
Deficiency in DNA Damage Repair Proteins Promotes Prostate Cancer Cell Migration through Oxidative Stress

 , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Transfections
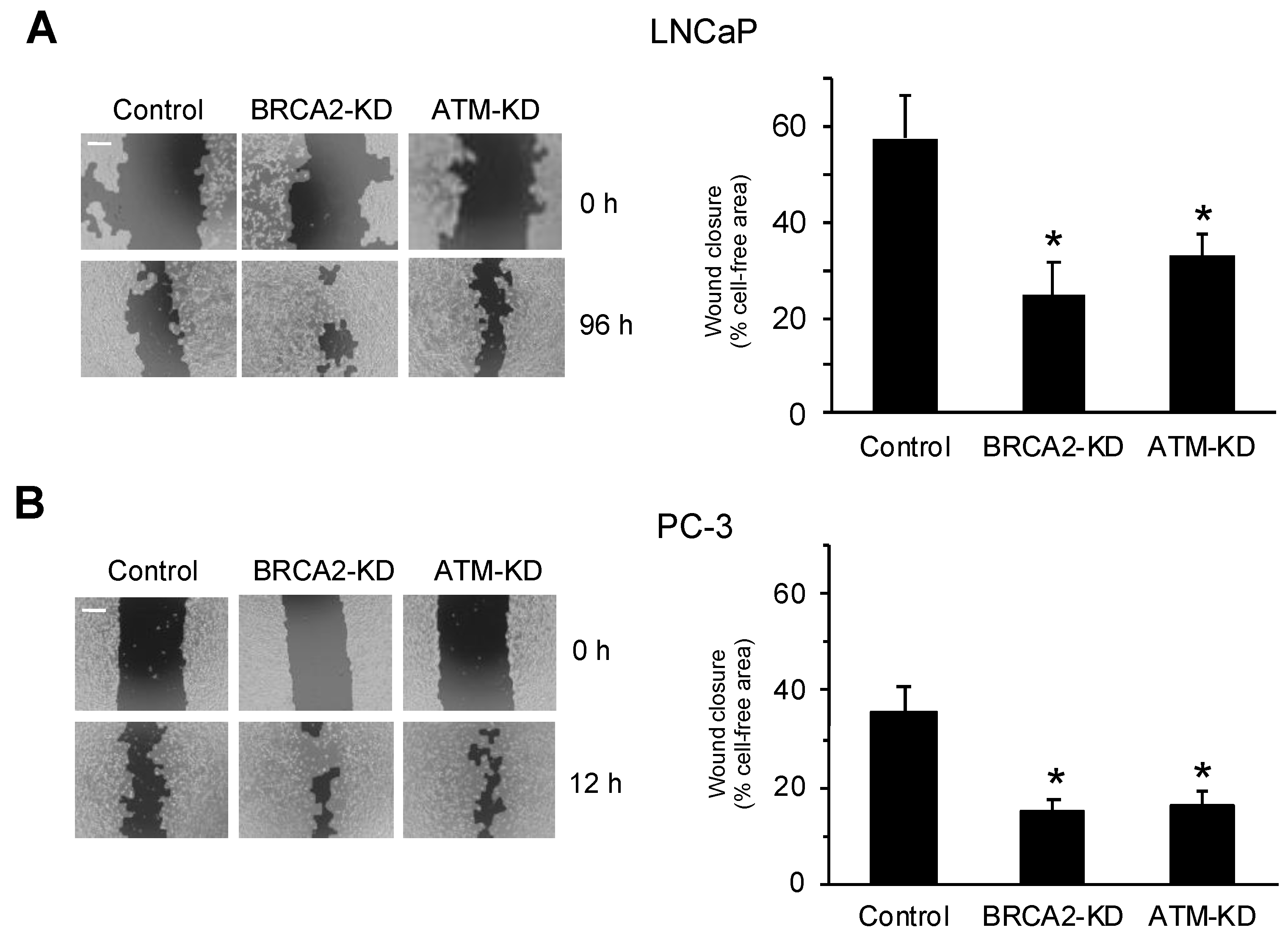
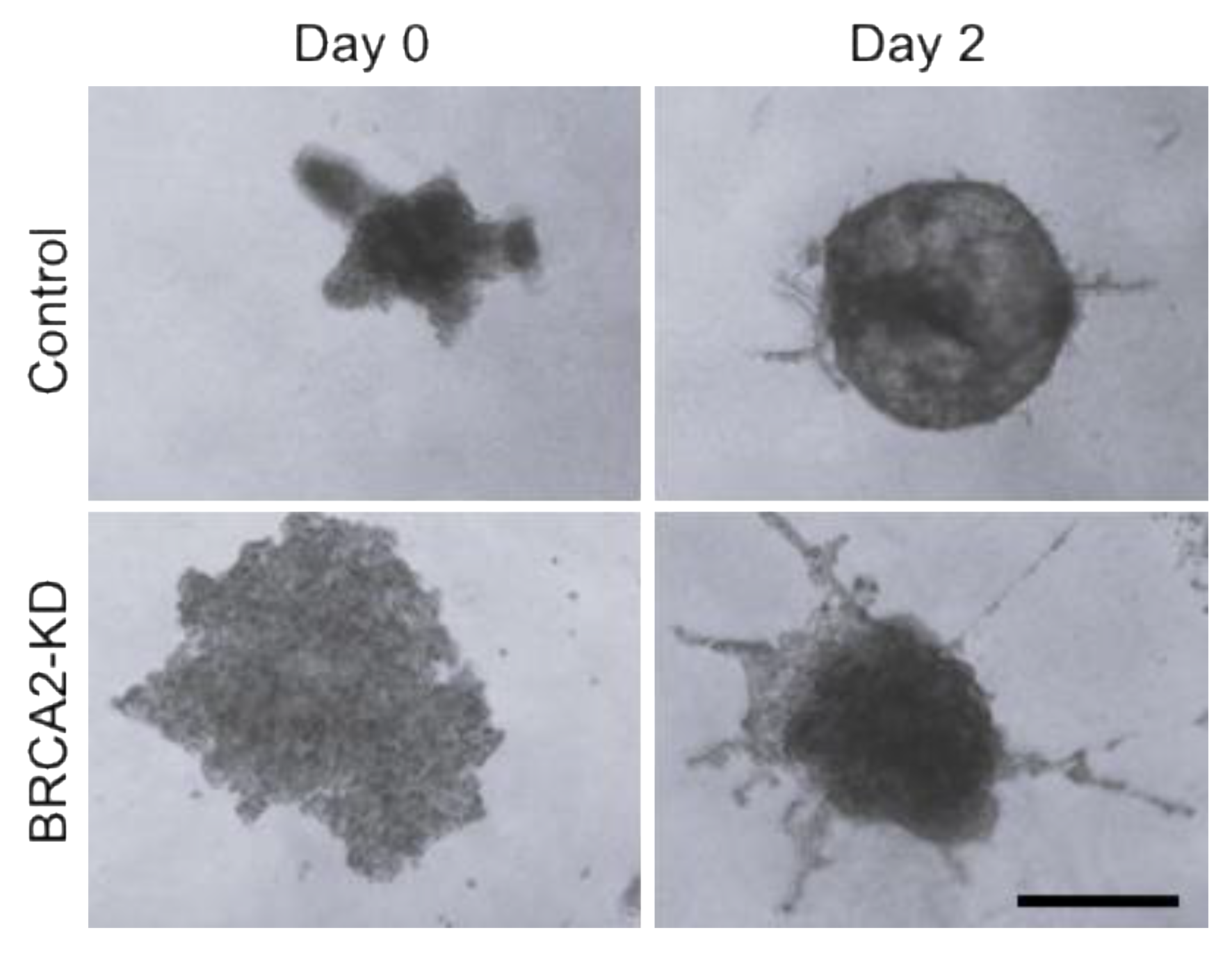
2.2. Wound Healing, Transwell Migration, and 3D Spheroid Invasion Assays
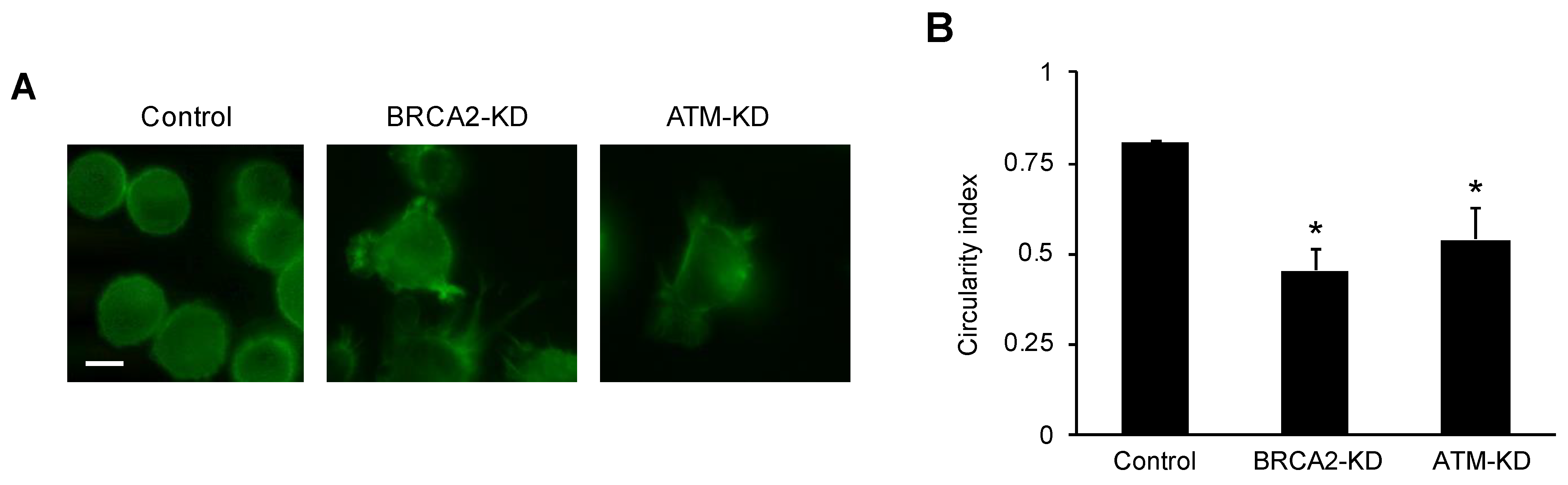
2.3. Fluorescence Microscopy and Cell Circularity Measurement
2.4. Measurement of Reactive Oxygen Species (ROS)
2.5. Immunohistochemistry
2.6. Statistical Analysis
3. Results
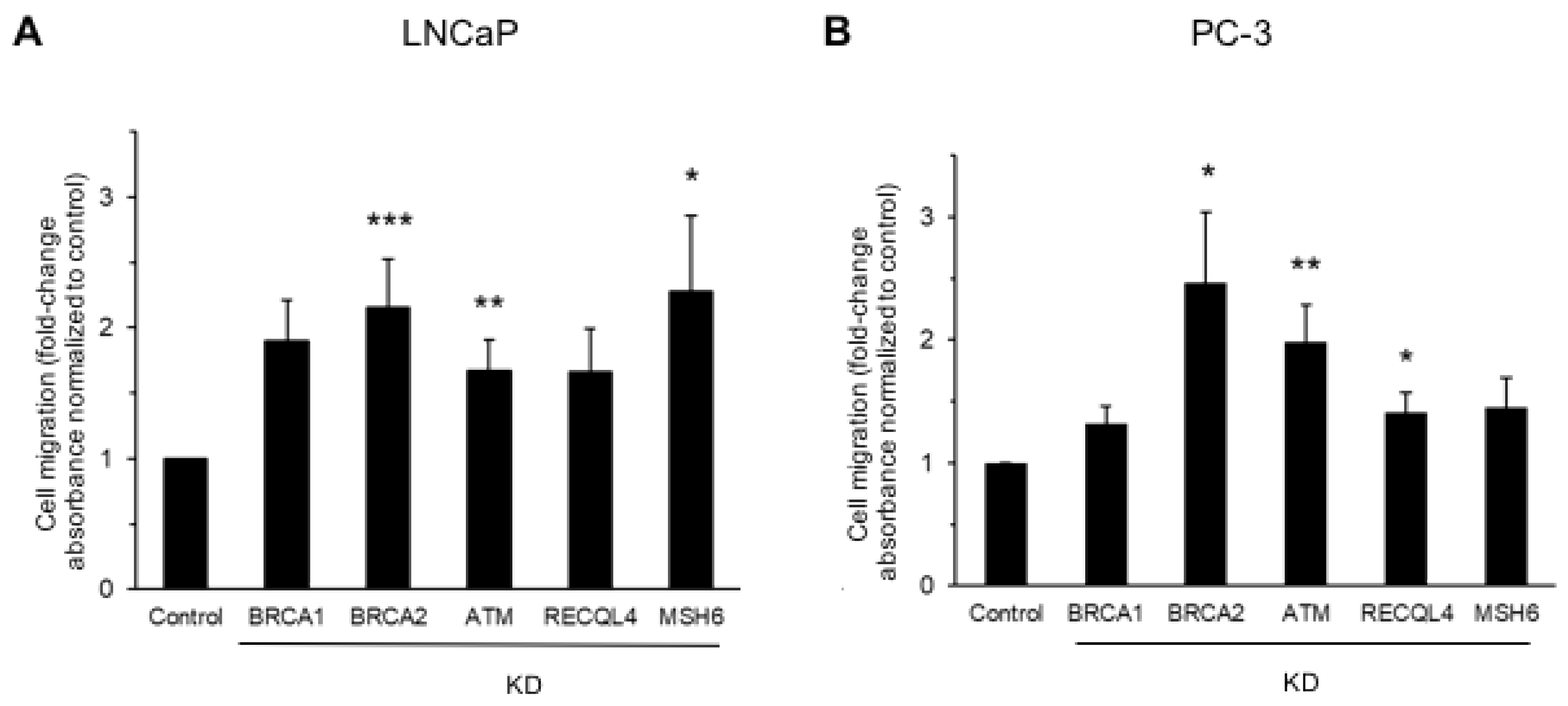
3.1. DNA Damage Repair Protein Deficiency Promotes Prostate Cancer Cell Migration
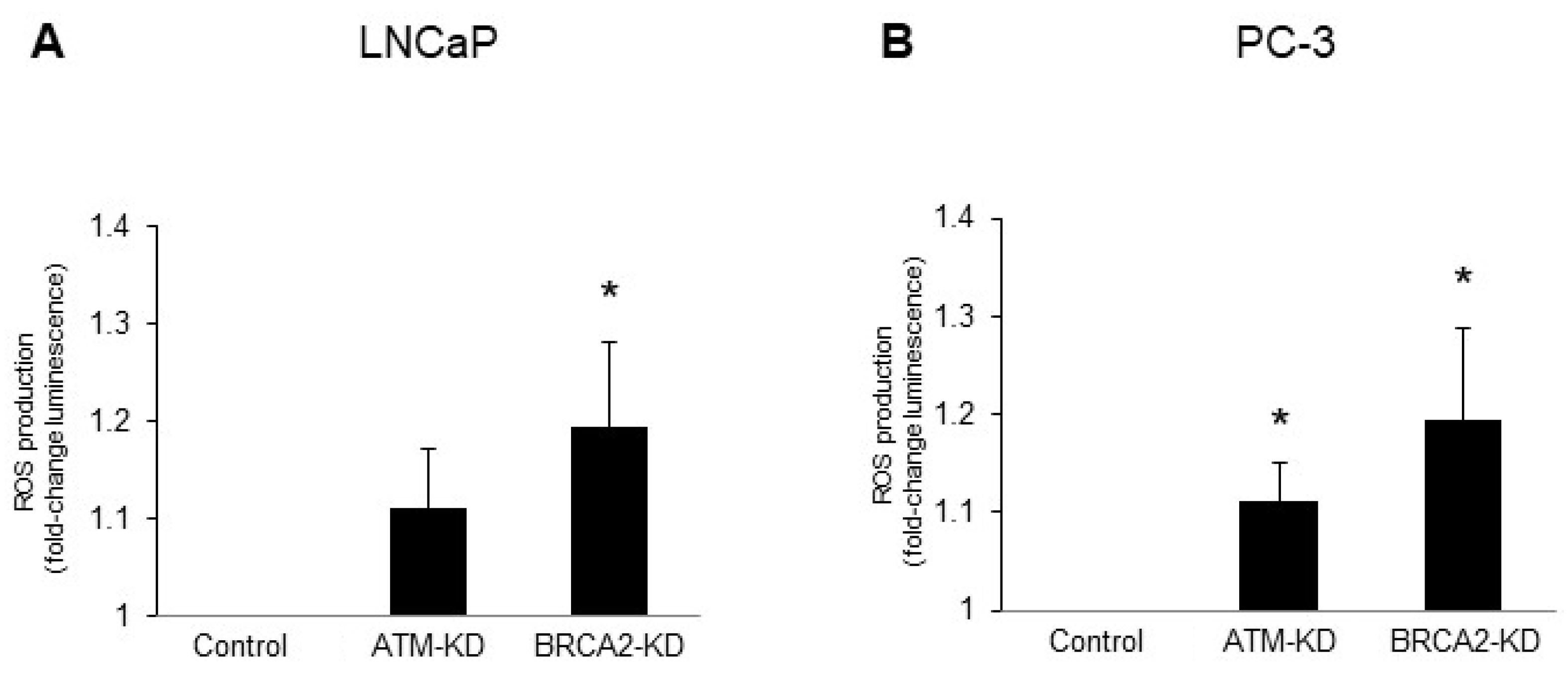
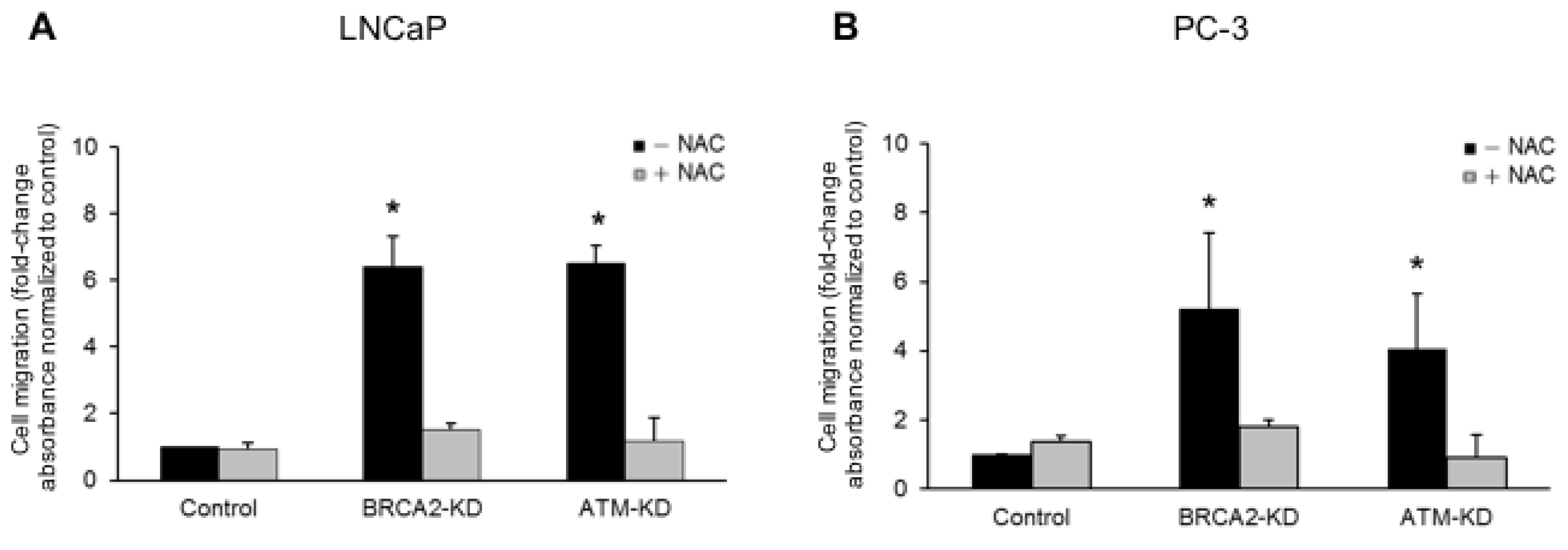
3.2. ATM or BRCA2 Deficiency Promotes Prostate Cancer Cell Migration through the Induction of Oxidative Stress
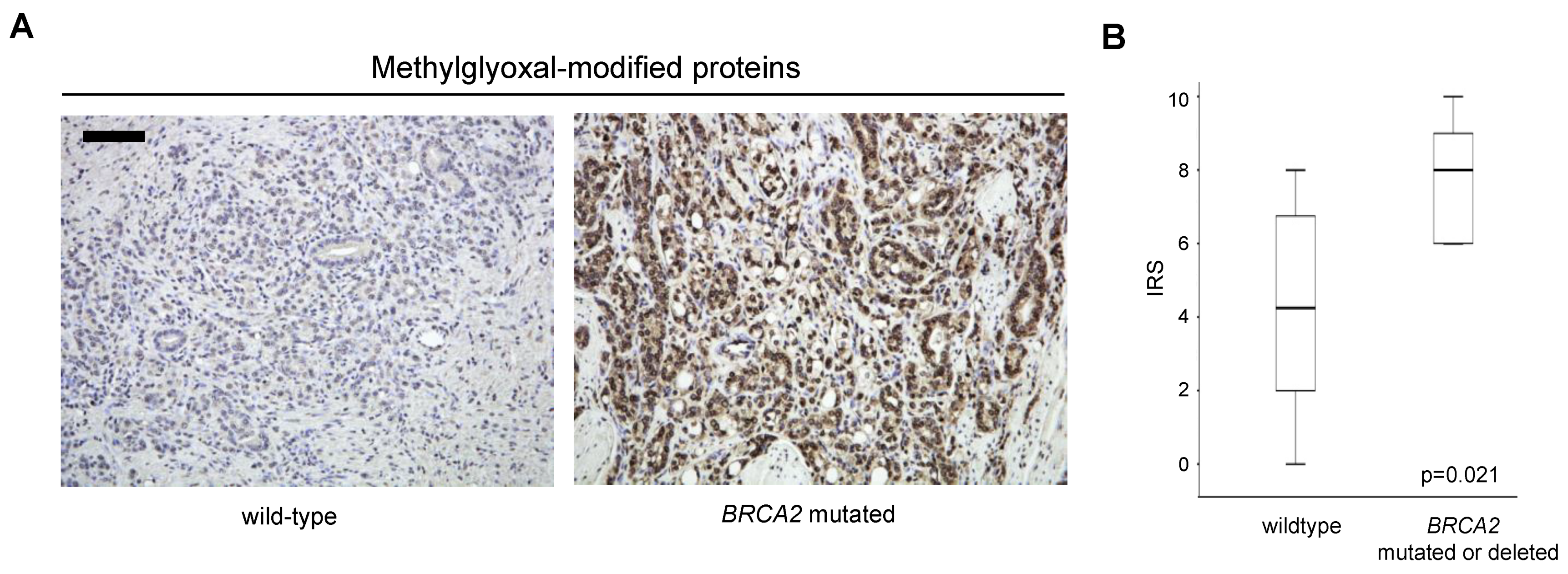
3.3. Increased Oxidative Stress in Prostate Cancer with BRCA2 Inactivation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Parker, C.; Eeles, R.A.; Schröder, F.; Tomlins, S.A.; Tannock, I.; Drake, C.G.; de Bono, J.S. Prostate cancer. Lancet 2016, 387, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Sartor, O.; de Bono, J.S. Metastatic Prostate Cancer. N. Engl. J. Med. 2018, 378, 1653–1654. [Google Scholar] [CrossRef]
