
Truth or Lie: Does the DNA Extraction Procedure Really Affect the Insight in Composition and Diversity of Microbial Communities in Saffron Cultivated Soils?

 , , , ,
, , , ,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sampling Site
2.2. Soil DNA Extraction, PCR Amplification, and Sequencing
2.3. Bioinformatic and Data Analysis
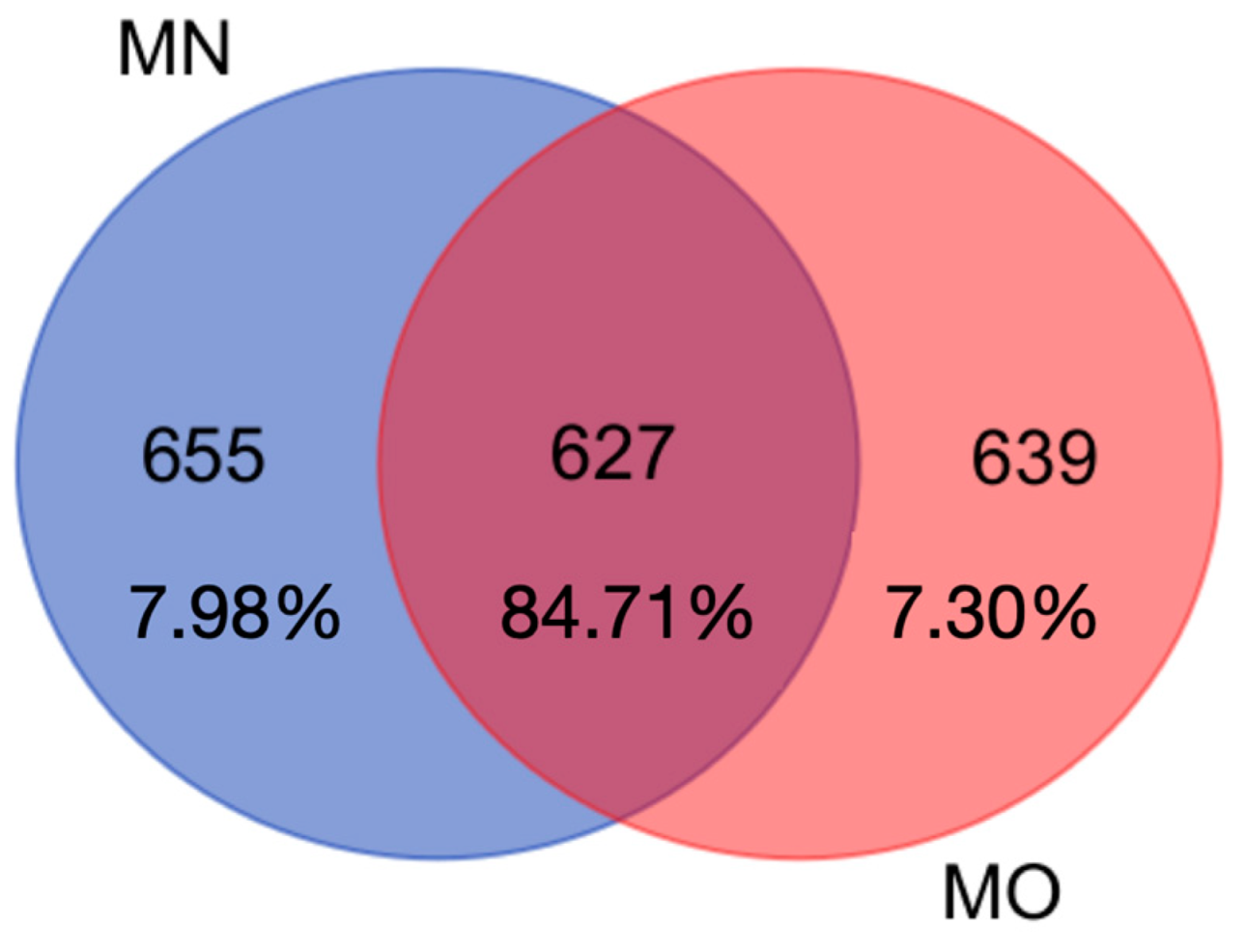
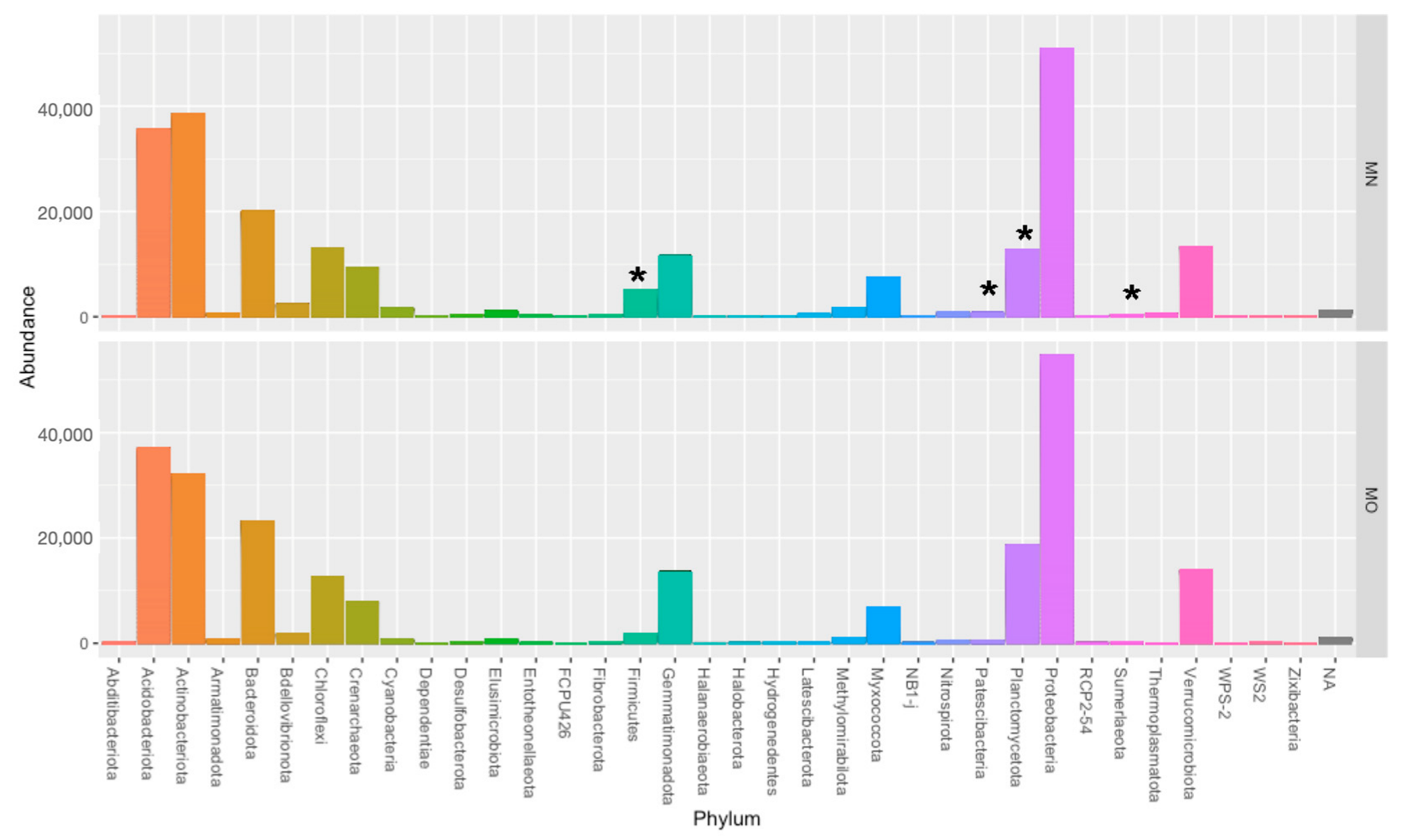
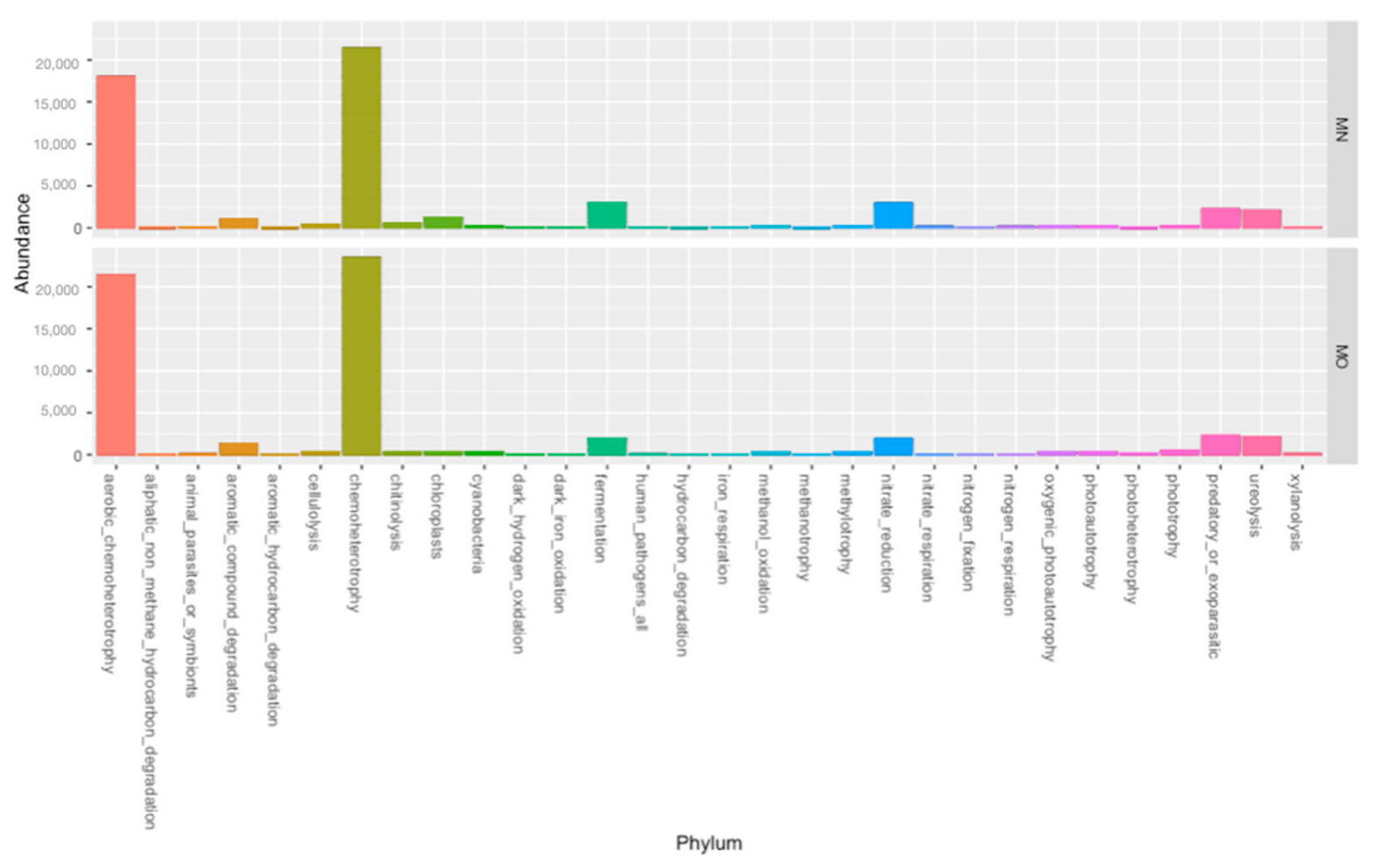
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pédron, J.; Guyon, L.; Lecomte, A.; Blottière, L.; Chandeysson, C.; Rochelle-Newall, E.; Raynaud, X.; Berge, O.; Barny, M.A. Comparison of Environmental and Culture-Derived Bacterial Communities through 16S Metabarcoding: A Powerful Tool to Assess Media Selectivity and Detect Rare Taxa. Microorganisms 2020, 8, 1129. [Google Scholar] [CrossRef] [PubMed]
- Foucher, A.; Evrard, O.; Ficetola, G.F.; Gielly, L.; Poulain, J.; Giguet-Covex, C.; Laceby, J.P.; Salvadores-Blanes, S.; Cerdan, O.; Poulenard, J. Persistence of environmental DNA in cultivated soils: Implication of this memory effect for reconstructing the dynamics of land use and cover changes. Sci. Rep. 2020, 10, 10502. [Google Scholar] [CrossRef] [PubMed]
- Taberlet, P.; Coissac, E.; Pompanon, F.; Brochmann, C.; Willerslev, E. Towards next-generation biodiversity assessment using DNA metabarcoding. Mol. Ecol. 2012, 21, 2045–2050. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, K.M.; Kline, R.J.; Rahman, M.S. Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: A systematic review in methods, monitoring, and applications of global eDNA. Glob. Ecol. Conserv. 2019, 17, e00547. [Google Scholar] [CrossRef]
