
Multiple Bio-Computational Tools Emerge as Valid Approach in the Assessment of Apolipoproteins Pathogenicity Related Mutations



Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Retrieval and Web Resources
2.2. Predictions Derived from Sequence Analysis
- Sorts Tolerant From Intolerant (SIFT): bioinformatics tool that predicts the potential impact of amino acid substitutions on protein function [16]. Advantage of SIFT, it allows the analysis of multiple variants, suitable for high-throughput genetic screening studies. A limitation of SIFT, however, is its dependence on multiple sequence alignments to assess the conservation of amino acid residues. When a protein lacks a sufficient number of homologous sequences, the algorithm may struggle to generate accurate predictions.
- PolyPhen-2: bioinformatics tool that predicts the possible effects of amino acid substitutions on protein structure and function [17]. It integrates comparative and physical properties of amino acids substitutions disrupting the native conformation of the protein. It gathers relevant information, which improves the robustness of predictions. The limit is the indirect prediction of disease risk.
- Functional Analysis Through Hidden Markov Models (FATHMM): bioinformatics tool used for functional impact of genetic variants prediction, particularly in coding regions of the genome [18]. The predictive accuracy depends heavily on the quality and size of the training dataset. For variants that are not well-represented in the training data, the predictions may be less reliable.
- SNPs&GO: web-based tool that utilizes a support vector machine (SVM) to identify deleterious single-nucleotide polymorphisms that lead to amino acid substitutions [19]. The SVM classifier integrates protein sequence data, functional profiles, and gene ontology (GO) annotations to differentiate between disease-associated and neutral variants. It applies machine learning algorithms trained on various features derived from protein sequence and structure to assess the potential effects of SNPs. A score greater than 0.5 suggests that a given substitution is likely to be pathogenic.
2.3. Predictions Derived from Structure Analysis
- Mutation Cutoff Scanning Matrix (mCSM): web-based tool used in structural bioinformatics to predict the effects of variants on protein stability [20]. It operates by evaluating single-point amino acid substitutions using a graph-based method. It can predict changes in protein stability. The limit is the indirect prediction of aminoacidic changes might affect the protein’s function or interactions, which may be critical in understanding disease mechanisms.
- Dynamut2: tool developed to predict the effects of amino acid substitutions on protein stability [21]. DynaMut2 employs a combination of Normal Mode Analysis (NMA) and molecular dynamics, providing a more detailed assessment of the effects of mutations on protein stability compared to other tools that rely solely on structural data. While DynaMut2 assesses protein stability, it does not directly predict the functional consequences of mutations, such as changes in enzymatic activity or alterations in protein–protein interactions.
- MAESTROweb: tool used to discern how variants impact protein stability, specifically focusing on the functional implications of genetic variations concerning human diseases [22]. By combining various computational tools, MAESTROweb provides insights into the consequences of variants on protein stability. While MAESTROweb assesses stability changes, the tool does not directly predict how mutations affect protein function, enzymatic activity, or molecular interactions.
- PremPS: tool that evaluates the impact of amino acid substitutions in proteins by employing multiple approaches, such as multiple sequence alignment (MSA), protein structure analysis, and a deep learning model [23] providing more accurate predictions compared to methods based solely on sequence or structure. One limitation is that predictions may be less reliable if no experimental protein structure is available, and homologous models are used instead.
2.4. Prediction of Pathogenic Potential
- MutPred2: web-based tool designed to predict the potential pathogenicity of amino acid substitutions. It does this by assessing the likelihood that a variant impacts protein function and structure [24]. MutPred2 provides insights into possible molecular mechanisms by which mutations can contribute to disease, such as altered protein–protein interactions, post-translational modifications, and structural changes. It evaluates both the structural and functional impact of amino acid substitutions, providing insights into potential disease mechanisms. While it generates hypotheses, experimental validation is necessary to confirm the predicted effects.
- PhD-SNP: tool designed to predict the impact of single nucleotide polymorphisms (SNPs) on protein function and their association with disease [25]. PhD-SNP is generally accessible through web-based interfaces, facilitating its use by researchers without requiring advanced computational expertise. However, as with many predictive tools, there exists the potential for both false positives and false negatives.
2.5. Profiling of Aggregation Propensity and Residual Frustration Analysis
- Camsol: tool method begins by using a linear combination of biophysical properties, to determine a solubility score for each individual residue [26]. The protein’s overall solubility score, derived from its intrinsic solubility profile, reflects the contributions of both poorly and highly soluble regions [29]. CamSol applies to diverse proteins, including those in disease and biotechnology, but excludes external factors like protein interactions or post-translational modifications.
- Aggrescan3D 2.0: web-based tool for predicting aggregation-prone regions, assessing mutation effects, and comparing protein aggregation profiles [27]. It quantifies intrinsic aggregation propensities of amino acids, providing insights into residue-specific contributions [26,27]. While valuable, the model may not fully capture complex interactions or folding dynamics influencing aggregation in biological contexts.
