

Depleted-MLH1 Expression Predicts Prognosis and Immunotherapeutic Efficacy in Uterine Corpus Endometrial Cancer: An In Silico Approach



Abstract
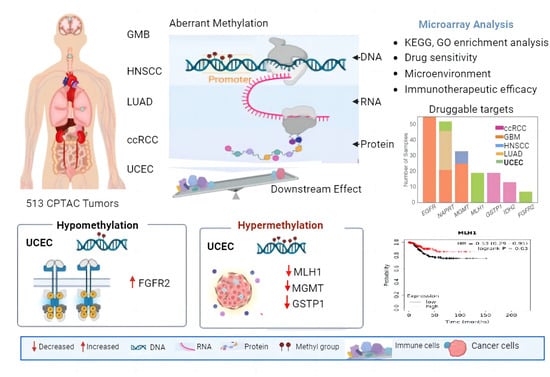
:
1. Introduction
2. Results
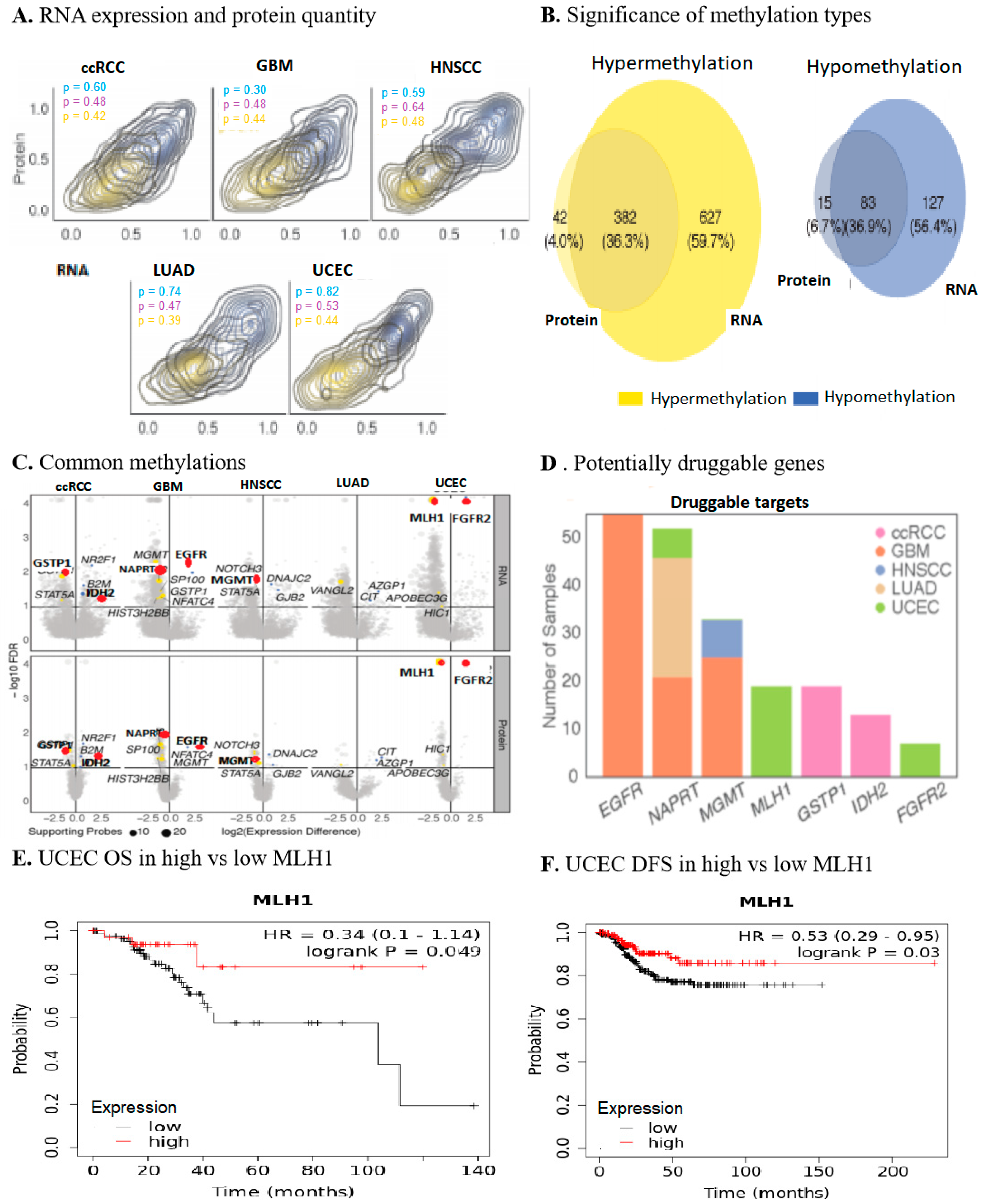
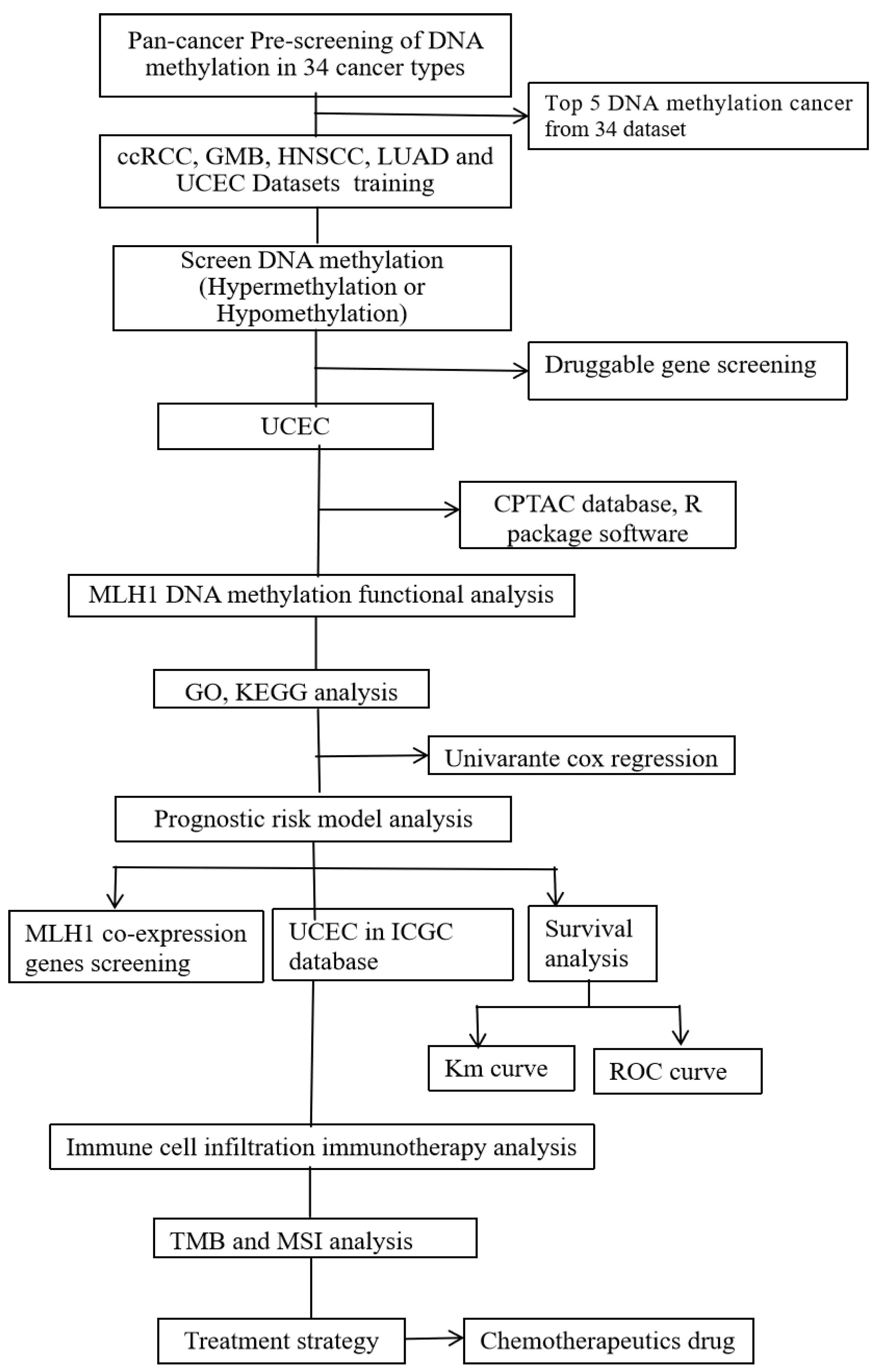
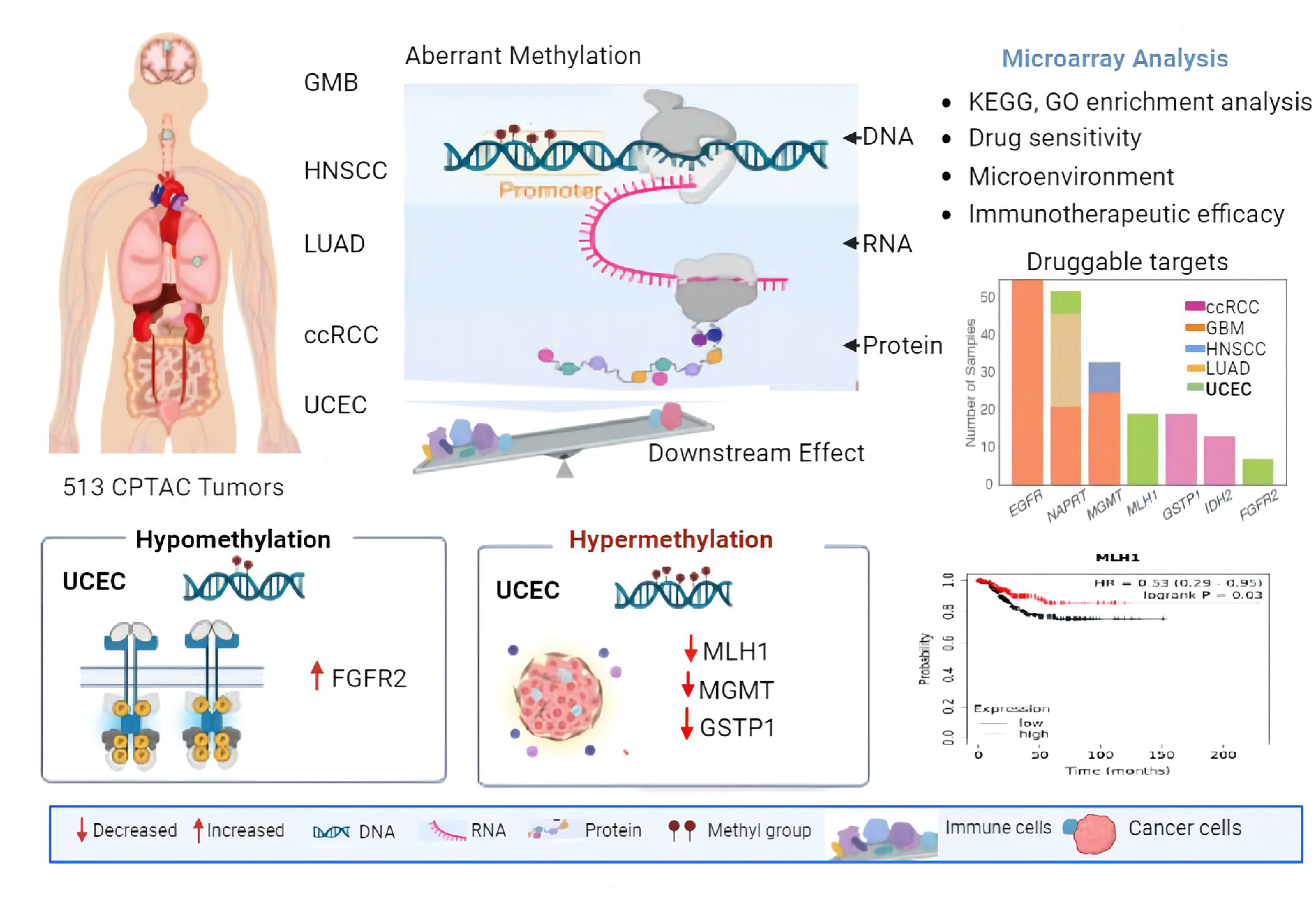
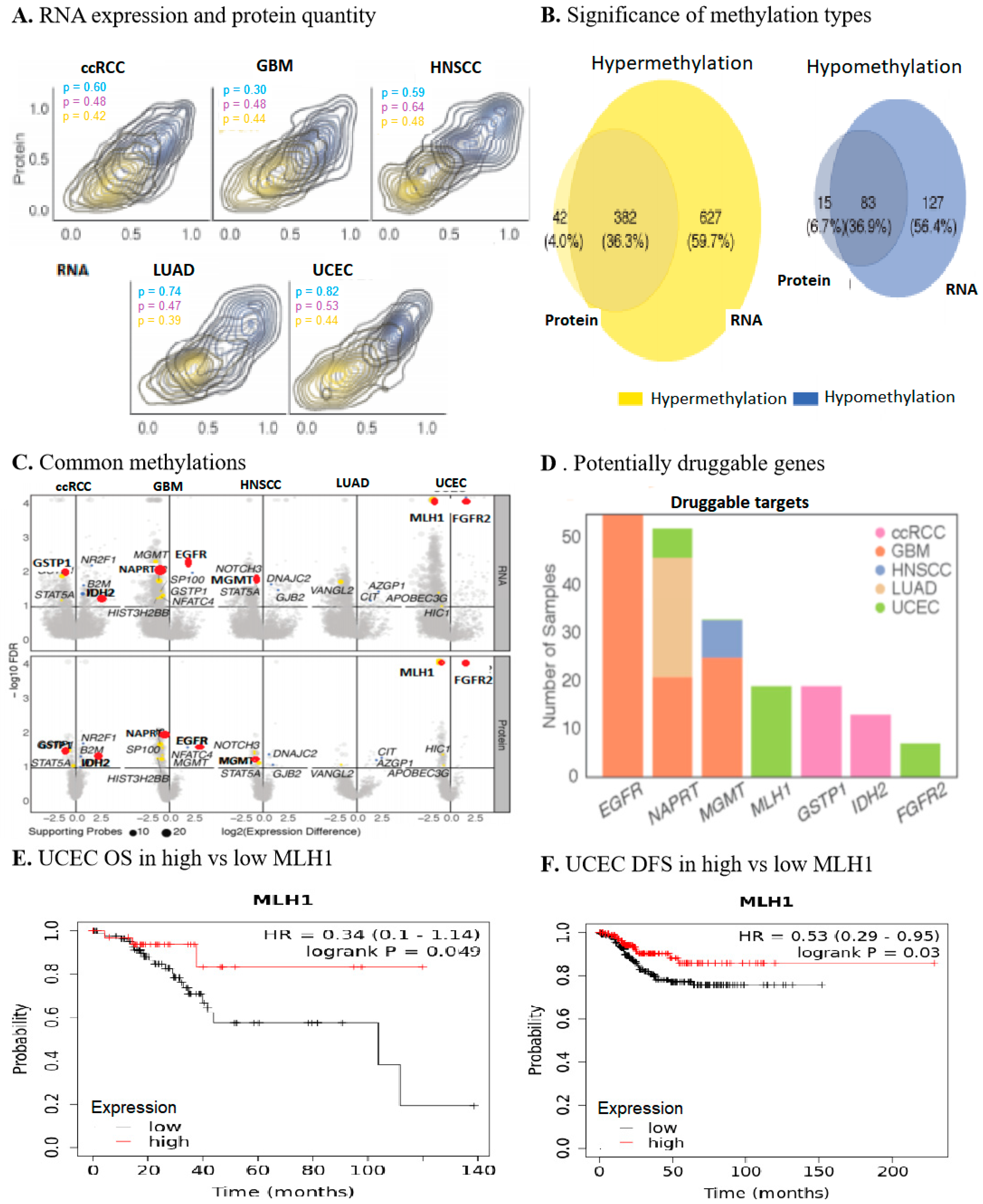
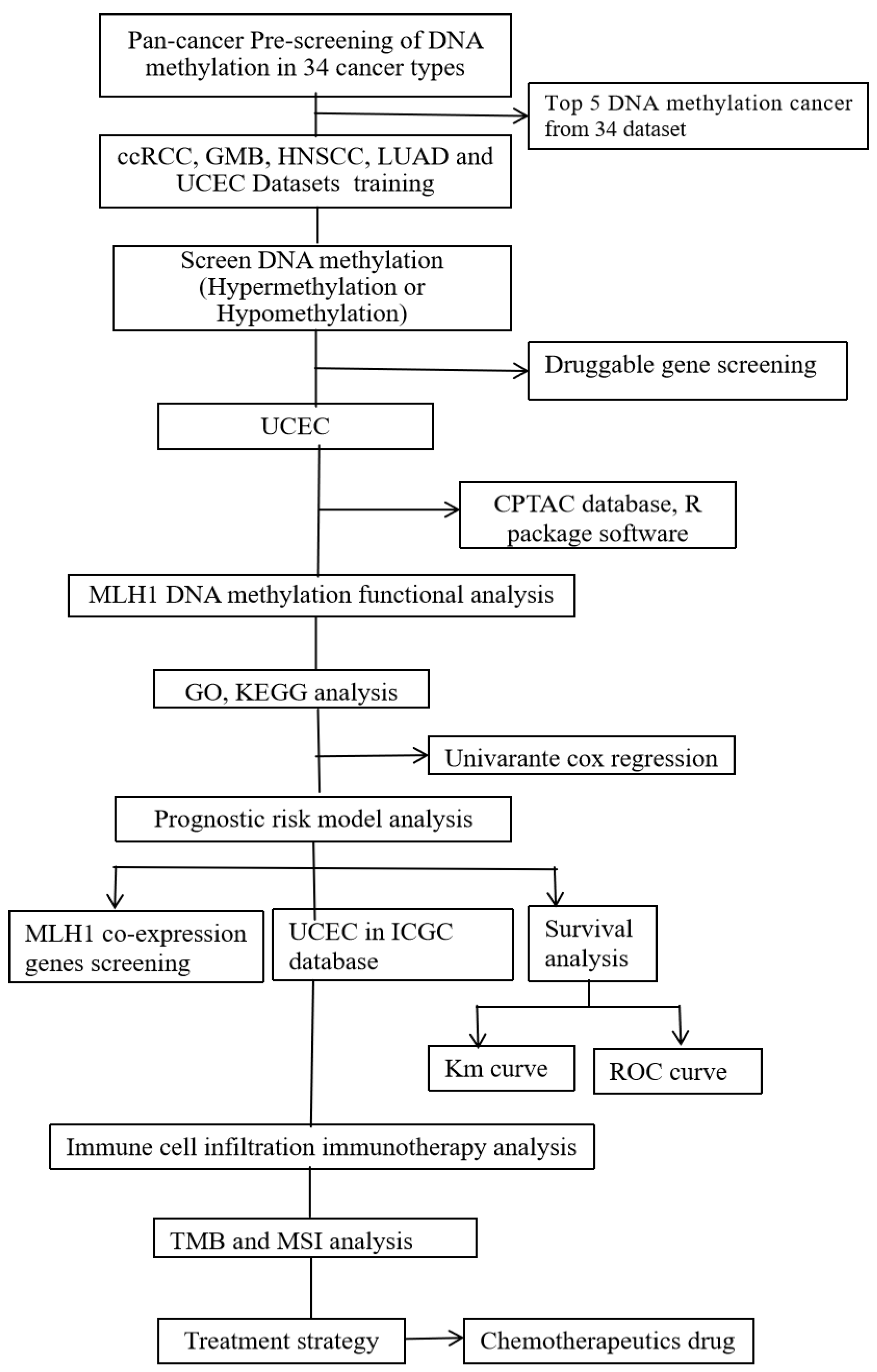
2.1. Multi-Omics Approach Identified MLH1 as a Druggable Target in UCEC
2.2. MLH1 Depletion in UCEC Positively Correlated with Poorer OS and DFS
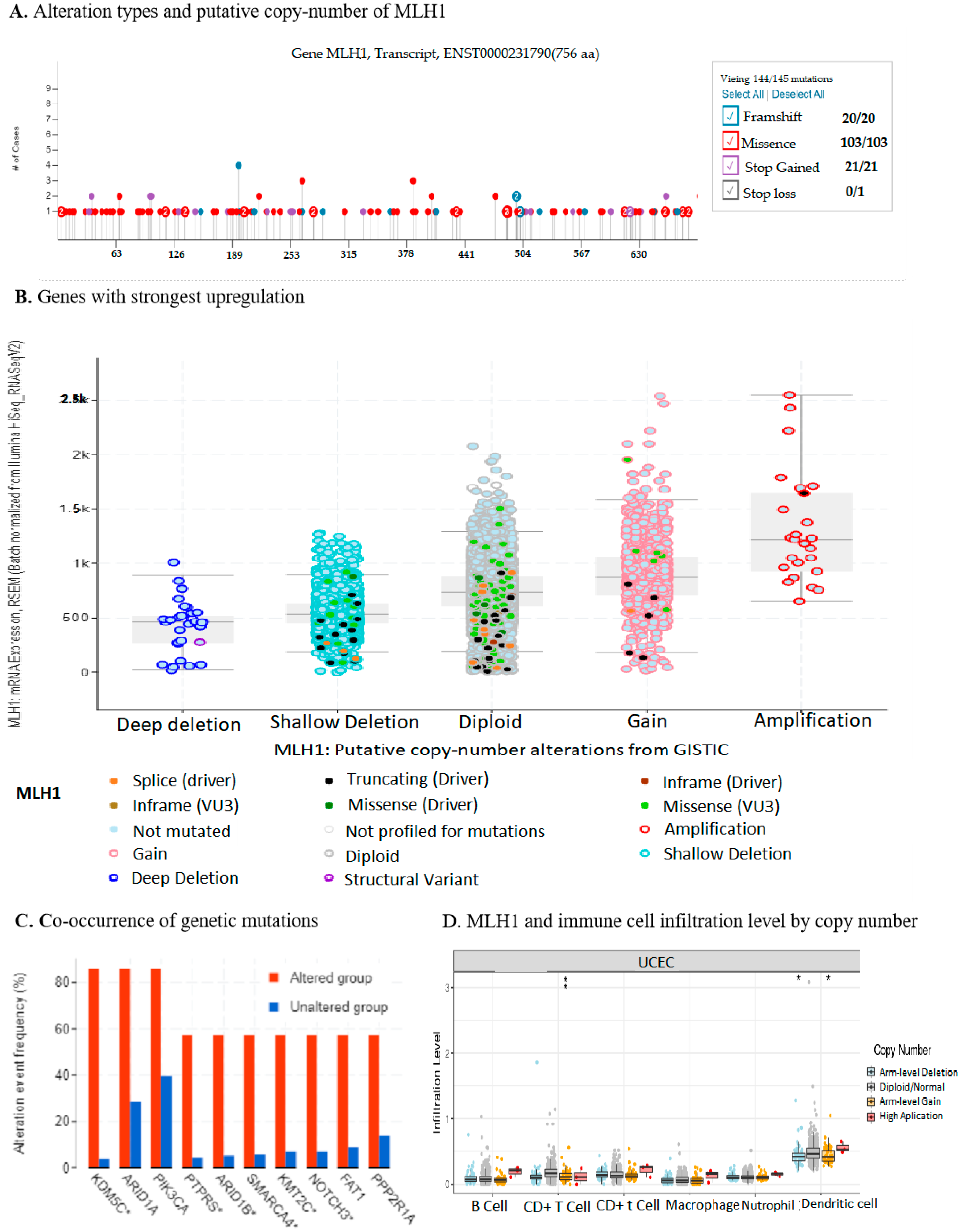
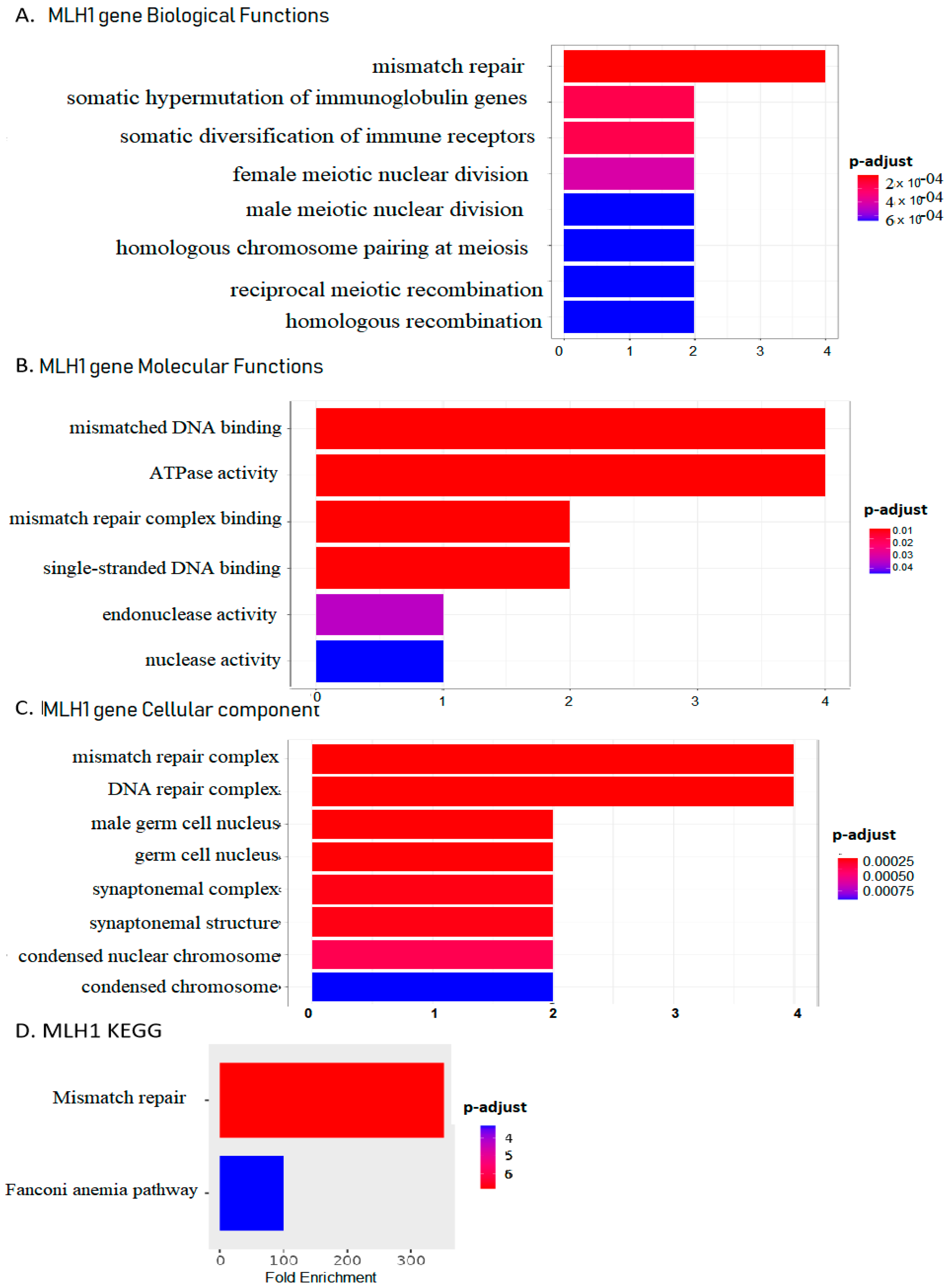
2.3. MLH1 Plays Roles in Mismatch Repair, Fanconi Anemia Pathways, ATPase Activity, and Endonuclease Activity in UCEC
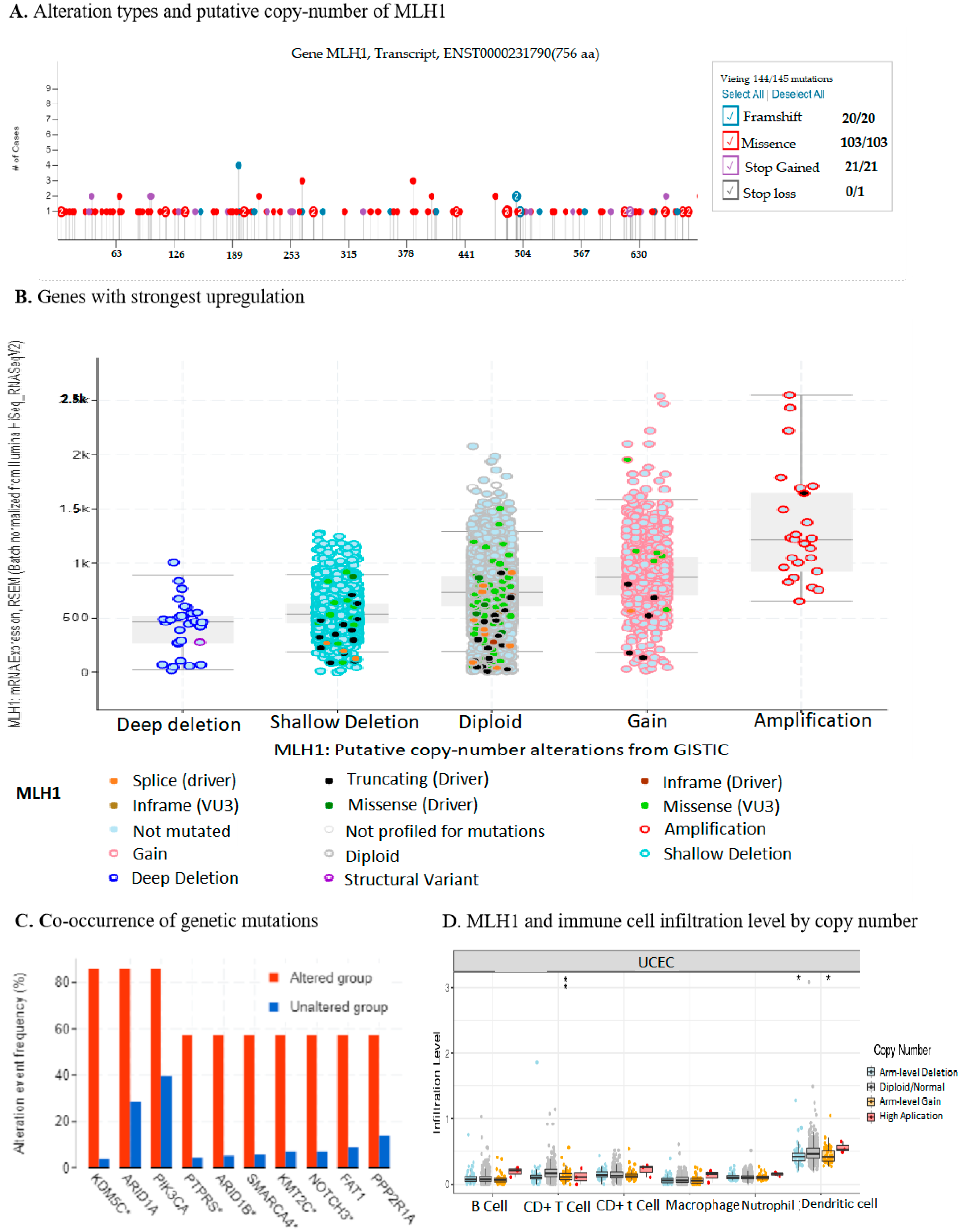
2.4. Downregulation of MLH1 Enhanced TMB and Worsened the Prognosis of UCEC
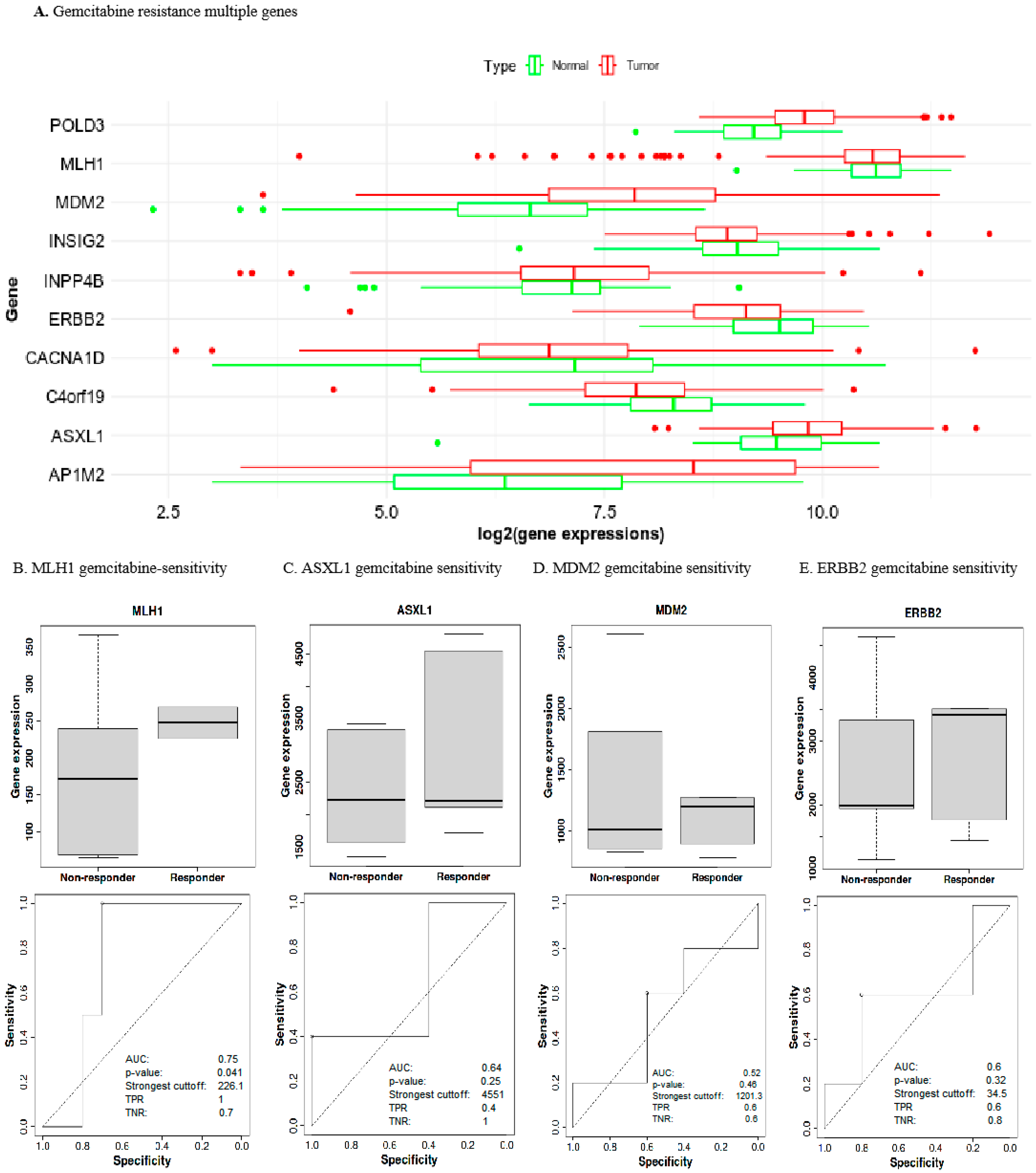
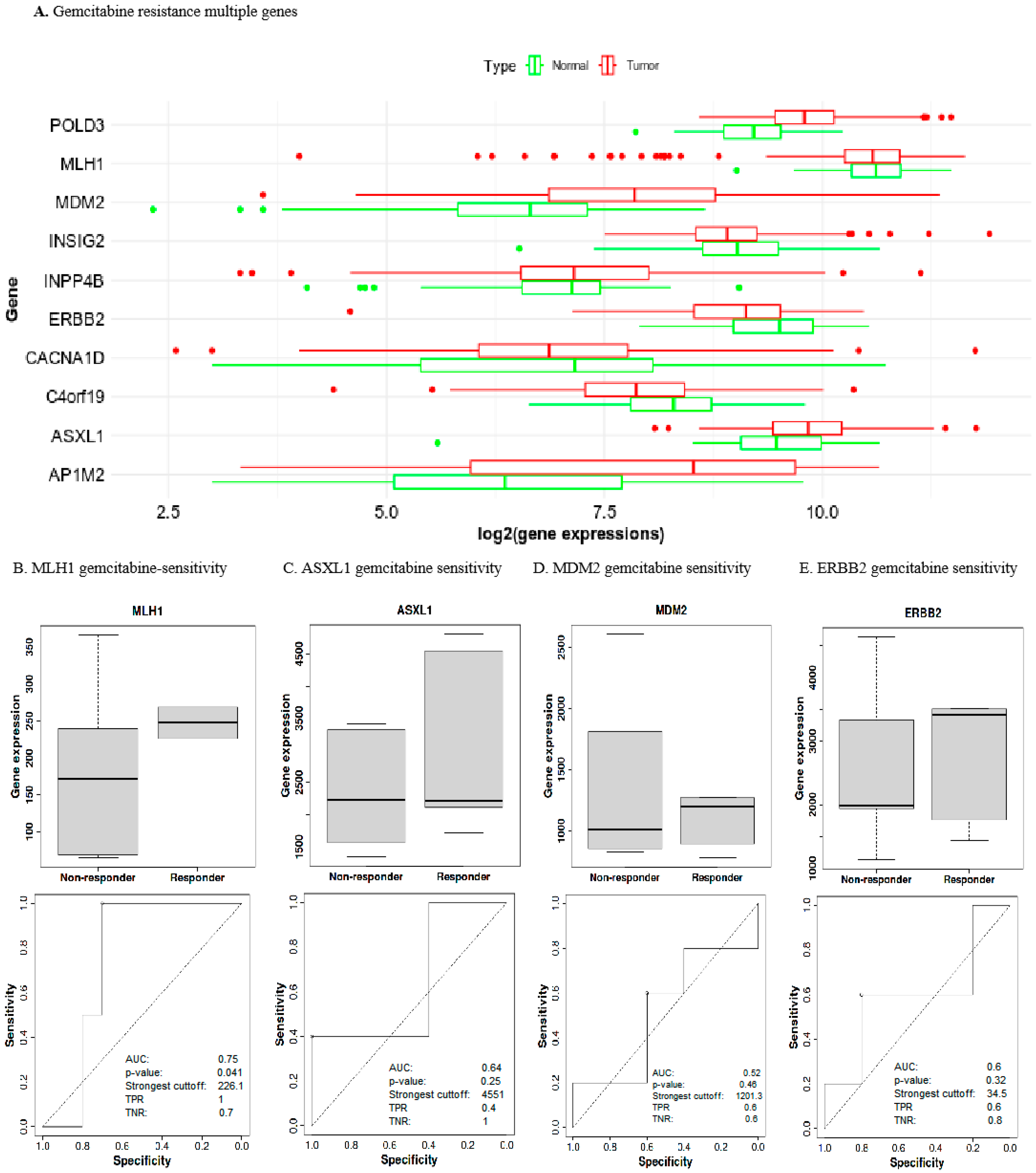
2.5. Prognostic Gene Expression Levels Associated with Gemcitabine Chemoresistance in UCEC
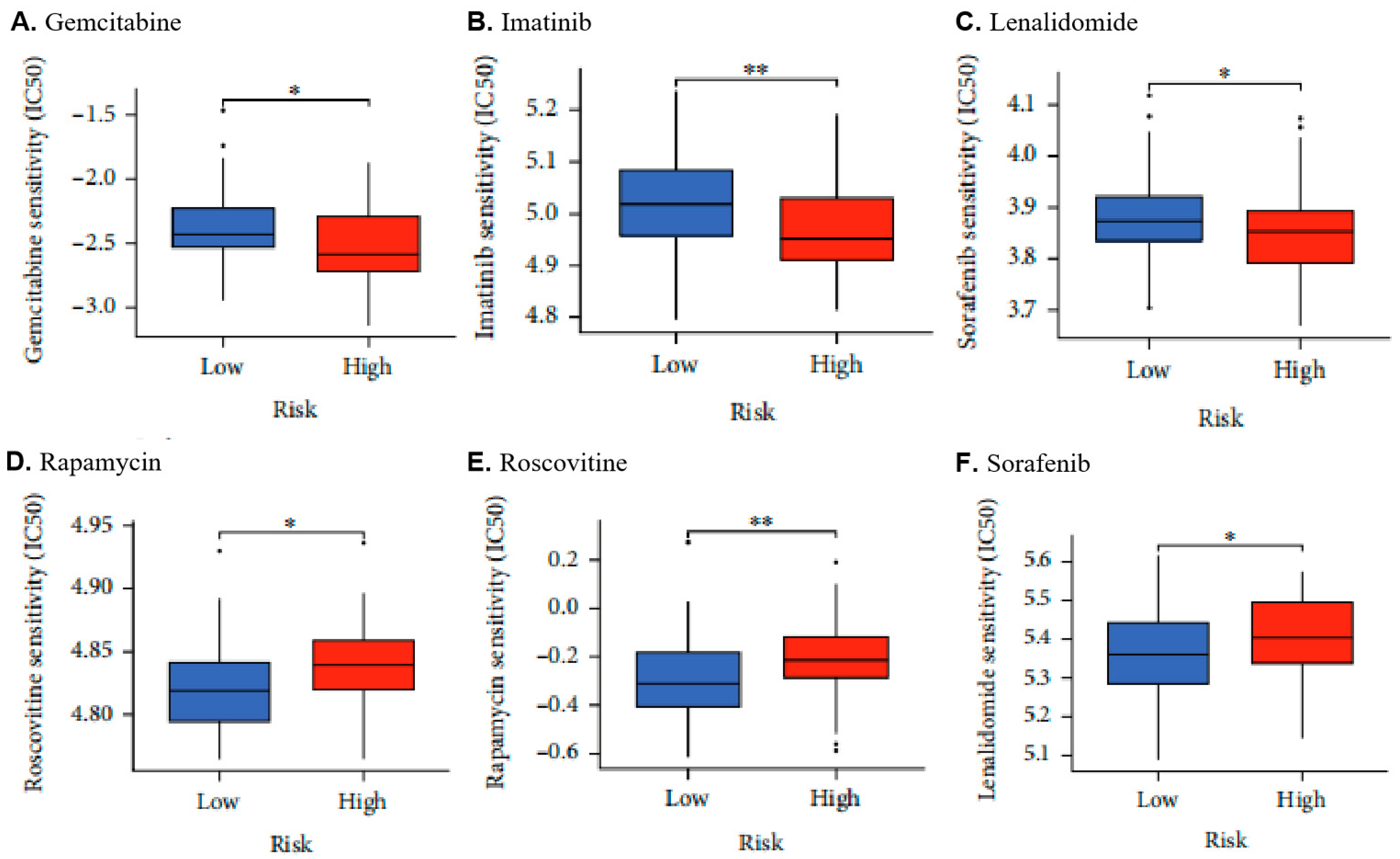
2.6. Low MLH1 Expression Decreased Drug Sensitivity in UCEC
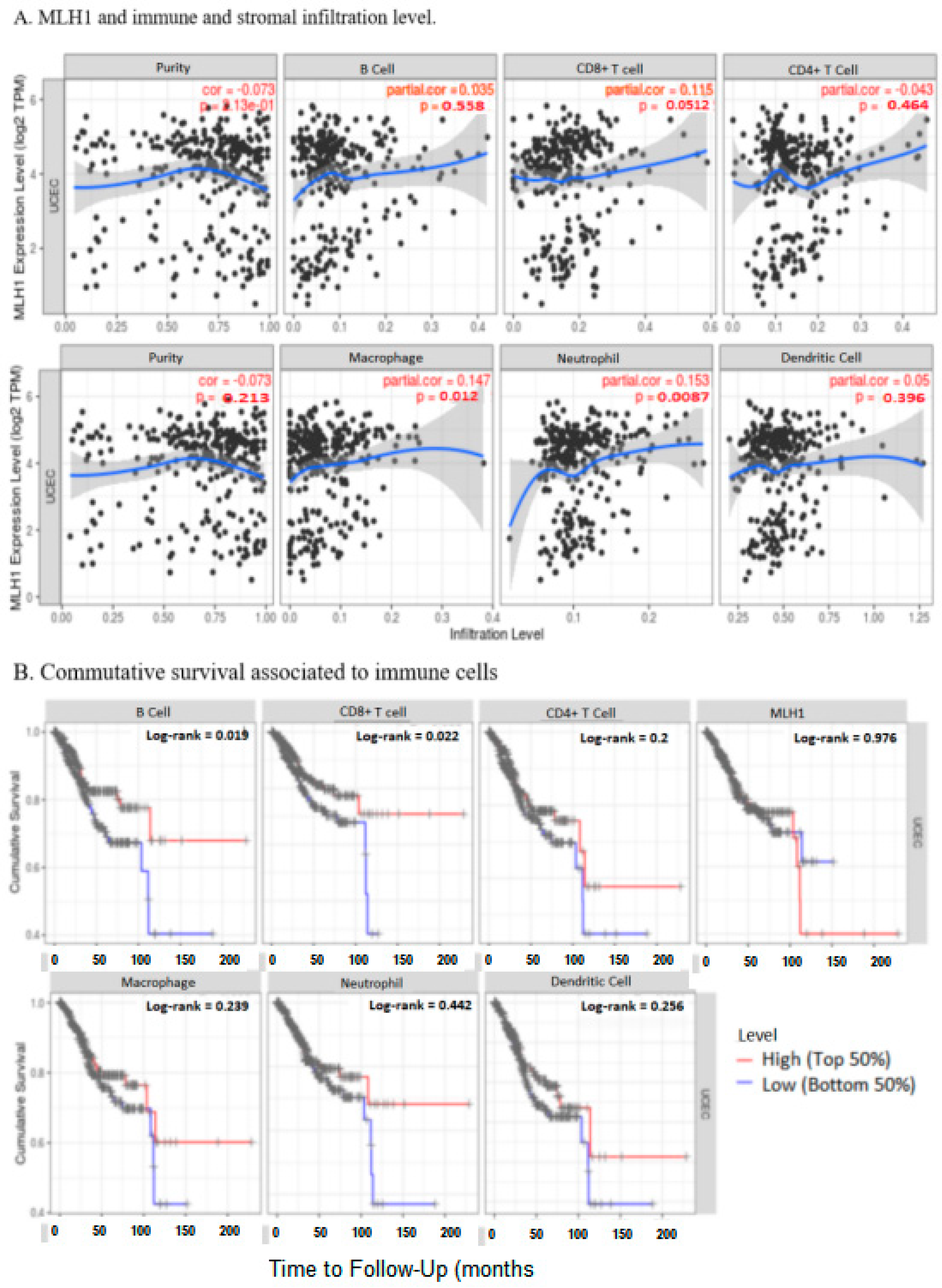
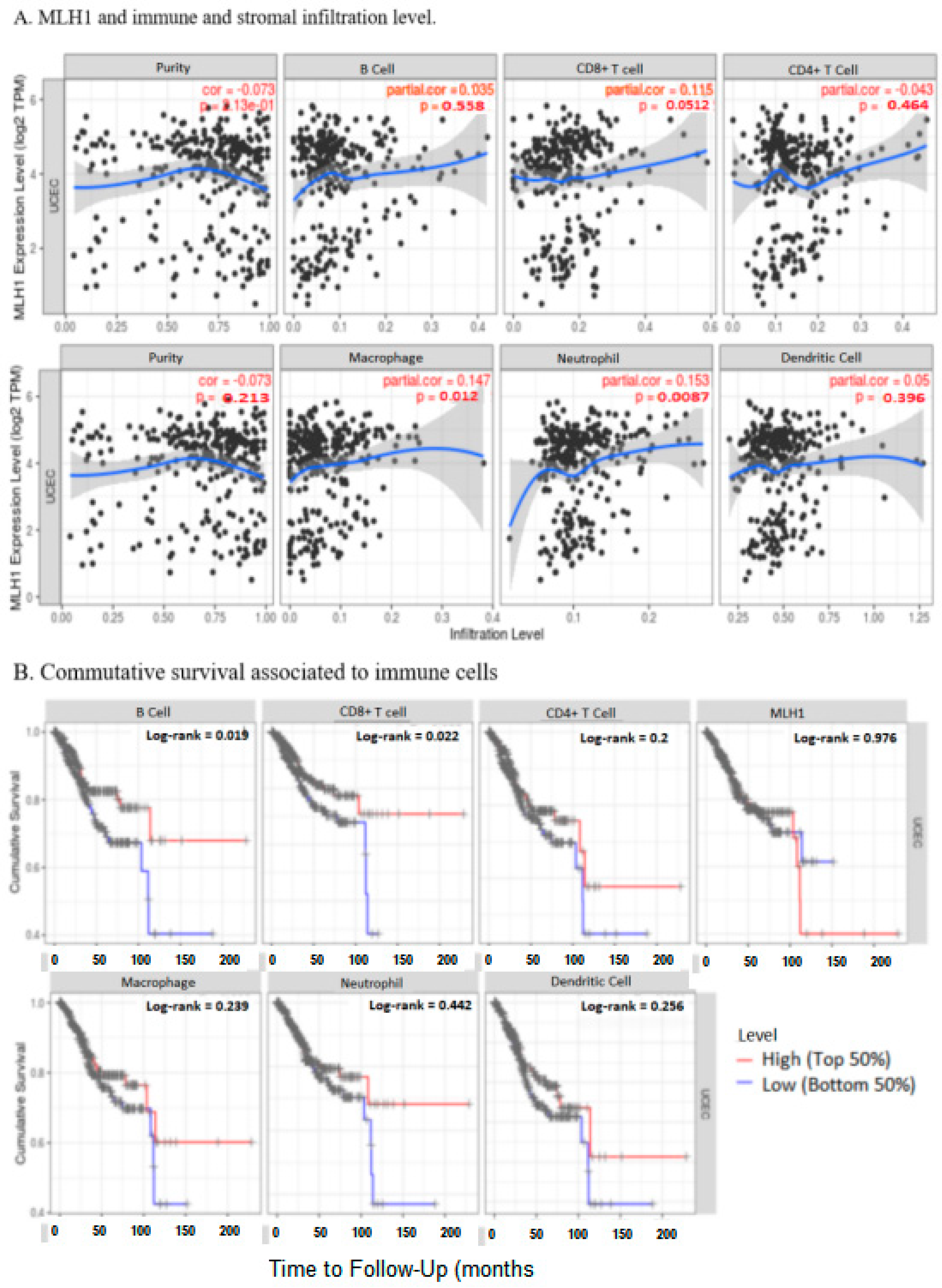
2.7. MLH1 Expression Negatively Correlated with the Tumor Immune Microenvironment in UCEC
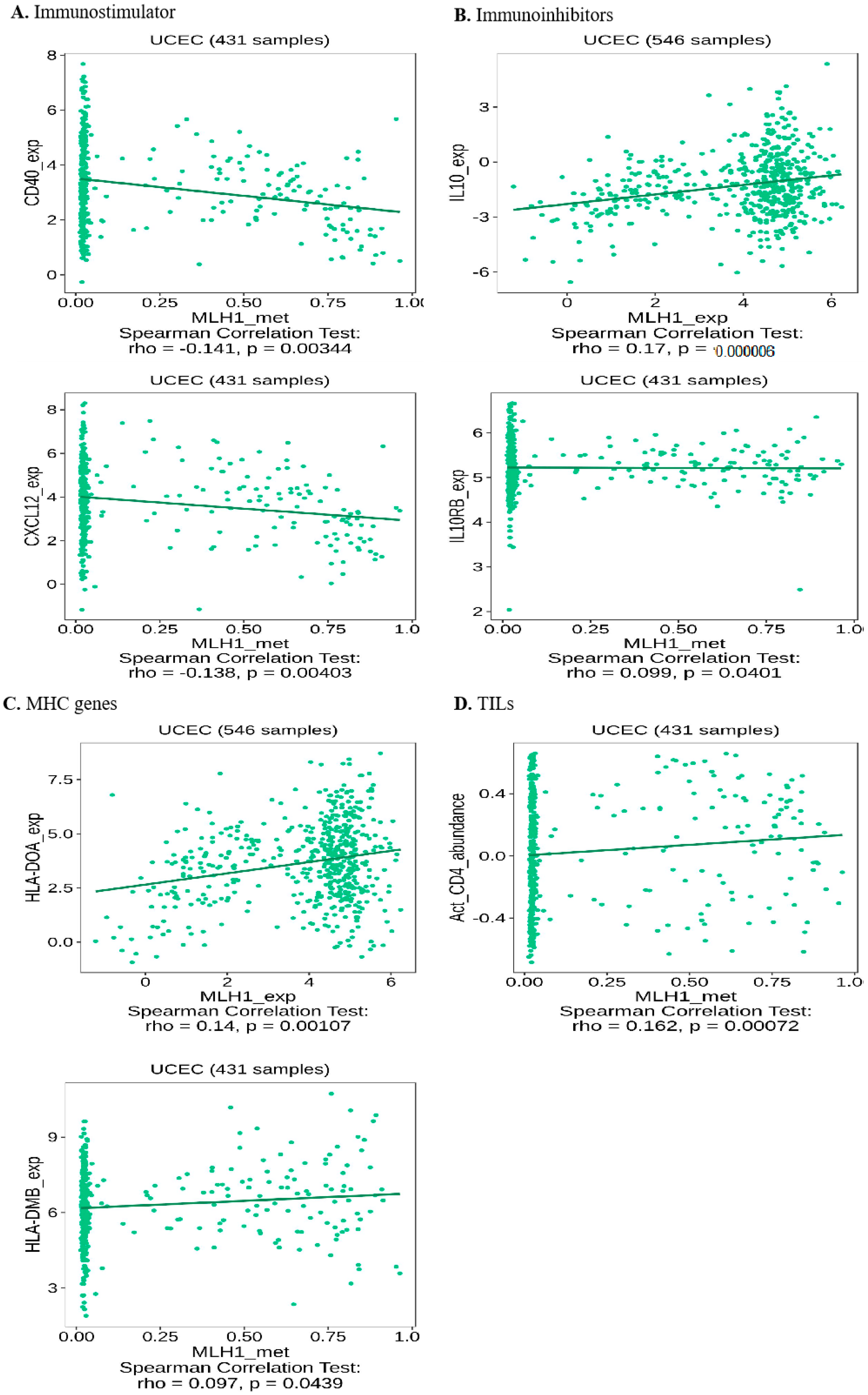
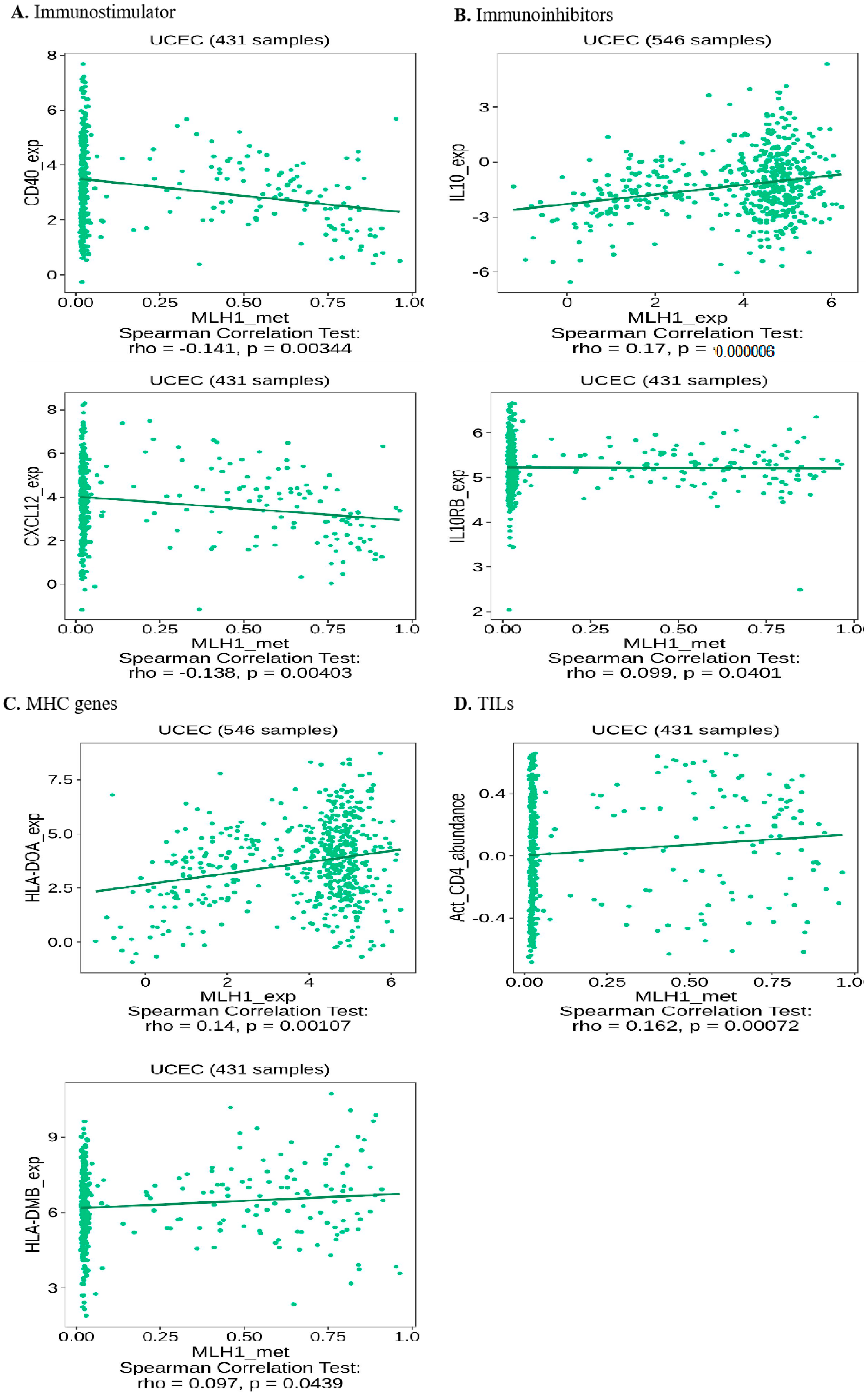
2.8. Depleted MLH1 Expression Negatively Correlated with Immunotherapeutic Efficacy in UCEC
3. Discussion
4. Materials and Methods
4.1. Data Collection
4.2. Prognostic Analysis of Deferentially Expressed Genes (DEGs)
4.3. Identification of Key Genes Regulating Gemcitabine Sensitivity
4.4. Investigation of MLH1 in Predicting Drug Sensitivity
4.5. Differential Gene Analysis and Functional Enrichment
4.6. Biological Function of MLH1 in UCEC
4.7. Genetic Alternation Analysis of MLH1
4.8. Analysis of Immune Cell Infiltration in UCEC
4.9. Immune Microenvironment Assessment
4.10. Investigation of MLH1 in Predicting Immunotherapeutic Efficacy
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bergeron, C.; Amant, F.; Ferenczy, A. Pathology and physiopathology of adenomyosis. Best Pract. Res. Clin. Obs. Gynaecol. 2006, 20, 511–521. [Google Scholar] [CrossRef]