- Bardgett, R.D.; van der Putten, W.H. Belowground biodiversity and ecosystem functioning. Nature 2014, 515, 505–511. [Google Scholar] [CrossRef]
- Orgiazzi, A.; Dunbar, M.B.; Panagos, P.; de Groot, A.G.; Lemanceau, P. Soil biodiversity and DNA barcodes: Opportunities and challenges. Soil Biol. Biochem. 2015, 80, 244–250. [Google Scholar] [CrossRef]
- Bloor, J.M.G.; Si-Moussi, S.; Taberlet, P.; Carrère, P.; Hedde, M. Analysis of complex trophic networks reveals the signature of land-use intensification on soil communities in agroecosystems. Sci. Rep. 2021, 11, 18260. [Google Scholar] [CrossRef]
- Banerjee, S.; Walder, F.; Büchi, L.; Marcel Meyer, M.; Held, A.Y.; Gattinger, A.; Keller, T.; Charles, R.; van der Heijden, M.G.A. Agricultural intensification reduces microbial network complexity and the abundance of keystone taxa in roots. ISME J. 2019, 13, 1722–1736. [Google Scholar] [CrossRef] [Green Version]
- Keck, F.; Blackman, R.C.; Bossart, R.; Brantschen, J.; Couton, M.; Hürlemann, S.; Kirschner, D.; Locher, N.; Zhang, H.; Altermatt, F. Meta-analysis shows both congruence and complementarity of DNA and eDNA metabarcoding to traditional methods for biological community assessment. Mol. Ecol. 2022, 31, 1820–1835. [Google Scholar] [CrossRef]