- Frustratometer: server was used to calculate individual and configurational residual indices for the structure of ApoE. This analysis provides valuable insights into a protein’s propensity to aggregate, which is useful for understanding how specific variants within the protein influence its tendency to form aggregates [28]. The frustration index might require careful interpretation; as high frustration does not necessarily correlate with aggregation in all contexts. Experimental validation is needed to confirm predictions.
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Basavaraju, P.; Balasubramani, R.; Kathiresan, D.S.; Devaraj, I.; Babu, K.; Alagarsamy, V.; Puthamohan, V.M. Genetic Regulatory Networks of Apolipoproteins and Associated Medical Risks. Front. Cardiovasc. Med. 2022, 8, 788852. [Google Scholar] [CrossRef]
- Liu, S.; Liu, J.; Weng, R.; Gu, X.; Zhong, Z. Apolipoprotein E gene polymorphism and the risk of cardiovascular disease and type 2 diabetes. BMC Cardiovasc. Disord. 2019, 19, 213. [Google Scholar] [CrossRef]
- Ismail, A.B.; Balcıoğlu, Ö.; Özcem, B.; Ergoren, M.Ç. APOE Gene Variation’s Impact on Cardiovascular Health: A Case-Control Study. Biomedicines 2024, 12, 695. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, M.; Dibello, V.; Sardone, R.; Castellana, F.; Zupo, R.; Lampignano, L.; Bortone, I.; Daniele, A.; Bellomo, A.; Solfrizzi, V.; et al. The Impact of Apolipoprotein E (APOE) Epigenetics on Aging and Sporadic Alzheimer’s Disease. Biology 2023, 12, 1529. [Google Scholar] [CrossRef]
- Saraceno, G.F.; Abrego-Guandique, D.M.; Cannataro, R.; Caroleo, M.C.; Cione, E. Machine Learning Approach to Identify Case-Control Studies on ApoE Gene Mutations Linked to Alzheimer’s Disease in Italy. BioMedInformatics 2024, 4, 600–622. [Google Scholar] [CrossRef]
- Roberts, B.R.; Laffoon, S.B.; Roberts, A.M.; Porter, T.; Fowler, C.; Masters, C.L.; Dratz, E.A.; Laws, S.M. Discovery of a Missense Mutation (Q222K) of the APOE Gene from the Australian Imaging, Biomarker and Lifestyle Study. J. Alzheimer’s Dis. Rep. 2023, 7, 165–172. [Google Scholar] [CrossRef]
- Perera, S.D.; Hegele, R.A. Genetic variation in apolipoprotein A-V in hypertriglyceridemia. Curr. Opin. Infect. Dis. 2024, 35, 66–77. [Google Scholar] [CrossRef]
- Dedoussis, G.V. Apolipoprotein polymorphisms and familial hypercholesterolemia. Pharmacogenomics 2007, 8, 1179–1189. [Google Scholar] [CrossRef]
- Zhou, F.; Guo, T.; Zhou, L.; Zhou, Y.; Yu, D. Variants in the APOB gene was associated with Ischemic Stroke susceptibility in Chinese Han male population. Oncotarget 2017, 9, 2249–2254. [Google Scholar] [CrossRef]