- de Haydu, C.; Black, J.D.; Schwab, C.L.; English, D.P.; Santin, A.D. An update on the current pharmacotherapy for endometrial cancer. Expert. Opin. Pharmacother. 2016, 17, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Morice, P.; Leary, A.; Creutzberg, C.; Abu-Rustum, N.; Darai, E. Endometrial cancer. Lancet 2016, 387, 1094–1108. [Google Scholar] [CrossRef] [PubMed]
- Mathur, P.; Sathishkumar, K.; Chaturvedi, M.; Das, P.; Sudarshan, K.L.; Santhappan, S.; Nallasamy, V.; John, A.; Narasimhan, S.; Roselind, F.S.; et al. Cancer Statistics, 2020: Report From National Cancer Registry Programme, India. JCO Glob. Oncol. 2020, 6, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, J.N.; Temkin, S.M.; Mackay, H.J. Endometrial cancer: Not your grandmother’s cancer. Cancer 2016, 122, 2787–2798. [Google Scholar] [CrossRef]
- Rutten, H.; Verhoef, C.; van Weelden, W.J.; Smits, A.; Dhanis, J.; Ottevanger, N.; Pijnenborg, J.M.A. Recurrent Endometrial Cancer: Local and Systemic Treatment Options. Cancers 2021, 13, 6275. [Google Scholar] [CrossRef]
- Miller, D.S.; Filiaci, V.L.; Mannel, R.S.; Cohn, D.E.; Matsumoto, T.; Tewari, K.S.; DiSilvestro, P.; Pearl, M.L.; Argenta, P.A.; Powell, M.A.; et al. Carboplatin and Paclitaxel for Advanced Endometrial Cancer: Final Overall Survival and Adverse Event Analysis of a Phase III Trial (NRG Oncology/GOG0209). J. Clin. Oncol. 2020, 38, 3841–3850. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Favier, L.; Weber, B.; Roemer-Becuwe, C.; Bougnoux, P.; Fabbro, M.; Floquet, A.; Joly, F.; Plantade, A.; Paraiso, D.; et al. Everolimus as second- or third-line treatment of advanced endometrial cancer: ENDORAD, a phase II trial of GINECO. Br. J. Cancer 2013, 108, 1771–1777. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Leal, A.; Overman, M.J. Clinical Development of Immunotherapy for Deficient Mismatch Repair Colorectal Cancer. Clin. Color. Cancer 2020, 19, 73–81. [Google Scholar] [CrossRef] [PubMed]
- MacKay, H.J.; Freixinos, V.R.; Fleming, G.F. Therapeutic Targets and Opportunities in Endometrial Cancer: Update on Endocrine Therapy and Nonimmunotherapy Targeted Options. Am. Soc. Clin. Oncol. Educ. Book. 2020, 40, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.P.N.; Pinto, B.; Monteiro, L.; Silva, P.M.A.; Bousbaa, H. Combination Therapy as a Promising Way to Fight Oral Cancer. Pharmaceutics 2023, 15, 1653. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Wells, A.; Clark, A.M. The pan-therapeutic resistance of disseminated tumor cells: Role of phenotypic plasticity and the metastatic microenvironment. Semin. Cancer Biol. 2020, 60, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Moxley, K.M.; McMeekin, D.S. Endometrial carcinoma: A review of chemotherapy, drug resistance, and the search for new agents. Oncologist 2010, 15, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Akgul, S.; Patch, A.M.; D’Souza, R.C.J.; Mukhopadhyay, P.; Nones, K.; Kempe, S.; Kazakoff, S.H.; Jeffree, R.L.; Stringer, B.W.; Pearson, J.V.; et al. Intratumoural Heterogeneity Underlies Distinct Therapy Responses and Treatment Resistance in Glioblastoma. Cancers 2019, 11, 190. [Google Scholar] [CrossRef]
- Belachew, E.B.; Sewasew, D.T. Molecular Mechanisms of Endocrine Resistance in Estrogen-Positive Breast Cancer. Front. Endocrinol. 2021, 12, 599586. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef]
- Jurkovicova, D.; Neophytou, C.M.; Gasparovic, A.C.; Goncalves, A.C. DNA Damage Response in Cancer Therapy and Resistance: Challenges and Opportunities. Int. J. Mol. Sci. 2022, 23, 14672. [Google Scholar] [CrossRef]
- Crucitta, S.; Cucchiara, F.; Mathijssen, R.; Mateo, J.; Jager, A.; Joosse, A.; Passaro, A.; Attili, I.; Petrini, I.; van Schaik, R.; et al. Treatment-driven tumour heterogeneity and drug resistance: Lessons from solid tumours. Cancer Treat. Rev. 2022, 104, 102340. [Google Scholar] [CrossRef]
- Batis, N.; Brooks, J.M.; Payne, K.; Sharma, N.; Nankivell, P.; Mehanna, H. Lack of predictive tools for conventional and targeted cancer therapy: Barriers to biomarker development and clinical translation. Adv. Drug Deliv. Rev. 2021, 176, 113854. [Google Scholar] [CrossRef]
- Wajapeyee, N.; Gupta, R. Epigenetic Alterations and Mechanisms That Drive Resistance to Targeted Cancer Therapies. Cancer Res. 2021, 81, 5589–5595. [Google Scholar] [CrossRef]
- Al Aboud, N.M.; Tupper, C.; Jialal, I. Genetics, Epigenetic Mechanism. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef]
- Yang, Y.; Li, X.; Wang, T.; Guo, Q.; Xi, T.; Zheng, L. Emerging agents that target signaling pathways in cancer stem cells. J. Hematol. Oncol. 2020, 13, 60. [Google Scholar] [CrossRef]