- Weiss, F.; Lauffenburger, D.; Friedl, P. Towards targeting of shared mechanisms of cancer metastasis and therapy resistance. Nat. Rev. Cancer 2022, 22, 157–173. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Nientiedt, C.; Duensing, A.; Zschäbitz, S.; Jäger, D.; Hohenfellner, M.; Stenzinger, A.; Duensing, S. PARP inhibition in prostate cancer. Genes Chromosom. Cancer 2021, 60, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Bednarz, N.; Eltze, E.; Semjonow, A.; Rink, M.; Andreas, A.; Mulder, L.; Hannemann, J.; Fisch, M.; Pantel, K.; Weier, H.-U.G.; et al. BRCA1 loss preexisting in small subpopulations of prostate cancer is associated with advanced disease and metastatic spread to lymph nodes and peripheral blood. Clin. Cancer Res. 2010, 16, 3340–3348. [Google Scholar] [CrossRef]
- Castro, E.; Goh, C.; Olmos, D.; Saunders, E.; Leongamornlert, D.; Tymrakiewicz, M.; Mahmud, N.; Dadaev, T.; Govindasami, K.; Guy, M.; et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J. Clin. Oncol. 2013, 31, 1748–1757. [Google Scholar] [CrossRef]
- Nientiedt, C.; Budczies, J.; Endris, V.; Kirchner, M.; Schwab, C.; Jurcic, C.; Behnisch, R.; Hoveida, S.; Lantwin, P.; Kaczorowski, A.; et al. Mutations in TP53 or DNA damage repair genes define poor prognostic subgroups in primary prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2022, 40, 8.e11–8.e18. [Google Scholar] [CrossRef]
- Taylor, R.A.; Fraser, M.; Livingstone, J.; Espiritu, S.M.G.; Thorne, H.; Huang, V.; Lo, W.; Shiah, Y.-J.; Yamaguchi, T.N.; Sliwinski, A.; et al. Germline BRCA2 mutations drive prostate cancers with distinct evolutionary trajectories. Nat. Commun. 2017, 8, 13671. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Yokomizo, A.; Tada, Y.; Inokuchi, J.; Kashiwagi, E.; Masubuchi, D.; Eto, M.; Uchiumi, T.; Naito, S. Castration resistance of prostate cancer cells caused by castration-induced oxidative stress through Twist1 and androgen receptor overexpression. Oncogene 2010, 29, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Decker, B.; Karyadi, D.M.; Davis, B.W.; Karlins, E.; Tillmans, L.S.; Stanford, J.L.; Thibodeau, S.N.; Ostrander, E.A. Biallelic BRCA2 Mutations Shape the Somatic Mutational Landscape of Aggressive Prostate Tumors. Am. J. Hum. Genet. 2016, 98, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, P.; Bandlamudi, C.; Cheng, M.L.; Srinivasan, P.; Chavan, S.S.; Friedman, N.D.; Rosen, E.Y.; Richards, A.L.; Bouvier, N.; Selcuklu, S.D.; et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature 2019, 571, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Holloman, W.K. Unraveling the mechanism of BRCA2 in homologous recombination. Nat. Struct. Mol. Biol. 2011, 18, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science 2014, 343, 1470–1475. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. Functions of BRCA1 and BRCA2 in the biological response to DNA damage. J. Cell Sci. 2001, 114, 3591–3598. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Koike, A.; Takeshita, T.; Ohta, T. The ubiquitin E3 ligase activity of BRCA1 and its biological functions. Cell Div. 2008, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Mateo, J.; Seed, G.; Bertan, C.; Rescigno, P.; Dolling, D.; Figueiredo, I.; Miranda, S.; Nava Rodrigues, D.; Gurel, B.; Clarke, M.; et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J. Clin. Investig. 