- Terrat, S.; Plassart, P.; Bourgeois, E.; Ferreira, S.; Dequiedt, S.; Adele-Dit-De-Renseville, N.; Lemanceau, P.; Bispo, A.; Chabbi, A.; Maron, P.A.; et al. Meta-barcoded evaluation of the ISO standard 11063 DNA extraction procedure to characterize soil bacterial and fungal community diversity and composition. Microb. Biotechnol. 2015, 8, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Zielińska, S.; Radkowski, P.; Blendowska, A.; Ludwig-Gałęzowska, A.; Łoś, J.M.; Łoś, M. The choice of the DNA extraction method may influence the outcome of the soil microbial community structure analysis. MicrobiologyOpen 2017, 6, e00453. [Google Scholar] [CrossRef] [PubMed]
- Soliman, T.; Yang, S.-Y.; Yamazaki, T.; Jenke-Kodama, H. Profiling soil microbial communities with next-generation sequencing: The influence of DNA kit selection and technician technical expertise. PeerJ 2017, 5, e4178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caser, M.; Victorino, Í.M.M.; Demasi, S.; Berruti, A.; Donno, D.; Lumini, E.; Bianciotto, V.; Scariot, V. Saffron Cultivation in Marginal Alpine Environments: How AMF Inoculation Modulates Yield and Bioactive Compounds. Agronomy 2019, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Victorino, Í.; Voyron, S.; Caser, M.; Orgiazzi, A.; Demasi, S.; Berruti, A.; Scariot, V.; Bianciotto, V.; Lumini, E. Metabarcoding of Soil Fungal Communities Associated with Alpine Field-Grown Saffron (Crocus sativus L.) Inoculated with AM Fungi. J. Fungi 2021, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2015, 18, 1403–1414. [Google Scholar] [CrossRef]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Bates, S.T.; Cropsey, G.W.G.; Caporaso, J.G.; Knight, R.; Fierer, N. Bacterial communities associated with the lichen symbiosis. Appl. Environ. Microbiol. 