- Sun, L.; Fang, J. Epigenetic regulation of epithelial-mesenchymal transition. Cell Mol. Life Sci. 2016, 73, 4493–4515. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Wainwright, E.N.; Scaffidi, P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer 2017, 3, 372–386. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, S. Cancer′s epigenetic drugs: Where are they in the cancer medicines? Pharmacogenomics J. 2020, 20, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, Y.; Nephew, K.P. Epigenetic Crosstalk between the Tumor Microenvironment and Ovarian Cancer Cells: A Therapeutic Road Less Traveled. Cancers 2018, 10, 295. [Google Scholar] [CrossRef]
- Bell, C.C.; Gilan, O. Principles and mechanisms of non-genetic resistance in cancer. Br. J. Cancer 2020, 122, 465–472. [Google Scholar] [CrossRef]
- Das Ghosh, D.; Roy Chowdhury, R.; Dutta, R.; Mukhopadhyay, I.; Mukhopadhyay, A.; Roychoudhury, S. In-silico analysis of TCGA data showing multiple POLE-like favourable subgroups overlapping with TP53 mutated endometrial cancer: Implications for clinical practice in low and middle-income countries. Gynecol. Oncol. Rep. 2023, 47, 101209. [Google Scholar] [CrossRef] [PubMed]
- Candido, S.; Tomasello, B.M.R.; Lavoro, A.; Falzone, L.; Gattuso, G.; Libra, M. Novel Insights into Epigenetic Regulation of IL6 Pathway: In Silico Perspective on Inflammation and Cancer Relationship. Int. J. Mol. Sci. 2021, 22, 10172. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Rawal, R.M. Genetic and Epigenetic Modulation of Drug Resistance in Cancer: Challenges and Opportunities. Curr. Drug Metab. 2019, 20, 1114–1131. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Kim, J.M. Multi-omics integration strategies for animal epigenetic studies—A review. Anim. Biosci. 2021, 34, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Khanam Irin, A.; Kodamullil, A.T.; Gundel, M.; Hofmann-Apitius, M. Computational Modelling Approaches on Epigenetic Factors in Neurodegenerative and Autoimmune Diseases and Their Mechanistic Analysis. J. Immunol. Res. 2015, 2015, 737168. [Google Scholar] [CrossRef]
- Yang, M.; Hlady, R.A.; Zhou, D.; Ho, T.H.; Robertson, K.D. In silico DNA methylation analysis identifies potential prognostic biomarkers in type 2 papillary renal cell carcinoma. Cancer Med. 2019, 8, 5760–5768. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef]
- Hatzimichael, E.; Lagos, K.; Sim, V.R.; Briasoulis, E.; Crook, T. Epigenetics in diagnosis, prognostic assessment and treatment of cancer: An update. EXCLI J. 2014, 13, 954–976. [Google Scholar]