- Brunzell, J.D.; Davidson, M.; Furberg, C.D.; Goldberg, R.B.; Howard, B.V.; Stein, J.H.; Witztum, J.L. Lipoprotein management in patients with cardiometabolic risk: Consensus statement from the American Diabetes Association and the American College of Cardiology Foundation. Diabetes Care 2008, 31, 811–822. [Google Scholar] [CrossRef]
- Emerging Risk Factors Collaboration; Di Angelantonio, E.; Sarwar, N.; Perry, P.; Kaptoge, S.; Ray, K.K.; Thompson, A.; Wood, A.M.; Lewington, S.; Sattar, N.; et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009, 302, 1993–2000. [Google Scholar] [CrossRef]
- Katsonis, P.; Koire, A.; Wilson, S.J.; Hsu, T.; Lua, R.C.; Wilkins, A.D.; Lichtarge, O. Single nucleotide variations: Biological impact and theoretical interpretation. Protein Sci. 2014, 23, 1650–1666. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.; Sirotkin, K. dbSNP—Database for Single Nucleotide Polymorphisms and Other Classes of Minor Genetic Variation. Genome Res. 1999, 9, 677–679. [Google Scholar] [CrossRef]
- Harrison, P.W.; Amode, M.R.; Austine-Orimoloye, O.; Azov, A.G.; Barba, M.; Barnes, I.; Becker, A.; Bennett, R.; Berry, A.; Bhai, J.; et al. Ensembl 2024. Nucleic Acids Res. 2024, 52, D891–D899. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.F.; Shihab, H.A.; Mort, M.; Cooper, D.N.; Gaunt, T.R.; Campbell, C. FATHMM-XF: Accurate prediction of pathogenic point mutations via extended features. Bioinformatics 2018, 34, 511–513. [Google Scholar] [CrossRef]
- Capriotti, E.; Calabrese, R.; Fariselli, P.; Martelli, P.L.; Altman, R.B.; Casadio, R. WS-SNPs&GO: A web server for predicting the deleterious effect of human protein variants using functional annotation. BMC Genom. 2013, 14 (Suppl. S3), S6. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 2021, 30, 60–69. [Google Scholar] [CrossRef]
- Laimer, J.; Hiebl-Flach, J.; Lengauer, D.; Lackner, P. MAESTROweb: A web server for structure-based protein stability prediction. Bioinformatics 2016, 32, 1414–1416. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, H.; Zhang, N.; Zhu, Z.; Wang, S.; Li, M. PremPS: Predicting the impact of missense mutations on protein stability. PLoS Comput. Biol. 2020, 16, e1008543. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P. PhD-SNPg: A webserver and lightweight tool for scoring single nucleotide variants. Nucleic Acids Res. 2017, 45, W247–W252. [Google Scholar] [CrossRef]
- Sormanni, P.; Aprile, F.A.; Vendruscolo, M. The CamSol method of rational design of protein mutants with enhanced solubility. J. Mol. Biol. 2015, 427, 478–490. [Google Scholar] [CrossRef]
- Zambrano, R.; Jamroz, M.; Szczasiuk, A.; Pujols, J.; Kmiecik, S.; Ventura, S. AGGRESCAN3D (A3D): Server for prediction of aggregation properties of protein structures. Nucleic Acids Res. 2015, 43, W306–W313. [Google Scholar] [CrossRef]