2011, 77, 1309–1314. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, G.T.; Bates, S.T.; Eilers, K.G.; Lauber, C.L.; Caporaso, J.G.; Walters, W.A.; Knight, R.; Fierer, N. The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol. Biochem. 2011, 43, 1450–1455. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Tanabe, S.H.; Yang, C.; Zhang, W.; Sun, J. Bacterial Community Composition of South China Sea Sediments through Pyrosequencing-Based Analysis of 16S rRNA Genes. PLoS ONE 2013, 8, e78501. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, S.; Robeson, M.S., II; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible sequence taxonomy reference database management. PLoS Comput. Biol. 2021, 17, e1009581. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development for R. RStudio, PBC, Boston, MA. 10/01/2020. Available online: http://www.rstudio.com/ (accessed on 3 June 2022).
- Bisanz, J.E. qiime2R: Importing QIIME2 Artifacts and Associated Data into R Sessions. 2018. Available online: https://github.com/jbisanz/qiime2R (accessed on 3 June 2022).
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.5-4. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 3 June 2022).
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Oliveros, J.C. Venny. An interactive Tool for Comparing Lists with Venn’s Diagrams 2007–2015. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 3 June 2022).
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef]
- Changey, F.; Blaud, A.; Pando, A.; Herrmann, A.M.; Lerch, T.Z. Monitoring soil microbial communities using molecular tools: DNA extraction methods may offset long-term management effects. Eur. J. Soil Sci. 2021, 72, 1026–1041. [Google Scholar] [CrossRef]
- Dai, Z.; Webster, T.M.; Enders, A.; Hanley, K.L.; Xu, J.; Thies, J.E.; Lehmann, J. DNA extraction efficiency from soil as affected by pyrolysis temperature and extractable organic carbon of high-ash biochar. Soil Biol. Biochem. 2017, 115, 129–136. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voyron, S.; Maxaieie Victorino, Í.M.; Caser, M.; Demasi, S.; Scariot, V.; Bianciotto, V.; Ghignone, S.; Lumini, E. Truth or Lie: Does the DNA Extraction Procedure Really Affect the Insight in Composition and Diversity of Microbial Communities in Saffron Cultivated Soils? Appl. Microbiol. 2022, 2, 492-501. https://doi.org/10.3390/applmicrobiol2030038
Voyron S, Maxaieie Victorino ÍM, Caser M, Demasi S, Scariot V, Bianciotto V, Ghignone S, Lumini E. Truth or Lie: Does the DNA Extraction Procedure Really Affect the Insight in Composition and Diversity of Microbial Communities in Saffron Cultivated Soils? Applied Microbiology. 2022; 2(3):492-501. https://doi.org/10.3390/applmicrobiol2030038
Chicago/Turabian StyleVoyron, Samuele, Íris Marisa Maxaieie Victorino, Matteo Caser, Sonia Demasi, Valentina Scariot, Valeria Bianciotto, Stefano Ghignone, and Erica Lumini. 2022. "Truth or Lie: Does the DNA Extraction Procedure Really Affect the Insight in Composition and Diversity of Microbial Communities in Saffron Cultivated Soils?" Applied Microbiology 2, no. 3: 492-501. https://doi.org/10.3390/applmicrobiol2030038