- Feehley, T.; O’Donnell, C.W.; Mendlein, J.; Karande, M.; McCauley, T. Drugging the epigenome in the age of precision medicine. Clin. Epigenetics 2023, 15, 6. [Google Scholar] [CrossRef]
- Rodriguez, H.; Zenklusen, J.C.; Staudt, L.M.; Doroshow, J.H.; Lowy, D.R. The next horizon in precision oncology: Proteogenomics to inform cancer diagnosis and treatment. Cell 2021, 184, 1661–1670. [Google Scholar] [CrossRef]
- Srivastava, A.; Murugaiyan, J.; Garcia, J.A.L.; De Corte, D.; Hoetzinger, M.; Eravci, M.; Weise, C.; Kumar, Y.; Roesler, U.; Hahn, M.W.; et al. Combined Methylome, Transcriptome and Proteome Analyses Document Rapid Acclimatization of a Bacterium to Environmental Changes. Front. Microbiol. 2020, 11, 544785. [Google Scholar] [CrossRef]
- Sepulveda, A.R.; Jones, D.; Ogino, S.; Samowitz, W.; Gulley, M.L.; Edwards, R.; Levenson, V.; Pratt, V.M.; Yang, B.; Nafa, K.; et al. CpG methylation analysis--current status of clinical assays and potential applications in molecular diagnostics: A report of the Association for Molecular Pathology. J. Mol. Diagn. 2009, 11, 266–278. [Google Scholar] [CrossRef]
- Bader, A.S.; Bushell, M. Damage-Net: A program for DNA repair meta-analysis identifies a network of novel repair genes that facilitate cancer evolution. DNA Repair 2021, 105, 103158. [Google Scholar] [CrossRef]
- Li, Z.; Jia, Y.; Zhu, H.; Yuan, H.; Xing, X.; Xin, Y.; Ma, T.; Pang, F.; Zhang, Y.; Hu, Y.; et al. Genomic landscape of microsatellite instability in Chinese tumors: A comparison of Chinese and TCGA cohorts. Int. J. Cancer 2022, 151, 1382–1393. [Google Scholar] [CrossRef] [PubMed]
- Russell, H.; Kedzierska, K.; Buchanan, D.D.; Thomas, R.; Tham, E.; Mints, M.; Keranen, A.; Giles, G.G.; Southey, M.C.; Milne, R.L.; et al. The MLH1 polymorphism rs1800734 and risk of endometrial cancer with microsatellite instability. Clin. Epigenetics 2020, 12, 102. [Google Scholar] [CrossRef]
- Damaso, E.; Castillejo, A.; Arias, M.D.M.; Canet-Hermida, J.; Navarro, M.; Del Valle, J.; Campos, O.; Fernandez, A.; Marin, F.; Turchetti, D.; et al. Primary constitutional MLH1 epimutations: A focal epigenetic event. Br. J. Cancer 2018, 119, 978–987. [Google Scholar] [CrossRef] [PubMed]
