


Excited-State Dynamics of Carbazole and tert-Butyl-Carbazole in Thin Films


Abstract

1. Introduction
2. Materials and Methods
2.1. Preparation of Thin Films
2.2. Atomic Force Microscopy and X-ray Diffraction Experiments
2.3. Steady-State Absorption and Photoluminescence
2.4. Time-Correlated Single-Photon Counting (TCSPC)
2.5. Broadband Transient Absorption Spectroscopy
3. Results
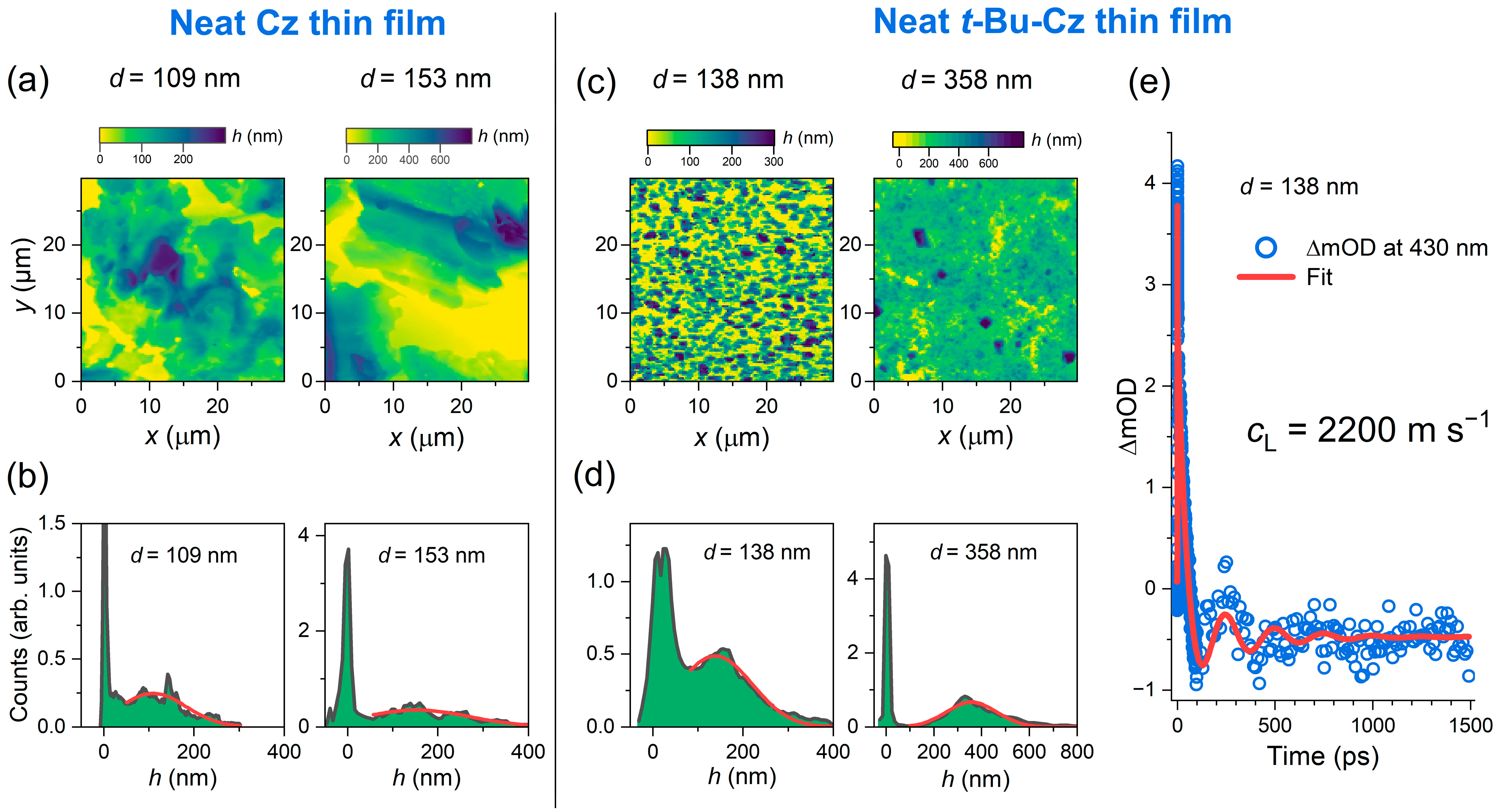
3.1. AFM Images and Picosecond Ultrasonics of Cz and t-Bu-Cz Thin Films on Quartz
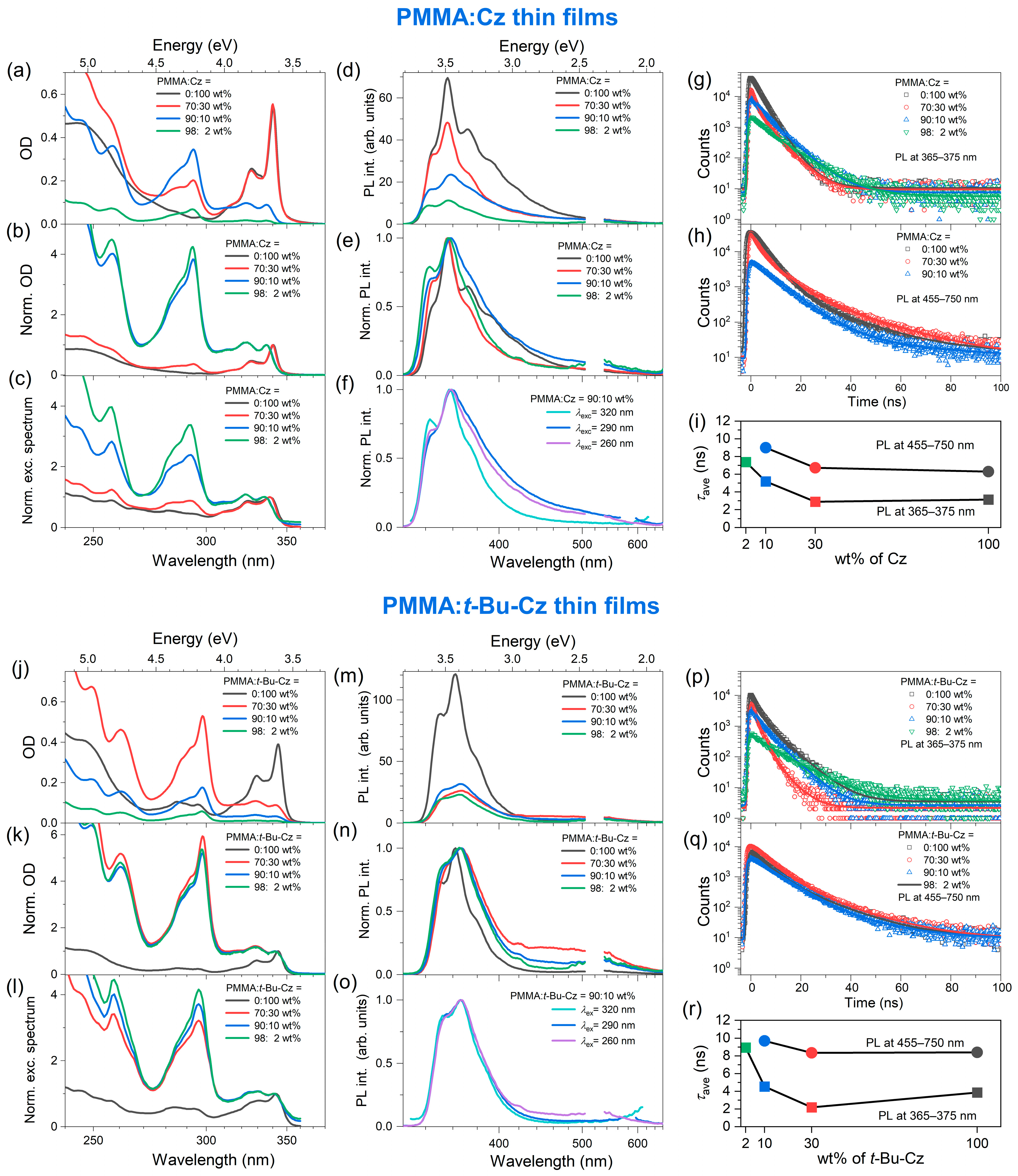
3.2. Steady-State Absorption, Photoluminescence and Transient Emission of the Thin Films
3.3. Femtosecond and Nanosecond UV-Vis-NIR Transient Absorption Spectra
3.4. Kinetic Analysis of Singlet–Singlet Annihilation Processes in Cz and t-Bu-Cz Films
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, J.; Grimsdale, A.C. Carbazole-based polymers for organic photovoltaic devices. Chem. Soc. Rev. 2010, 39, 2399–2410. [Google Scholar] [CrossRef] [PubMed]
- Oner, S.; Bryce, M.R. A review of fused-ring carbazole derivatives as emitter and/or host materials in organic light emitting diode (OLED) applications. Mater. Chem. Front. 2023, 7, 4304–4338. [Google Scholar] [CrossRef]
- Radhakrishna, K.; Manjunath, S.B.; Devadiga, D.; Chetri, R.; Nagaraja, A.T. Review on Carbazole-Based Hole Transporting Materials for Perovskite Solar Cell. ACS Appl. Electron. Mater. 2023, 6, 3635–3664. [Google Scholar] [CrossRef]
- Ledwon, P. Recent advances of donor-acceptor type carbazole-based molecules for light emitting applications. Org. Electron. 2019, 75, 105422. [Google Scholar] [CrossRef]
- Godumala, M.; Choi, S.; Cho, M.J.; Choi, D.H. Recent breakthroughs in thermally activated delayed fluorescence organic light emitting diodes containing non-doped emitting layers. J. Mater. Chem. C 2019, 7, 2172–2198. [Google Scholar] [CrossRef]
- Nakanotani, H.; Sasabe, H.; Adachi, C. Singlet-singlet and singlet-heat annihilations in fluorescence-based organic light-emitting diodes under steady-state high current density. Appl. Phys. Lett. 2005, 86, 213506. [Google Scholar] [CrossRef]
- Hasan, M.; Shukla, A.; Ahmad, V.; Sobus, J.; Bencheikh, F.; McGregor, S.K.M.; Mamada, M.; Adachi, C.; Lo, S.-C.; Namdas, E.B. Exciton–Exciton Annihilation in Thermally Activated Delayed Fluorescence Emitter. Adv. Funct. Mater. 2020, 30, 2000580. [Google Scholar] [CrossRef]