- Parra, R.G.; Schafer, N.P.; Radusky, L.G.; Tsai, M.-Y.; Guzovsky, A.B.; Wolynes, P.G.; Ferreiro, D.U. Protein Frustratometer 2: A tool to localize energetic frustration in protein molecules, now with electrostatics. Nucleic Acids Res. 2016, 44, W356–W360. [Google Scholar] [CrossRef]
- Sormanni, P.; Amery, L.; Ekizoglou, S.; Vendruscolo, M.; Popovic, B. Rapid and accurate in silico solubility screening of a monoclonal antibody library. Sci. Rep. 2017, 7, 8200. [Google Scholar] [CrossRef]
- Breslow, J.L.; Ross, D.; McPherson, J.; Williams, H.; Kurnit, D.; Nussbaum, A.L.; Karathanasis, S.K.; Zannis, V.I. Isolation and characterization of cDNA clones for human apolipoprotein A-I. Proc. Natl. Acad. Sci. USA 1982, 79, 6861–6865. [Google Scholar] [CrossRef]
- Brewer, H.B.; Lux, S.E.; Ronan, R.; John, K.M. Amino acid sequence of human apoLp-Gln-II (apoA-II), an apolipoprotein isolated from the high-density lipoprotein complex. Proc. Natl. Acad. Sci. USA 1972, 69, 1304–1308. [Google Scholar] [CrossRef]
- Qu, J.; Ko, C.-W.; Tso, P.; Bhargava, A. Apolipoprotein A-IV: A Multifunctional Protein Involved in Protection against Atherosclerosis and Diabetes. Cells 2019, 8, 319. [Google Scholar] [CrossRef]
- Pennacchio, L.A.; Rubin, E.M. Apolipoprotein A5, a newly identified gene that affects plasma triglyceride levels in humans and mice. Arter. Thromb. Vasc. Biol. 2003, 23, 529–534. [Google Scholar] [CrossRef]
- Kane, J.P.; Hardman, D.A.; Paulus, H.E. Heterogeneity of apolipoprotein B: Isolation of a new species from human chylomicrons. Proc. Natl. Acad. Sci. USA 1980, 77, 2465–2469. [Google Scholar] [CrossRef]
- Fuior, E.V.; Gafencu, A.V. Apolipoprotein C1: Its Pleiotropic Effects in Lipid Metabolism and Beyond. Int. J. Mol. Sci. 2019, 20, 5939. [Google Scholar] [CrossRef]
- Wolska, A.; Dunbar, R.L.; Freeman, L.A.; Ueda, M.; Amar, M.J.; Sviridov, D.O.; Remaley, A.T. Apolipoprotein C-II: New findings related to genetics, biochemistry, and role in triglyceride metabolism. Atherosclerosis 2017, 267, 49–60. [Google Scholar] [CrossRef]
- Vaith, P.; Assmann, G.; Uhlenbruck, G. Characterization of the oligosaccharide side chain of apolipoprotein C-III from human plasma very low density lipoproteins. Biochim. Biophys. Acta (BBA) Gen. Subj. 1978, 541, 234–240. [Google Scholar] [CrossRef]
- Allan, C.M.; Walker, D.; Segrest, J.P.; Taylor, J.M. Identification and characterization of a new human gene (APOC4) in the apolipoprotein E, C-I, and C-II gene locus. Genomics 1995, 28, 291–300. [Google Scholar] [CrossRef]
- McConathy, W.; Alaupovic, P. Isolation and partial characterization of apolipoprotein D: A new protein moiety of the human plasma lipoprotein system. FEBS Lett. 1973, 37, 178–182. [Google Scholar] [CrossRef]
