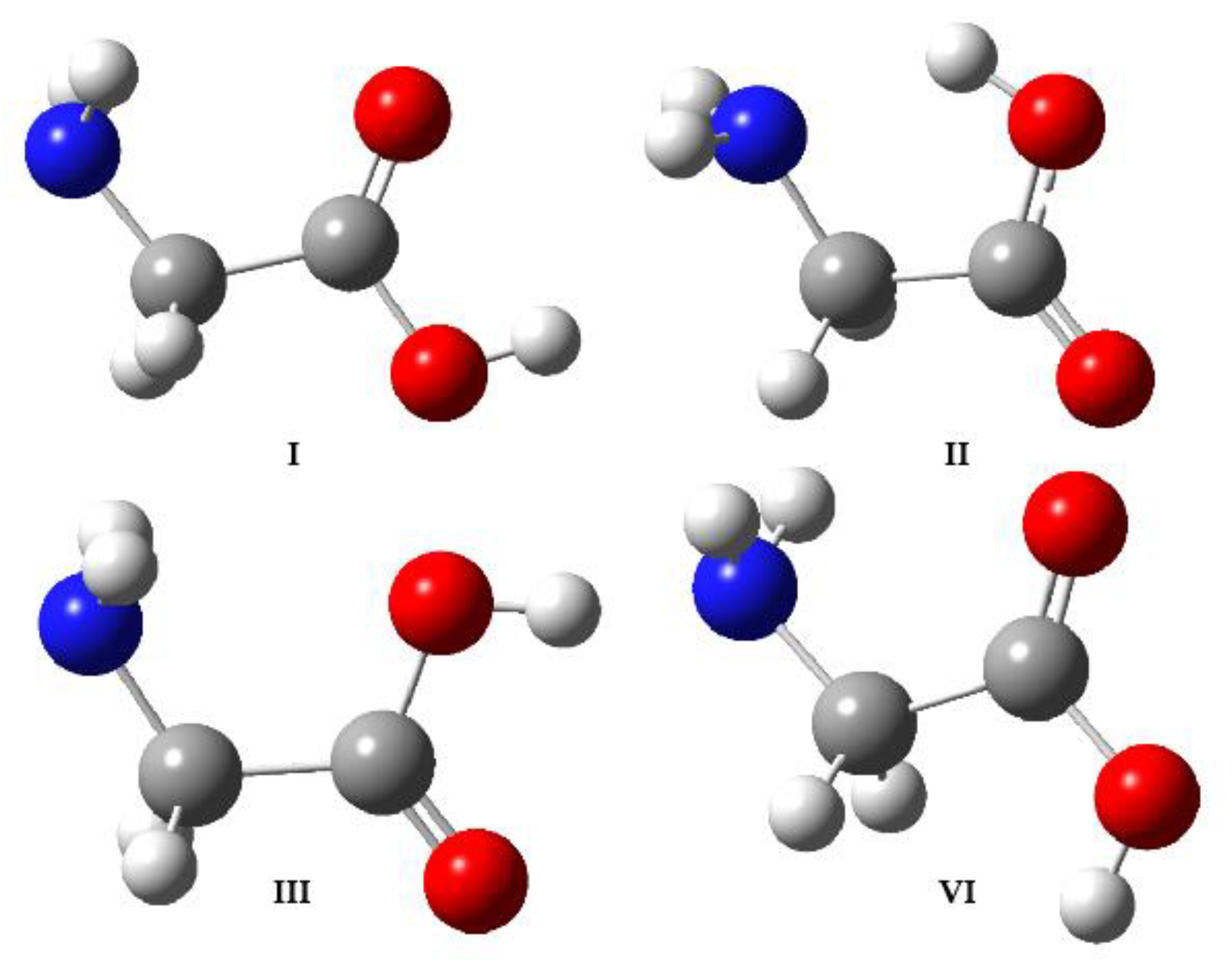

{kind=link}
{kind=link}
{kind=link}
| FA | AA | ||||
|---|---|---|---|---|---|
| Ar (6929.9 cm−1) | Ar (6934.7 cm−1) | n-H2 (6937.6 cm−1) | p-H2 (6940.0 cm−1) | Ar (6957.9 cm−1) | |
| (cm−2) | (2.6 ± 0.4) × 1016 | (1.7 ± 0.5) × 1016 | (1.8 ± 0.3) × 1016 | (1.7 ± 0.1) × 1016 | (5.8 ± 0.9) × 1016 |
| (cm−2) | (5.7 ± 0.3) × 1015 | (1.9 ± 0.1) × 1016 | (2.5 ± 0.1) × 1015 | (3 ± 1) × 1014 | (5.1 ± 0.4) × 1015 |
| (s−1) | (5.7 ± 0.4) × 10−4 | (3 ± 1) × 10−3 | (9.4 ± 0.2) × 10−4 | (4 ± 4) × 10−3 | (2.2 ± 0.1) × 10−3 |
| (cm−2) | (2.8 ± 0.4) × 1016 | (2.8 ± 0.5) × 1016 | (2.0 ± 0.3) × 1016 | (1.7 ± 0.1) × 1016 | (6.3 ± 0.9) × 1016 |
| 0.0108 | 0.0148 | 0.0025 | 0.0025 | 0.0183 | |
| (cm2) | (3.9 ± 0.6) × 10−19 | (5 ± 1) × 10−19 | (1.3 ± 0.2) × 10−19 | (1.5 ± 0.1) × 10−19 | (2.9 ± 0.5) × 10−19 |
| (W) | 7.4 × 10−3 a | 7.4 × 10−3 a | 2.4 × 10−2 | 2.4 × 10−2 | 2.4 × 10−2 |
| (J) | 1.4 × 10−19 | 1.4 × 10−19 | 1.4 × 10−19 | 1.4 × 10−19 | 1.4 × 10−19 |
| (cm−2 s−1) | 5.3 × 1016 | 5.3 × 1016 | 1.7 × 1017 | 1.7 × 1017 | 1.7 × 1017 |
| 2.8 × 10−2 | 1.1 × 10−1 | 4.3 × 10−2 | 1.6 × 10−1 | 4.5 × 10−2 | |
| (s−1) | 1.6 × 1013 | 8.4 × 1013 | 1.9 × 1013 | 7.0 × 1013 | 1.4 × 1014 |
| (s−1) | 1.3 × 1015 | 1.8 × 1015 | 9.8 × 1014 | 9.8 × 1014 | 7.0 × 1015 |
| 1.2 × 10−2 | 4.7 × 10−2 | 1.9 × 10−2 | 7.1 × 10−2 | 2.0 × 10−2 | |
| (2.0 ± 0.8) × 10−2 | (8 ± 3) × 10−2 | (3 ± 1) × 10−2 | (1.2 ± 0.5) × 10−1 | (3 ± 1) × 10−2 | |
| Glycine | |||
|---|---|---|---|
| Ar (6968.8 cm−1) | n-H2 (6963.2 cm−1) a | p-H2 (6968.1 cm−1) a | |
| (s−1) | (4.8 ± 0.1) × 10−4 | (3.5 ± 0.1) × 10−4 | (3.4 ± 0.1) × 10−4 |
| (cm−2) | 6.0 × 1016 | 5.8 × 1016 | 5.8 × 1016 |
| 0.0043 | 0.0056 | 0.0184 | |
| (cm2) | 7.2 × 10−20 | 9.7 × 10−20 | 3.2 × 10−19 |
| (W) | 0.021 ± 0.002 | 0.023 ± 0.001 | 0.021 ± 0.002 |