2020, 130, 1743–1751. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Ledet, E.M.; Antonarakis, E.S.; Isaacs, W.B.; Lotan, T.L.; Pritchard, C.; Sartor, A.O. Germline BLM mutations and metastatic prostate cancer. Prostate 2020, 80, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.T.; Bernstein, K.A. Role and Regulation of the RECQL4 Family during Genomic Integrity Maintenance. Genes 2021, 12, 1919. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [PubMed]
- Kaczorowski, A.; Tolstov, Y.; Falkenstein, M.; Vasioukhin, V.; Prigge, E.-S.; Geisler, C.; Kippenberger, M.; Nientiedt, C.; Ratz, L.; Kuryshev, V.; et al. Rearranged ERG confers robustness to prostate cancer cells by subverting the function of p53. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 736.e1–736.e10. [Google Scholar] [CrossRef] [PubMed]
- Gebäck, T.; Schulz, M.M.P.; Koumoutsakos, P.; Detmar, M. TScratch: A Novel and Simple Software Tool for Automated Analysis of Monolayer Wound Healing Assays. BioTechniques 2009, 46, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Cox, E.P. A method of assigning numerical and percentage values to the degree of roundness of sand grains. J. Paleontol. 1927, 1, 179–183. [Google Scholar]
- Nientiedt, C.; Endris, V.; Jenzer, M.; Mansour, J.; Pour Sedehi, N.T.; Pecqueux, C.; Volckmar, A.-L.; Leichsenring, J.; Neumann, O.; Kirchner, M.; et al. High prevalence of DNA damage repair gene defects and TP53 alterations in men with treatment-naïve metastatic prostate cancer –Results from a prospective pilot study using a 37 gene panel. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 637.e17–637.e27. [Google Scholar] [CrossRef]