- Utermann, G.; Jaeschke, M.; Menzel, J. Familial hyperlipoproteinemia type III: Deficiency of a specific apolipoprotein (apo E-III) in the very-low-density lipoproteins. FEBS Lett. 1975, 56, 352–355. [Google Scholar] [CrossRef]
- Fidge, N.H.; Nestel, P.J. Identification of apolipoproteins involved in the interaction of human high density lipoprotein3 with receptors on cultured cells. J. Biol. Chem. 1985, 260, 3570–3575. [Google Scholar] [CrossRef]
- Polz, E.; Kostner, G.M. The binding of β2-glycoprotein-I to human serum lipoproteins: Distribution among density fractions. FEBS Lett. 1979, 102, 183–186. [Google Scholar] [CrossRef]
- Fritz, I.B.; Burdzy, K.; Sétchell, B.; Blaschuk, O. Ram rete testis fluid contains a protein (clusterin) which influences cell-cell interactions in vitro. Biol. Reprod. 1983, 28, 1173–1188. [Google Scholar] [CrossRef]
- Duchateau, P.N.; Pullinger, C.R.; Orellana, R.E.; Kunitake, S.T.; Naya-Vigne, J.; O’Connor, P.M.; Malloy, M.J.; Kane, J.P. Apolipoprotein L, a new human high density lipoprotein apolipoprotein expressed by the pancreas. Identification, cloning, characterization, and plasma distribution of apolipoprotein L. J. Biol. Chem. 1997, 272, 25576–25582. [Google Scholar] [CrossRef]
- Monajemi, H.; Fontijn, R.D.; Pannekoek, H.; Horrevoets, A.J. The apolipoprotein L gene cluster has emerged recently in evolution and is expressed in human vascular tissue. Genomics 2002, 79, 539–546. [Google Scholar] [CrossRef]
- Xu, N.; Dahlbäck, B. A Novel Human Apolipoprotein (apoM). J. Biol. Chem. 1999, 274, 31286–31290. [Google Scholar] [CrossRef]
- Lamant, M.; Smih, F.; Harmancey, R.; Philip-Couderc, P.; Pathak, A.; Roncalli, J.; Galinier, M.; Collet, X.; Massabuau, P.; Senard, J.-M.; et al. ApoO, a novel apolipoprotein, is an original glycoprotein up-regulated by diabetes in human heart. J. Biol. Chem. 2006, 281, 36289–36302. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Wolk, D.A.; Dickerson, B.C.; Initiative, T.A.D.N.; Weiner, M.; Aiello, M.; Aisen, P.; Albert, M.S.; Alexander, G.; Anderson, H.S.; Anderson, K.; et al. Apolipoprotein E (APOE) genotype has dissociable effects on memory and attentional–executive network function in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 10256–10261. [Google Scholar] [CrossRef]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E: Structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J. Lipid Res. 2009, 50, S183–S188. [Google Scholar] [CrossRef]
- Marais, A.D.; Solomon, G.A.E.; Blom, D.J. Dysbetalipoproteinaemia: A mixed hyperlipidaemia of remnant lipoproteins due to mutations in apolipoprotein E. Crit. Rev. Clin. Lab. Sci. 2014, 51, 46–62. [Google Scholar] [CrossRef]