- Bree, A.; Zwarich, R. Vibrational Assignment of Carbazole from Infrared, Raman, and Fluorescence Spectra. J. Chem. Phys. 1968, 49, 3344–3355. [Google Scholar] [CrossRef]
- Chakravorty, S.C.; Ganguly, S.C. Polarized Absorption Spectra of Carbazole Single Crystal. J. Chem. Phys. 1970, 52, 2760–2762. [Google Scholar] [CrossRef]
- Tanaka, M. Electronic States of Fluorene, Carbazole and Dibenzofuran. Bull. Chem. Soc. Jpn. 1976, 49, 3382–3388. [Google Scholar] [CrossRef]
- Nakhimovsky, L.A.; Fuchs, R.; Martin, D.; Small, G.J. Optical Properties of Carbazole Thin Monocrystalline Films. Mol. Cryst. Liq. Cryst. 1988, 154, 89–105. [Google Scholar] [CrossRef]
- Nguyen, D.D.; Trunk, J.; Nakhimovsky, L.; Spanget-Larsen, J. Electronic transitions of fluorene, dibenzofuran, carbazole, and dibenzothiophene: From the onset of absorption to the ionization threshold. J. Mol. Spectrosc. 2010, 264, 19–25. [Google Scholar] [CrossRef]
- Morgenroth, M.; Lenzer, T.; Oum, K. Understanding Excited-State Relaxation in 1,3-Bis(N-carbazolyl)benzene, a Host Material for Organic Light-Emitting Diodes. J. Phys. Chem. C 2023, 127, 4582–4593. [Google Scholar] [CrossRef]
- Scully, S.R.; McGehee, M.D. Effects of optical interference and energy transfer on exciton diffusion length measurements in organic semiconductors. J. Appl. Phys. 2006, 100, 034907. [Google Scholar] [CrossRef]
- Lewis, A.J.; Ruseckas, A.; Gaudin, O.P.M.; Webster, G.R.; Burn, P.L.; Samuel, I.D.W. Singlet exciton diffusion in MEH-PPV films studied by exciton–exciton annihilation. Org. Electron. 2006, 7, 452–456. [Google Scholar] [CrossRef]
- Shaw, P.E.; Ruseckas, A.; Samuel, I.D.W. Exciton Diffusion Measurements in Poly(3-hexylthiophene). Adv. Mater. 2008, 20, 3516–3520. [Google Scholar] [CrossRef]
- Ruseckas, A.; Ribierre, J.C.; Shaw, P.E.; Staton, S.V.; Burn, P.L.; Samuel, I.D.W. Singlet energy transfer and singlet-singlet annihilation in light-emitting blends of organic semiconductors. Appl. Phys. Lett. 2009, 95, 183305. [Google Scholar] [CrossRef]
- Lunt, R.R.; Giebink, N.C.; Belak, A.A.; Benziger, J.B.; Forrest, S.R. Exciton diffusion lengths of organic semiconductor thin films measured by spectrally resolved photoluminescence quenching. J. Appl. Phys. 2009, 105, 053711. [Google Scholar] [CrossRef]
- Cook, S.; Furube, A.; Katoh, R.; Han, L. Estimate of singlet diffusion lengths in PCBM films by time-resolved emission studies. Chem. Phys. Lett. 2009, 478, 33–36. [Google Scholar] [CrossRef]