- van Roon, E.H.; van Puijenbroek, M.; Middeldorp, A.; van Eijk, R.; de Meijer, E.J.; Erasmus, D.; Wouters, K.A.; van Engeland, M.; Oosting, J.; Hes, F.J.; et al. Early onset MSI-H colon cancer with MLH1 promoter methylation, is there a genetic predisposition? BMC Cancer 2010, 10, 180. [Google Scholar] [CrossRef] [PubMed]
- Schafer, A.; Schomacher, L.; Barreto, G.; Doderlein, G.; Niehrs, C. Gemcitabine functions epigenetically by inhibiting repair mediated DNA demethylation. PLoS ONE 2010, 5, e14060. [Google Scholar] [CrossRef]
- Umene, K.; Banno, K.; Kisu, I.; Yanokura, M.; Nogami, Y.; Tsuji, K.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Yamagami, W.; et al. New candidate therapeutic agents for endometrial cancer: Potential for clinical practice (review). Oncol. Rep. 2013, 29, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Mendez, D.; Mendoza, L.; Villarreal, C.; Huerta, L. Continuous Modeling of T CD4 Lymphocyte Activation and Function. Front. Immunol. 2021, 12, 743559. [Google Scholar] [CrossRef]
- Gasser, S.; Raulet, D. The DNA damage response, immunity and cancer. Semin. Cancer Biol. 2006, 16, 344–347. [Google Scholar] [CrossRef]
- Pineda, M.; Mur, P.; Iniesta, M.D.; Borras, E.; Campos, O.; Vargas, G.; Iglesias, S.; Fernandez, A.; Gruber, S.B.; Lazaro, C.; et al. MLH1 methylation screening is effective in identifying epimutation carriers. Eur. J. Hum. Genet. 2012, 20, 1256–1264. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, G. IL22RA1/JAK/STAT Signaling Acts As a Cancer Target Through Pan-Cancer Analysis. Front. Immunol. 2022, 13, 915246. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ramzan, F.; Zhong, G. Investigating novel biomarkers in uterine corpus endometrial carcinoma: In silico analysis and clinical specimens validation via RT-qPCR and immunohistochemistry. Am. J. Cancer Res. 2023, 13, 4376–4400. [Google Scholar] [PubMed]
- Lawlor, R.T.; Mattiolo, P.; Mafficini, A.; Hong, S.M.; Piredda, M.L.; Taormina, S.V.; Malleo, G.; Marchegiani, G.; Pea, A.; Salvia, R.; et al. Tumor Mutational Burden as a Potential Biomarker for Immunotherapy in Pancreatic Cancer: Systematic Review and Still-Open Questions. Cancers 2021, 13, 3119. [Google Scholar] [CrossRef]