- McNeill, E.; Channon, K.M.; Greaves, D.R. Inflammatory cell recruitment in cardiovascular disease: Murine models and potential clinical applications. Clin. Sci. 2010, 118, 641–655. [Google Scholar] [CrossRef]
- Deary, I.J.; Whiteman, M.C.; Pattie, A.; Starr, J.M.; Hayward, C.; Wright, A.F.; Carothers, A.; Whalley, L.J. Cognitive change and the APOE ε4 allele. Nature 2002, 418, 932. [Google Scholar] [CrossRef]
- Jacobs, E.G.; Kroenke, C.; Lin, J.; Epel, E.S.; Kenna, H.A.; Blackburn, E.H.; Rasgon, N.L. Accelerated cell aging in female APOE-ε4 carriers: Implications for hormone therapy use. PLoS ONE 2013, 8, e54713. [Google Scholar] [CrossRef]
- Stefl, S.; Nishi, H.; Petukh, M.; Panchenko, A.R.; Alexov, E. Molecular mechanisms of disease-causing missense mutations. J. Mol. Biol. 2013, 425, 3919–3936. [Google Scholar] [CrossRef]
- Bhardwaj, T.; Giri, R. Potential of ADAM 17 Signal Peptide to Form Amyloid Aggregates in Vitro. ACS Chem. Neurosci. 2023, 14, 3818–3825. [Google Scholar] [CrossRef]
- Invernizzi, G.; Papaleo, E.; Sabate, R.; Ventura, S. Protein aggregation: Mechanisms and functional consequences. Int. J. Biochem. Cell Biol. 2012, 44, 1541–1554. [Google Scholar] [CrossRef]
- Pan, Q.; Parra, G.B.; Myung, Y.; Portelli, S.; Nguyen, T.B.; Ascher, D.B. AlzDiscovery: A computational tool to identify Alzheimer’s disease-causing missense mutations using protein structure information. Protein Sci. 2024, 33, e5147. [Google Scholar] [CrossRef]
- Redler, R.L.; Das, J.; Diaz, J.R.; Dokholyan, N.V. Protein Destabilization as a Common Factor in Diverse Inherited Disorders. J. Mol. Evol. 2016, 82, 11–16. [Google Scholar] [CrossRef]
- Garai, K.; Frieden, C. The association−dissociation behavior of the ApoE proteins: Kinetic and equilibrium studies. Biochemistry 2010, 49, 9533–9541. [Google Scholar] [CrossRef]
- Abrego-Guandique, D.M.; Saraceno, G.F.; Cannataro, R.; de Burnside, M.M.; Caroleo, M.C.; Cione, E. Apolipoprotein E and Alzheimer’s Disease in Italian Population: Systematic Review and Meta-Analysis. Brain Sci. 2024, 14, 908. [Google Scholar] [CrossRef]
- Nemergut, M.; Marques, S.M.; Uhrik, L.; Vanova, T.; Nezvedova, M.; Gadara, D.C.; Jha, D.; Tulis, J.; Novakova, V.; Planas-Iglesias, J.; et al. Domino-like effect of C112R mutation on ApoE4 aggregation and its reduction by Alzheimer’s Disease drug candidate. Mol. Neurodegener. 2023, 18, 38. [Google Scholar] [CrossRef]
- Owji, H.; Eslami, M.; Nezafat, N.; Ghasemi, Y. In Silico Elucidation of Deleterious Non-synonymous SNPs in SHANK3, the Autism Spectrum Disorder Gene. J. Mol. Neurosci. 2020, 70, 1649–1667. [Google Scholar] [CrossRef]