- Cook, S.; Liyuan, H.; Furube, A.; Katoh, R. Singlet Annihilation in Films of Regioregular Poly(3-hexylthiophene): Estimates for Singlet Diffusion Lengths and the Correlation between Singlet Annihilation Rates and Spectral Relaxation. J. Phys. Chem. C 2010, 114, 10962–10968. [Google Scholar] [CrossRef]
- Shaw, P.E.; Ruseckas, A.; Peet, J.; Bazan, G.C.; Samuel, I.D.W. Exciton-Exciton Annihilation in Mixed-Phase Polyfluorene Films. Adv. Funct. Mater. 2010, 20, 155–161. [Google Scholar] [CrossRef]
- Masri, Z.; Ruseckas, A.; Emelianova, E.V.; Wang, L.; Bansal, A.K.; Matheson, A.; Lemke, H.T.; Nielsen, M.N.; Nguyen, H.; Coulembier, O.; et al. Molecular Weight Dependence of Exciton Diffusion in Poly(3-hexylthiophene). Adv. Energy Mater. 2013, 3, 1445–1453. [Google Scholar] [CrossRef]
- Lin, J.D.A.; Mikhnenko, O.V.; Chen, J.; Masri, Z.; Ruseckas, A.; Mikhailovsky, A.; Raab, R.P.; Liu, J.; Blom, P.W.M.; Loi, M.A.; et al. Systematic study of exciton diffusion length in organic semiconductors by six experimental methods. Mater. Horiz. 2014, 1, 280–285. [Google Scholar] [CrossRef]
- Bardeen, C.J. The Structure and Dynamics of Molecular Excitons. Annu. Rev. Phys. Chem. 2014, 65, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Gulbinas, V.; Valkunas, L.; Kuciauskas, D.; Katilius, E.; Liuolia, V.; Zhou, W.; Blankenship, R.E. Singlet-Singlet Annihilation and Local Heating in FMO Complexes. J. Phys. Chem. 1996, 100, 17950–17956. [Google Scholar] [CrossRef]
- Stevens, M.A.; Silva, C.; Russell, D.M.; Friend, R.H. Exciton dissociation mechanisms in the polymeric semiconductors poly(9,9-dioctylfluorene) and poly(9,9-dioctylfluorene-co-benzothiadiazole). Phys. Rev. B 2001, 63, 165213. [Google Scholar] [CrossRef]
- King, S.M.; Dai, D.; Rothe, C.; Monkman, A.P. Exciton annihilation in a polyfluorene: Low threshold for singlet-singlet annihilation and the absence of singlet-triplet annihilation. Phys. Rev. B 2007, 76, 085204. [Google Scholar] [CrossRef]
- Zaushitsyn, Y.; Jespersen, K.G.; Valkunas, L.; Sundström, V.; Yartsev, A. Ultrafast dynamics of singlet-singlet and singlet-triplet exciton annihilation in poly(3-2′-methoxy-5′-octylphenyl)thiophene films. Phys. Rev. B 2007, 75, 195201. [Google Scholar] [CrossRef]
- Völker, S.F.; Schmiedel, A.; Holzapfel, M.; Renziehausen, K.; Engel, V.; Lambert, C. Singlet−Singlet Exciton Annihilation in an Exciton-Coupled Squaraine-Squaraine Copolymer: A Model toward Hetero-J-Aggregates. J. Phys. Chem. C 2014, 118, 17467–17482. [Google Scholar] [CrossRef]