- Brito, A.B.C.; Camandaroba, M.P.G.; de Lima, V.C.C. Anti-PD1 versus anti-PD-L1 immunotherapy in first-line therapy for advanced non-small cell lung cancer: A systematic review and meta-analysis. Thorac. Cancer 2021, 12, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Kientz, C.; Prieur, F.; Clemenson, A.; Joly, M.O.; Stachowicz, M.L.; Auclair, J.; Attignon, V.; Schiappa, R.; Wang, Q. MLH1 promoter hypermethylation: Are you absolutely sure about the absence of MLH1 germline mutation? About a new case. Fam. Cancer 2020, 19, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Furman, C.M.; Elbashir, R.; Alani, E. Expanded roles for the MutL family of DNA mismatch repair proteins. Yeast 2021, 38, 39–53. [Google Scholar] [CrossRef]
- O’Brien, V.; Brown, R. Signalling cell cycle arrest and cell death through the MMR System. Carcinogenesis 2006, 27, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.M.; Plug, A.W.; Prolla, T.A.; Bronner, C.E.; Harris, A.C.; Yao, X.; Christie, D.M.; Monell, C.; Arnheim, N.; Bradley, A.; et al. Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat. Genet. 1996, 13, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Lu, C.; Jin, Q.; Lu, H.; Chen, X.; Tian, L.; Zhang, Y.; Ortega, J.; Zhang, J.; Siteni, S.; et al. MLH1 Deficiency-Triggered DNA Hyperexcision by Exonuclease 1 Activates the cGAS-STING Pathway. Cancer Cell 2021, 39, 109–121.e105. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, N.; Yoshida, H.; Miwa, M.; Fujiwara, K. MLH1 Is a Prognostic Biomarker for Serous Ovarian Cancer Treated With Platinum- and Taxane-based Chemotherapy. Anticancer. Res. 2019, 39, 5505–5513. [Google Scholar] [CrossRef] [PubMed]
- Romeo, F.; Falbo, L.; Di Sanzo, M.; Misaggi, R.; Faniello, M.C.; Viglietto, G.; Cuda, G.; Costanzo, F.; Quaresima, B. BRCA1 is required for hMLH1 stabilization following doxorubicin-induced DNA damage. Int. J. Biochem. Cell Biol. 2011, 43, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Hinrichsen, I.; Ernst, B.P.; Nuber, F.; Passmann, S.; Schafer, D.; Steinke, V.; Friedrichs, N.; Plotz, G.; Zeuzem, S.; Brieger, A. Reduced migration of MLH1 deficient colon cancer cells depends on SPTAN1. Mol. Cancer 2014, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, A.; Schrecker, C.; Bon, D.; Friedrichs, N.; Bankov, K.; Wild, P.; Plotz, G.; Zeuzem, S.; Herrmann, E.; Hansmann, M.L.; et al. Downregulation of SPTAN1 is related to MLH1 deficiency and metastasis in colorectal cancer. PLoS ONE 2019, 14, e0213411. [Google Scholar] [CrossRef]