- Petrosino, M.; Novak, L.; Pasquo, A.; Chiaraluce, R.; Turina, P.; Capriotti, E.; Consalvi, V. Analysis and Interpretation of the Impact of Missense Variants in Cancer. Int. J. Mol. Sci. 2021, 22, 5416. [Google Scholar] [CrossRef]
- Mustafa, M.I.; Mohammed, Z.O.; Murshed, N.S.; Elfadol, N.M.; Abdelmoneim, A.H.; Hassan, M.A. In Silico Genetics Revealing 5 Mutations in CEBPA Gene Associated with Acute Myeloid Leukemia. Cancer Inform. 2019, 18, 1176935119870817. [Google Scholar] [CrossRef]
- Shafie, A.; Ashour, A.A.; Anjum, F.; Shamsi, A.; Hassan, I. Elucidating the Impact of Deleterious Mutations on IGHG1 and Their Association with Huntington’s Disease. J. Pers. Med. 2024, 14, 380. [Google Scholar] [CrossRef]
- Medway, C.W.; Abdul-Hay, S.; Mims, T.; Ma, L.; Bisceglio, G.; Zou, F.; Pankratz, S.; Sando, S.B.; Aasly, J.O.; Barcikowska, M.; et al. ApoE variant p.V236E is associated with markedly reduced risk of Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 11. [Google Scholar] [CrossRef]
- Rasmussen, K.L.; Tybjærg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. APOE and dementia—resequencing and genotyping in 105,597 individuals. Alzheimer’s Dement. 2020, 16, 1624–1637. [Google Scholar] [CrossRef]
- Rasmussen, K.L.; Tybjærg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. Plasma levels of apolipoprotein E and risk of dementia in the general population. Ann. Neurol. 2015, 77, 301–311. [Google Scholar] [CrossRef]
- Rasmussen, K.L.; Tybjærg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. Plasma apolipoprotein E levels and risk of dementia: A Mendelian randomization study of 106,562 individuals. Alzheimer’s Dement. 2018, 14, 71–80. [Google Scholar] [CrossRef]
- Kalra, M.; Pal, P.; Kaushal, R.; Amin, R.S.; Dolan, L.M.; Fitz, K.; Kumar, S.; Sheng, X.; Guha, S.; Mallik, J.; et al. Association of ApoE genetic variants with obstructive sleep apnea in children. Sleep Med. 2008, 9, 260–265. [Google Scholar] [CrossRef] [PubMed]
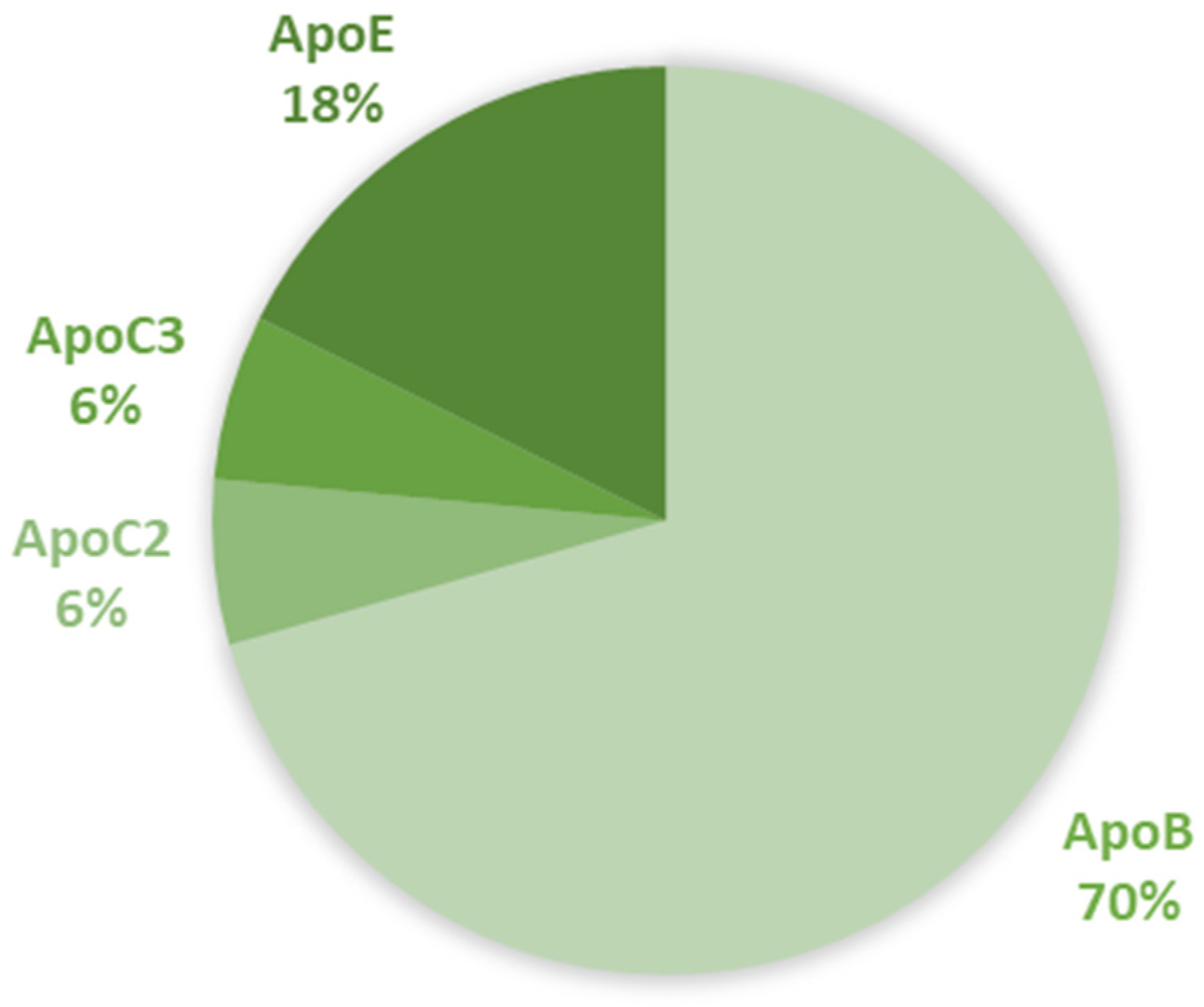
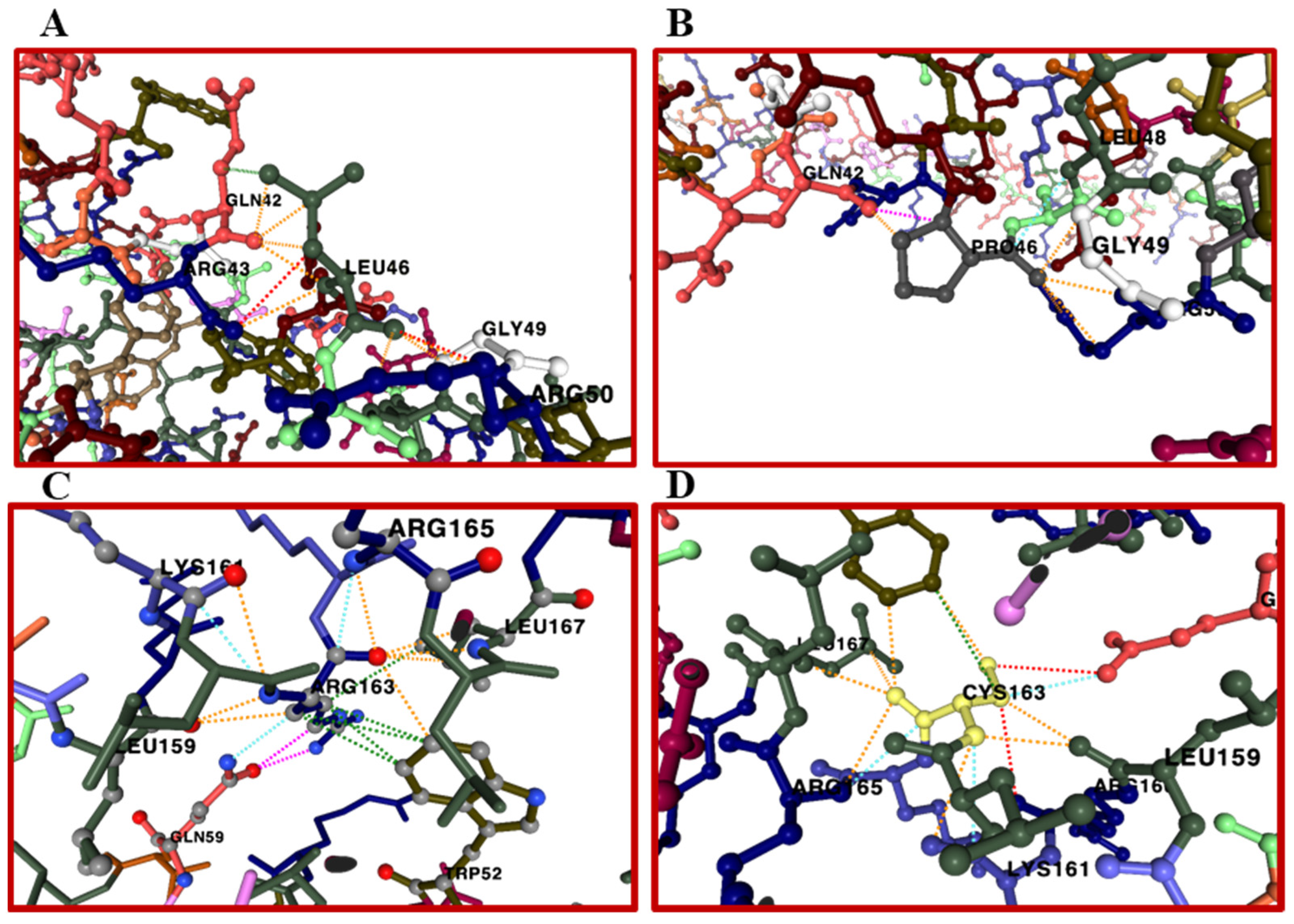
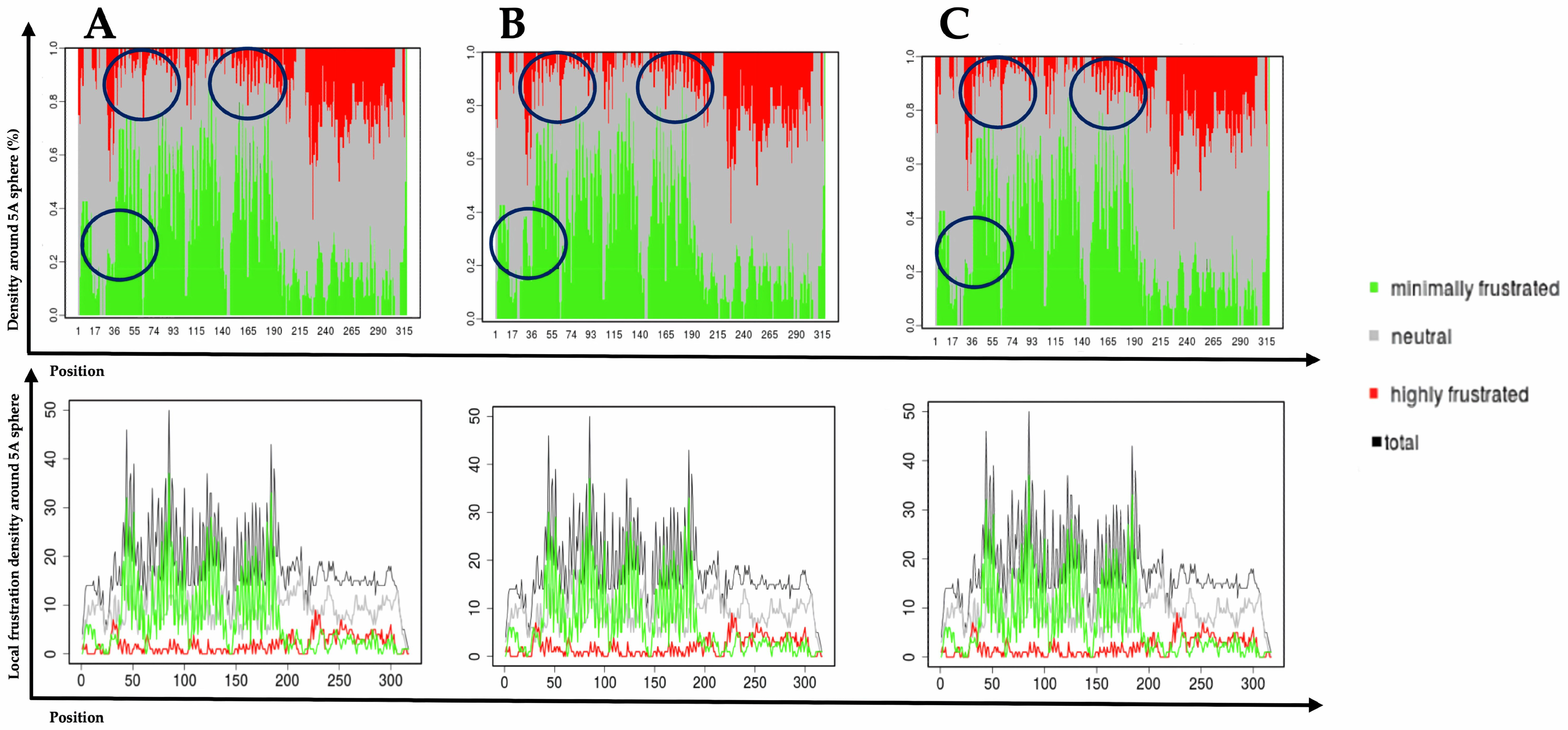

{kind=link}
{kind=link}
{kind=link}
| Gene | aa Replacement | SNPs ID (rs) | SIFT Class 1 | PolyPhen-2 2 | FATHMM 3 | SNPs&GO 4 | Prediction Score |
|---|---|---|---|---|---|---|---|
| ApoB | T191A | rs1318040190 | Tolerated | Benign | Tolerated | Neutral | 0/4 |
| ApoB | F299V | rs72653060 | Tolerated | Probably Damaging | Tolerated | Neutral | 1/4 |
| ApoB | N902K | rs1801700 | Deleterious | Possibly Damaging | Tolerated | Neutral | 2/4 |
| ApoB | R1128H | rs12713843 | Deleterious | Possibly Damaging | Tolerated | Neutral | 2/4 |
| ApoB | S1203G | rs78875649 | Tolerated | Possibly Damaging | Tolerated | Neutral | 1/4 |
| ApoB | G2540V | rs571626569 | Tolerated | Possibly Damaging | Tolerated | Neutral | 1/4 |