- Aslan, M.; Hsu, E.-C.; Liu, S.; Stoyanova, T. Quantifying the invasion and migration ability of cancer cells with a 3D Matrigel drop invasion assay. Biol. Methods Protoc. 2021, 6, bpab014. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Castro, E.; Goh, C.; Leongamornlert, D.; Saunders, E.; Tymrakiewicz, M.; Dadaev, T.; Govindasami, K.; Guy, M.; Ellis, S.; Frost, D.; et al. Effect of BRCA Mutations on Metastatic Relapse and Cause-specific Survival After Radical Treatment for Localised Prostate Cancer. Eur. Urol. 2015, 68, 186–193. [Google Scholar] [CrossRef]
- Gau, D.M.; Lesnock, J.L.; Hood, B.L.; Bhargava, R.; Sun, M.; Darcy, K.; Luthra, S.; Chandran, U.; Conrads, T.P.; Edwards, R.P.; et al. BRCA1 deficiency in ovarian cancer is associated with alteration in expression of several key regulators of cell motility—A proteomics study. Cell Cycle 2015, 14, 1884–1892. [Google Scholar] [CrossRef] [PubMed]
- Tolbert, C.E.; Beck, M.V.; Kilmer, C.E.; Srougi, M.C. Loss of ATM positively regulates Rac1 activity and cellular migration through oxidative stress. Biochem. Biophys. Res. Commun. 2019, 508, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Moro, L.; Arbini, A.A.; Yao, J.L.; di Sant’agnese, P.A.; Marra, E.; Greco, M. Loss of BRCA2 promotes prostate cancer cell invasion through up-regulation of matrix metalloproteinase-9. Cancer Sci. 2008, 99, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Renaudin, X.; Lee, M.; Shehata, M.; Surmann, E.