- Fredriksson, K.; Van Itallie, C.M.; Aponte, A.; Gucek, M.; Tietgens, A.J.; Anderson, J.M. Proteomic analysis of proteins surrounding occludin and claudin-4 reveals their proximity to signaling and trafficking networks. PLoS ONE 2015, 10, e0117074. [Google Scholar] [CrossRef]
- Chow, R.D.; Michaels, T.; Bellone, S.; Hartwich, T.M.P.; Bonazzoli, E.; Iwasaki, A.; Song, E.; Santin, A.D. Distinct Mechanisms of Mismatch-Repair Deficiency Delineate Two Modes of Response to Anti-PD-1 Immunotherapy in Endometrial Carcinoma. Cancer Discov. 2023, 13, 312–331. [Google Scholar] [CrossRef]
- Goltz, D.; Gevensleben, H.; Dietrich, J.; Dietrich, D. PD-L1 (CD274) promoter methylation predicts survival in colorectal cancer patients. Oncoimmunology 2017, 6, e1257454. [Google Scholar] [CrossRef]
- Goldman, M.; Craft, B.; Swatloski, T.; Cline, M.; Morozova, O.; Diekhans, M.; Haussler, D.; Zhu, J. The UCSC Cancer Genomics Browser: Update 2015. Nucleic Acids Res. 2015, 43, D812–D817. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Datasets | Tissue | Tumor | Normal | Platform |
|---|---|---|---|---|
| GSE115810 | Endometrium | 24 | 3 | GPL96 |
| GSE17025 | 91 | 12 | GPL570 | |
| GSE3689 | 13 | 7 | GPL96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolde, T.; Huang, J.; Huang, P.; Pandey, V.; Qin, P. Depleted-MLH1 Expression Predicts Prognosis and Immunotherapeutic Efficacy in Uterine Corpus Endometrial Cancer: An In Silico Approach. BioMedInformatics 2024, 4, 326-346. https://doi.org/10.3390/biomedinformatics4010019
Wolde T, Huang J, Huang P, Pandey V, Qin P. Depleted-MLH1 Expression Predicts Prognosis and Immunotherapeutic Efficacy in Uterine Corpus Endometrial Cancer: An In Silico Approach. BioMedInformatics. 2024; 4(1):326-346. https://doi.org/10.3390/biomedinformatics4010019
Chicago/Turabian StyleWolde, Tesfaye, Jing Huang, Peng Huang, Vijay Pandey, and Peiwu Qin. 2024. "Depleted-MLH1 Expression Predicts Prognosis and Immunotherapeutic Efficacy in Uterine Corpus Endometrial Cancer: An In Silico Approach" BioMedInformatics 4, no. 1: 326-346. https://doi.org/10.3390/biomedinformatics4010019
APA StyleWolde, T., Huang, J., Huang, P., Pandey, V., & Qin, P. (2024). Depleted-MLH1 Expression Predicts Prognosis and Immunotherapeutic Efficacy in Uterine Corpus Endometrial Cancer: An In Silico Approach. BioMedInformatics, 4(1), 326-346. https://doi.org/10.3390/biomedinformatics4010019