- Chen, C.; Chi, Z.; Chong, K.C.; Batsanov, A.S.; Yang, Z.; Mao, Z.; Yang, Z.; Liu, B. Carbazole isomers induce ultralong organic phosphorescence. Nat. Mater. 2021, 20, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chong, K.C.; Pan, Y.; Qi, G.; Xu, S.; Liu, B. Revisiting Carbazole: Origin, Impurity, and Properties. ACS Mater. Lett. 2021, 3, 1081–1087. [Google Scholar] [CrossRef]
- Lutterotti, L. Total pattern fitting for the combined size-strain-stress-texture determination in thin film diffraction. Nucl. Instrum. Methods Phys. Res. Sect. B 2010, 268, 334–340. [Google Scholar] [CrossRef]
- Dobryakov, A.L.; Kovalenko, S.A.; Weigel, A.; Pérez Lustres, J.L.; Lange, J.; Müller, A.; Ernsting, N.P. Femtosecond pump/supercontinuum-probe spectroscopy: Optimized setup and signal analysis for single-shot spectral referencing. Rev. Sci. Instrum. 2010, 81, 113106. [Google Scholar] [CrossRef] [PubMed]
- Oum, K.; Lenzer, T.; Scholz, M.; Jung, D.Y.; Sul, O.; Cho, B.J.; Lange, J.; Müller, A. Observation of Ultrafast Carrier Dynamics and Phonon Relaxation of Graphene from the Deep-Ultraviolet to the Visible Region. J. Phys. Chem. C 2014, 118, 6454–6461. [Google Scholar] [CrossRef]
- Flender, O.; Scholz, M.; Klein, J.R.; Oum, K.; Lenzer, T. Excited-state relaxation of the solar cell dye D49 in organic solvents and on mesoporous Al2O3 and TiO2 thin films. Phys. Chem. Chem. Phys. 2016, 18, 26010–26019. [Google Scholar] [CrossRef] [PubMed]
- Merker, A.; Scholz, M.; Morgenroth, M.; Lenzer, T.; Oum, K. Photoinduced Dynamics of (CH3NH3)4Cu2Br6 Thin Films Indicating Efficient Triplet Photoluminescence. J. Phys. Chem. Lett. 2021, 12, 2736–2741. [Google Scholar] [CrossRef]
- Ruello, P.; Gusev, V.E. Physical mechanisms of coherent acoustic phonons generation by ultrafast laser action. Ultrasonics 2015, 56, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, C.; Strait, J.; Vardeny, Z.; Maris, H.J.; Tauc, J.; Hauser, J.J. Coherent Phonon Generation and Detection by Picosecond Light Pulses. Phys. Rev. Lett. 1984, 53, 989–992. [Google Scholar] [CrossRef]
- Grahn, H.T.; Maris, H.J.; Tauc, J. Picosecond Ultrasonics. IEEE J. Quantum Electron. 1989, 25, 2562–2569. [Google Scholar] [CrossRef]
- Kanner, G.S.; Vardeny, Z.V.; Hess, B.C. Picosecond acoustics in polythiophene thin films. Phys. Rev. B 1990, 42, 5403–5406. [Google Scholar] [CrossRef] [PubMed]