| (J) | 1.4 × 10−19 | 1.4 × 10−19 | 1.4 × 10−19 |
| (cm−2 s−1) | (1.5 ± 0.1) × 1017 | (1.6 ± 0.1) × 1017 | (1.5 ± 0.1) × 1017 |
| 4.4 × 10−2 | 2.3 × 10−2 | 7.1 × 10−3 | |
| (s−1) | 2.9 × 1013 | 2.0 × 1013 | 2.0 × 1013 |
| (s−1) | 1.5 × 1015 | 2.0 × 1015 | 6.2 × 1015 |
| 1.9 × 10−2 | 1.0 × 10−2 | 3.2 × 10−3 | |
| (3 ± 1) × 10−2 | (1.7 ± 0.7) × 10−2 | (5 ± 3) × 10−3 | |
| Species | Matrix | Ref. | ||
|---|---|---|---|---|
| FA | Ne | - | ≈ 5 ± 1 a | [42] |
| Ar | (2.6 ± 0.5) × 10−3 | 270 ± 50 b | - | |
| (2.4 ± 0.1) × 10−3 | 290 ± 10 c | - | ||
| 2.3 × 10−3 | - | [2,5] | ||
| Kr | ≈1.5 × 10−3 | - | [2] | |
| Xe | ≈4 × 10−4 | - | [2] | |
| N2 | ≈2 × 10−5 | - | [6] | |
| AA | n-H2 | (6.8 ± 0.5) × 10−3 | 102 ± 8 | - |
| - | ≈ 140 a | [48] | ||
| p-H2 | (2.1 ± 0.3) × 10−1 | 3.3 ± 0.6 | - | |
| Ar | (2.5 ± 0.3) × 10−2 | 28 ± 3 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Góbi, S.; Ragupathy, G.; Bazsó, G.; Tarczay, G. Vibrational-Excitation-Induced and Spontaneous Conformational Changes in Solid Para-H2—Diminished Matrix Effects. Photochem 2022, 2, 563-579. https://doi.org/10.3390/photochem2030039
Góbi S, Ragupathy G, Bazsó G, Tarczay G. Vibrational-Excitation-Induced and Spontaneous Conformational Changes in Solid Para-H2—Diminished Matrix Effects. Photochem. 2022; 2(3):563-579. https://doi.org/10.3390/photochem2030039
Chicago/Turabian StyleGóbi, Sándor, Gopi Ragupathy, Gábor Bazsó, and György Tarczay. 2022. "Vibrational-Excitation-Induced and Spontaneous Conformational Changes in Solid Para-H2—Diminished Matrix Effects" Photochem 2, no. 3: 563-579. https://doi.org/10.3390/photochem2030039
APA StyleGóbi, S., Ragupathy, G., Bazsó, G., & Tarczay, G. (2022). Vibrational-Excitation-Induced and Spontaneous Conformational Changes in Solid Para-H2—Diminished Matrix Effects. Photochem, 2(3), 563-579. https://doi.org/10.3390/photochem2030039