-M.; Venkitaraman, A.R. BRCA2 deficiency reveals that oxidative stress impairs RNaseH1 function to cripple mitochondrial DNA maintenance. Cell Rep. 2021, 36, 109478. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Cai, H.; Wu, T.; Sobhian, B.; Huo, Y.; Alcivar, A.; Mehta, M.; Cheung, K.L.; Ganesan, S.; Kong, A.-N.T.; et al. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol. Cell. Biol. 2012, 32, 1506–1517. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.G.; Piskounova, E.; Morrison, S.J. Cancer, Oxidative Stress, and Metastasis. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 163–175. [Google Scholar] [CrossRef]
- Evensen, N.A.; Madhusoodhan, P.P.; Meyer, J.; Saliba, J.; Chowdhury, A.; Araten, D.J.; Nersting, J.; Bhatla, T.; Vincent, T.L.; Teachey, D.; et al. MSH6 haploinsufficiency at relapse contributes to the development of thiopurine resistance in pediatric B-lymphoblastic leukemia. Haematologica 2018, 103, 830–839. [Google Scholar] [CrossRef]
- Ding, N.; Bonham, E.M.; Hannon, B.E.; Amick, T.R.; Baylin, S.B.; O’Hagan, H.M. Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage. J. Mol. Cell Biol. 2016, 8, 244–254. [Google Scholar] [CrossRef]
- Lalonde, M.; Trauner, M.; Werner, M.; Hamperl, S. Consequences and Resolution of Transcription–Replication Conflicts. Life 2021, 11, 637. [Google Scholar] [CrossRef]