- Kaake, L.G.; Welch, G.C.; Moses, D.; Bazan, G.C.; Heeger, A.J. Influence of Processing Additives on Charge-Transfer Time Scales and Sound Velocity in Organic Bulk Heterojunction Films. J. Phys. Chem. Lett. 2012, 3, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Scholz, M.; Morgenroth, M.; Cho, M.J.; Choi, D.H.; Lenzer, T.; Oum, K. Coherent acoustic phonon dynamics in chiral copolymers. Struct. Dyn. 2019, 6, 064502. [Google Scholar] [CrossRef] [PubMed]
- Nowak, R.; Bernstein, E.R. On the phase transition in N-isopropylcarbazole. J. Chem. Phys. 1986, 85, 6858–6866. [Google Scholar] [CrossRef]
- Auty, A.R.; Jones, A.C.; Phillips, D. Spectroscopy and decay dynamics of jet-cooled carbazole and N-ethylcarbazole and their homocyclic analogues. Chem. Phys. 1976, 103, 163–182. [Google Scholar] [CrossRef]
- Bigelow, R.W.; Ceasar, G.P. Hydrogen Bonding and N-Alkylation Effects on the Electronic Structure of Carbazole. J. Phys. Chem. 1979, 83, 1790–1795. [Google Scholar] [CrossRef]
- Yi, J.T.; Alvarez-Valtierra, L.; Pratt, D.W. Rotationally resolved S1 ← S0 electronic spectra of fluorene, carbazole, and dibenzofuran: Evidence for Herzberg-Teller coupling with the S2 state. J. Chem. Phys. 2006, 124, 244302. [Google Scholar] [CrossRef] [PubMed]
- Ljubić, I.; Sabljić, A. CASSCF/CASPT2 and TD-DFT Study of Valence and Rydberg Electronic Transitions in Fluorene, Carbazole, Dibenzofuran, and Dibenzothiophene. J. Phys. Chem. A 2011, 115, 4840–4850. [Google Scholar] [CrossRef]
- Bonesi, S.M.; Erra-Balsells, R. Electronic spectroscopy of carbazole and N- and C-substituted carbazoles in homogeneous media and in solid matrix. J. Lumin. 2001, 93, 51–74. [Google Scholar] [CrossRef]
- Knötig, K.M.; Gust, D.; Lenzer, T.; Oum, K. Excited-State Dynamics of Carbazole and tert-Butyl-Carbazole in Organic Solvents. Photochem 2024, 4, 163–178. [Google Scholar] [CrossRef]
- Gajda, K.; Zarychta, B.; Kopka, K.; Daszkiewicz, Z.; Ejsmont, K. Substituent effects in nitro derivatives of carbazoles investigated by comparison of low-temperature crystallographic studies with density functional theory (DFT) calculations. Acta Crystallogr. Sect. C Struct. Chem. 2014, 70, 987–991. [Google Scholar] [CrossRef]
- Yan, Q.; Gin, E.; Wasinska-Kalwa, M.; Banwell, M.G.; Carr, P.D. A Palladium-Catalyzed Ullmann Cross-Coupling/Reductive Cyclization Route to the Carbazole Natural Products 3-Methyl-9H-carbazole, Glycoborine, Glycozoline, Clauszoline K, Mukonine, and Karapinchamine A. J. Org. Chem. 2017, 82, 4148–4159. [Google Scholar] [CrossRef] [PubMed]