| ApoB | E2566K | rs1801696 | Deleterious | Benign | Tolerated | Neutral | 1/4 |
| ApoB | N2785H | rs2163204 | Deleterious | Benign | Tolerated | Neutral | 1/4 |
| ApoB | P2821L | rs72653095 | Tolerated | Probably Damaging | Tolerated | Neutral | 1/4 |
| ApoB | Y3295H | rs186299244 | Tolerated | Probably Damaging | Tolerated | Neutral | 1/4 |
| ApoB | R3500W | rs144467873 | Tolerated | Probably Damaging | Damaging | Neutral | 2/4 |
| ApoB | R4385H | rs533755016 | Damaging | Benign | Tolerated | Neutral | 1/4 |
| ApoC2 | K41T | rs120074114 | Tolerated | Benign | Tolerated | Neutral | 0/4 |
| ApoC3 | A43T | rs147210663 | Tolerated | Probably Damaging | Damaging | Neutral | 2/4 |
| ApoE | L46P | rs769452 | Tolerated | Probably Damaging | Damaging | Disease | 3/4 |
| ApoE | C130R | rs429358 | Tolerated | Benign | Tolerated | Disease | 1/4 |
| ApoE | R163C | rs769455 | Damaging | Probably Damaging | Tolerated | Disease | 3/4 |
| aa Replacement | MAESTROweb 1 | PremPS 2 | mCSM 3 | DynaMut2 4 | Prediction Score |
|---|---|---|---|---|---|
| L46P | Destabilizing | Destabilizing | Destabilizing | Stabilizing | 3/4 |
| ΔΔG | −0.01 kcal/mol | 0.25 kcal/mol | −0.554 kcal/mol | 0.33 kcal/mol | |
| R163C | Destabilizing | Destabilizing | Destabilizing | Destabilizing | 4/4 |
| ΔΔG | −0.672 kcal/mol | 0.71 kcal/mol | −1.565 kcal/mol | −1.28 kcal/mol |
| aa Replacement | PhD-SNP 1 | Mutpred 2 | Pathogenicity Score |
|---|---|---|---|
| L46P | Disease | Neutral | 1/2 |
| R163C | Neutral | Neutral | 0/2 |
| aa Replacement | Camsol 1 | Aggrescan3D 2.0 2 |
|---|---|---|
| wild type | 1.819611 | −401.4035 |
| L46P | 1.841586 | −408.2955 |
| R163C | 1.731944 | −405.0661 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saraceno, G.F.; Cione, E. Multiple Bio-Computational Tools Emerge as Valid Approach in the Assessment of Apolipoproteins Pathogenicity Related Mutations. BioMedInformatics 2025, 5, 16. https://doi.org/10.3390/biomedinformatics5010016
Saraceno GF, Cione E. Multiple Bio-Computational Tools Emerge as Valid Approach in the Assessment of Apolipoproteins Pathogenicity Related Mutations. BioMedInformatics. 2025; 5(1):16. https://doi.org/10.3390/biomedinformatics5010016
Chicago/Turabian StyleSaraceno, Giorgia Francesca, and Erika Cione. 2025. "Multiple Bio-Computational Tools Emerge as Valid Approach in the Assessment of Apolipoproteins Pathogenicity Related Mutations" BioMedInformatics 5, no. 1: 16. https://doi.org/10.3390/biomedinformatics5010016
APA StyleSaraceno, G. F., & Cione, E. (2025). Multiple Bio-Computational Tools Emerge as Valid Approach in the Assessment of Apolipoproteins Pathogenicity Related Mutations. BioMedInformatics, 5(1), 16. https://doi.org/10.3390/biomedinformatics5010016