- Andrs, M.; Stoy, H.; Boleslavska, B.; Chappidi, N.; Kanagaraj, R.; Nascakova, Z.; Menon, S.; Rao, S.; Oravetzova, A.; Dobrovolna, J.; et al. Excessive reactive oxygen species induce transcription-dependent replication stress. Nat. Commun. 2023, 14, 1791. [Google Scholar] [CrossRef] [PubMed]
- Pani, G.; Galeotti, T.; Chiarugi, P. Metastasis: Cancer cell’s escape from oxidative stress. Cancer Metastasis Rev. 2010, 29, 351–378. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Zavadskiy, S.P.; Astakhov, D.V.; Terentiev, A.A. Lipid peroxidation: Reactive carbonyl species, protein/DNA adducts, and signaling switches in oxidative stress and cancer. Biochem. Biophys. Res. Commun. 2023, 687, 149167. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Storz, P. Detecting reactive oxygen species by immunohistochemistry. Methods Mol. Biol. 2015, 1292, 97–104. [Google Scholar]
- de Bari, L.; Scirè, A.; Minnelli, C.; Cianfruglia, L.; Kalapos, M.P.; Armeni, T. Interplay among Oxidative Stress, Methylglyoxal Pathway and S-Glutathionylation. Antioxidants 2020, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, T.P.; Thorpe, S.R.; Baynes, J.W. Chemical modification of proteins by methylglyoxal. Cell. Mol. Biol. 1998, 44, 1139–1145. [Google Scholar]
- Privat, M.; Radosevic-Robin, N.; Aubel, C.; Cayre, A.; Penault-Llorca, F.; Marceau, G.; Sapin, V.; Bignon, Y.-J.; Morvan, D. BRCA1 induces major energetic metabolism reprogramming in breast cancer cells. PLoS ONE 2014, 9, e102438. [Google Scholar] [CrossRef]