- Spano, F.C. The Spectral Signatures of Frenkel Polarons in H- and J-Aggregates. Acc. Chem. Res. 2010, 43, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Hernández, F.J.; Crespo-Otero, R. Excited state mechanisms in crystalline carbazole: The role of aggregation and isomeric defects. J. Mater. Chem. C 2021, 9, 11882–11892. [Google Scholar] [CrossRef]
- Dicke, R.H. Coherence in Spontaneous Radiation Processes. Phys. Rev. 1954, 93, 99–110. [Google Scholar] [CrossRef]
- Blach, D.D.; Lumsargis, V.A.; Clark, D.E.; Chuang, C.; Wang, K.; Dou, L.; Schaller, R.D.; Cao, J.; Li, C.W.; Huang, L. Superradiance and Exciton Delocalization in Perovskite Quantum Dot Superlattices. Nano Lett. 2022, 22, 7811–7818. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.E.; Mantulin, W.W.; Huber, J.R. Effect of Molecular Geometry on Spin-Orbit Coupling of Aromatic Amines in Solution. Diphenylamine, Iminobibenzyl, Acridan, and Carbazole. J. Am. Chem. Soc. 1973, 95, 5477–5481. [Google Scholar] [CrossRef]
- Martin, M.M.; Ware, W.R. Fluorescence Quenching of Carbazole by Pyridine and Substituted Pyridines. Radiationless Processes in the Carbazole-Amine Hydrogen Bonded Complex. J. Phys. Chem. 1978, 82, 2770–2776. [Google Scholar] [CrossRef]
- Förster, T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann. Phys. 1948, 437, 55–75. [Google Scholar] [CrossRef]
- Förster, T. Transfer Mechanisms of Electronic Excitation. Discuss. Faraday Soc. 1959, 27, 7–17. [Google Scholar] [CrossRef]
- Mikhnenko, O.V.; Blom, P.W.M.; Nguyen, T.-Q. Exciton diffusion in organic semiconductors. Energy Environ. Sci. 2015, 8, 1867–1888. [Google Scholar] [CrossRef]
- Stalindurai, K.; Krishnan, K.G.; Nagarajan, E.R.; Ramalingan, C. Experimental and theoretical studies on new 7-(3,6-di-tert-butyl-9H-carbazol-9-yl)-10-alkyl-10H-phenothiazine-3-carbaldehydes. J. Mol. Struct. 2017, 1130, 633–643. [Google Scholar] [CrossRef]
- Hofkens, J.; Cotlet, M.; Vosch, T.; Tinnefeld, P.; Weston, K.D.; Ego, C.; Grimsdale, A.; Müllen, K.; Beljonne, D.; Brédas, J.L.; et al. Revealing competitive Förster-type resonance energy-transfer pathways in single bichromophoric molecules. Proc. Natl. Acad. Sci. USA 2003, 100, 13146–13151. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.; St-Pierre, F.; Gong, Y.; Marshall, J.D.; Cranfill, P.J.; Baird, M.A.; McKeown, M.R.; Wiedenmann, J.; Davidson, M.W.; Schnitzer, M.J.; et al. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 2012, 9, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.M.; Galliardt, H.; Schneider, J.; Barisas, B.G.; Seidel, T. Quantification of Förster resonance energy transfer by monitoring sensitized emission in living plant cells. Front. Plant Sci. 2013, 4, 413. [Google Scholar] [CrossRef] [PubMed]
- Murawski, C.; Leo, K.; Gather, M.C. Efficiency Roll-Off in Organic Light-Emitting Diodes. Adv. Mater. 2013, 25, 6801–6827. [Google Scholar] [CrossRef] [PubMed]
- Morgenroth, M.; Scholz, M.; Cho, M.J.; Choi, D.H.; Oum, K.; Lenzer, T. Mapping the broadband circular dichroism of copolymer films with supramolecular chirality in time and space. Nat. Commun. 2022, 13, 210. [Google Scholar] [CrossRef] [PubMed]
- Morgenroth, M.; Scholz, M.; Guy, L.; Oum, K.; Lenzer, T. Spatiotemporal Mapping of Efficient Chiral Induction by Helicene-Type Additives in Copolymer Thin Films. Angew. Chem. Int. Ed. 2022, 61, e202203075. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.B.; Michl, J. Singlet Fission. Chem. Rev. 2010, 110, 6891–6936. [Google Scholar] [CrossRef] [PubMed]
- Wachsstock, D. Tenua 2.1—The Kinetics Simulator for Java. 2007. Available online: http://bililite.com/tenua/ (accessed on 5 January 2024).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thin Film Composition | Mixing Ratio (wt%:wt%) | λabs0–0 (nm) | λPL0–0 (nm) | λmaxPL (nm) | ∆λStokes (nm) 1 | Stokes (meV) 1 | Stokes (cm−1) 1 |
|---|---|---|---|---|---|---|---|
| PMMA:Cz | 0:100 | 341 2 | 344 | 356 | 3 | 32 | 260 |
| 70:30 | 340 2 | 344 | 356 | 4 | 42 | 340 | |
| 90:10 | 336 | 342 | 358 | 6 | 64 | 520 | |
| 98:2 | 336 | 341 | 357 | 5 | 55 | 440 | |
| PMMA:t-Bu-Cz | 0:100 | 344 2 | 342 | 349 | 5 | 52 | 420 |
| 70:30 | 342 | 341 | 351 | 9 | 93 | 750 | |
| 90:10 | 342 | 341 | 348 | 6 | 62 | 500 | |
| 98:2 | 342 | 341 | 349 | 7 | 73 | 590 |
| Thin Film Composition | Mixing Ratio (wt%:wt%) | λprobe (nm) | τ1 (ns) 1 | τ2 (ns) 1 | τ3 (ns) 1 | τ4 (ns) 1 | 〈τ〉 (ns) 1 |
|---|---|---|---|---|---|---|---|
| PMMA:Cz | 0:100 | 365–375 | 3.0 (97%) 1 | 5.7 (3%) | – | – | 3.1 |
| 455–750 | 1.1 (46%) | 2.9 (23%) | 4.9 (30%) | 16.66 (1%) | 4.5 | ||
| 70:30 | 365–375 | 0.6 (64%) | 2.2 (26%) | 5.2 (10%) | – | 2.9 | |
| 455–750 | 2.1 (67%) | 6.5 (30%) | 18.8 (3%) | – | 6.7 | ||
| 90:10 | 365–375 | 3.7 (62%) | 6.6 (38%) | – | – | 5.2 | |
| 455–750 | 1.0 (33%) | 7.2 (65%) | 20.5 (2%) | – | 8.1 | ||
| 98:2 | 365–375 | 3.9 (37%) | 8.3 (63%) | – | – | 7.4 | |
| 455–750 2 | – | – | – | – | – | ||
| PMMA:t-Bu-Cz | 0:100 | 365–375 | 1.0 (47%) | 3.1 (40%) | 6.5 (13%) | – | 3.9 |
| 455–750 | 0.3 (40%) | 5.4 (51%) | 14.2 (9%) | – | 8.0 | ||