- Nokin, M.-J.; Bellier, J.; Durieux, F.; Peulen, O.; Rademaker, G.; Gabriel, M.; Monseur, C.; Charloteaux, B.; Verbeke, L.; van Laere, S.; et al. Methylglyoxal, a glycolysis metabolite, triggers metastasis through MEK/ERK/SMAD1 pathway activation in breast cancer. Breast Cancer Res. 2019, 21, 11. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lantwin, P.; Kaczorowski, A.; Nientiedt, C.; Schwab, C.; Kirchner, M.; Schütz, V.; Görtz, M.; Hohenfellner, M.; Duensing, A.; Stenzinger, A.; et al. Deficiency in DNA Damage Repair Proteins Promotes Prostate Cancer Cell Migration through Oxidative Stress. Onco 2024, 4, 56-67. https://doi.org/10.3390/onco4020005
Lantwin P, Kaczorowski A, Nientiedt C, Schwab C, Kirchner M, Schütz V, Görtz M, Hohenfellner M, Duensing A, Stenzinger A, et al. Deficiency in DNA Damage Repair Proteins Promotes Prostate Cancer Cell Migration through Oxidative Stress. Onco. 2024; 4(2):56-67. https://doi.org/10.3390/onco4020005
Chicago/Turabian StyleLantwin, Philippa, Adam Kaczorowski, Cathleen Nientiedt, Constantin Schwab, Martina Kirchner, Viktoria Schütz, Magdalena Görtz, Markus Hohenfellner, Anette Duensing, Albrecht Stenzinger, and et al. 2024. "Deficiency in DNA Damage Repair Proteins Promotes Prostate Cancer Cell Migration through Oxidative Stress" Onco 4, no. 2: 56-67. https://doi.org/10.3390/onco4020005
APA StyleLantwin, P., Kaczorowski, A., Nientiedt, C., Schwab, C., Kirchner, M., Schütz, V., Görtz, M., Hohenfellner, M., Duensing, A., Stenzinger, A., & Duensing, S. (2024). Deficiency in DNA Damage Repair Proteins Promotes Prostate Cancer Cell Migration through Oxidative Stress. Onco, 4(2), 56-67. https://doi.org/10.3390/onco4020005