| 70:30 | 365–375 | 1.1 (58%) | 2.5 (39%) | 5.3 (3%) | – | 2.2 | |
| 455–750 | 0.8 (38%) | 6.3 (59%) | 17.2 (3%) | – | 7.4 | ||
| 90:10 | 365–375 | 1.3 (24%) | 3.6 (46%) | 6.0 (30%) | – | 4.5 | |
| 455–750 | 1.2 (26%) | 7.9 (71%) | 22.3 (3%) | – | 8.9 | ||
| 98:2 | 365–375 | 1.2 (14%) | 3.8 (22%) | 9.8 (64%) | – | 8.9 | |
| 455–750 2 | – | – | – | – | – |
| Expt. | λpump (nm) | PMMA:Cz (wt%:wt%) | λprobe (nm) | τ1 (ns) | τ2 (ns) | τ3 (ns) | τ4 (ns) |
|---|---|---|---|---|---|---|---|
| fs-TA 1 | 260 | 0:100 | global | 0.012 (40%) | 640 (59%) | 3 2 (1%) | – |
| 70:30 | global | 0.0001 (9%) | 0.007 (18%) | 0.107 (24%) | 0.580 (49%) | ||
| 90:10 | global | 0.001 (15%) | 0.020 (25%) | 0.209 (35%) | 3.7 2 (25%) | ||
| ns-TA 1 | 260 | 0:100 | global | 3 (47%) | 41 (37%) | 790 (16%) | – |
| 70:30 | global | 0.580 (24%) | 13 (20%) | 113 (49%) | 5100 (7%) | ||
| 90:10 | global | 3.7 (20%) | 223 (50%) | 7700 (30%) | – |
| Physical Quantity | Cz | t-Bu-Cz | Physical Quantity | Cz | t-Bu-Cz |
|---|---|---|---|---|---|
| Nx (cm−3) | 4.0 × 1018 | 3.8 × 1018 | F (mJ cm−2) 1 | 1.03 | 0.92 |
| kx (s−1) | 5.0 × 1012 | 5.0 × 1012 | τx (fs) | 200 | 200 |
| k1,IC+Fl (s−1) | 1.6 × 108 | 1.3 × 108 | τ1,IC+Fl (ns) | 6.3 | 7.7 |
| k1,ISC (s−1) | 1.6 × 108 | 1.3 × 108 | τ1,ISC (ns) | 6.3 | 7.7 |
| k2,ISC+Ph (s−1) | 1.25 × 106 | 1.25 × 106 | τ2,ISC+Ph (μs) | 0.8 | 0.8 |
| kCET (s−1) | 1.57 × 108 | 1.57 × 108 | τCET (ns) | 0.64 | 0.64 |
| kdiff (cm3 s−1) | 2.00 × 10−8 | 1.25 × 10−8 | kF,1 (cm6 s−1) 2 | 2.40 × 10−27 | 1.80 × 10−27 |
| kn (s−1) | 5.0 × 1013 | 5.0 × 1013 | τn (fs) | 20 | 20 |
| d (nm) 3 | 153 | 138 | A 4 | 0.397 | 0.371 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knötig, K.M.; Gust, D.; Oum, K.; Lenzer, T. Excited-State Dynamics of Carbazole and tert-Butyl-Carbazole in Thin Films. Photochem 2024, 4, 179-197. https://doi.org/10.3390/photochem4020011
Knötig KM, Gust D, Oum K, Lenzer T. Excited-State Dynamics of Carbazole and tert-Butyl-Carbazole in Thin Films. Photochem. 2024; 4(2):179-197. https://doi.org/10.3390/photochem4020011
Chicago/Turabian StyleKnötig, Konstantin Moritz, Domenic Gust, Kawon Oum, and Thomas Lenzer. 2024. "Excited-State Dynamics of Carbazole and tert-Butyl-Carbazole in Thin Films" Photochem 4, no. 2: 179-197. https://doi.org/10.3390/photochem4020011
APA StyleKnötig, K. M., Gust, D., Oum, K., & Lenzer, T. (2024). Excited-State Dynamics of Carbazole and tert-Butyl-Carbazole in Thin Films. Photochem, 4(2), 179-197. https://doi.org/10.3390/photochem4020011