SARS-CoV-2 Viroporins: A Multi-Omics Insight from Nucleotides to Amino Acids


Abstract
:1. Introduction
2. Materials and Methods
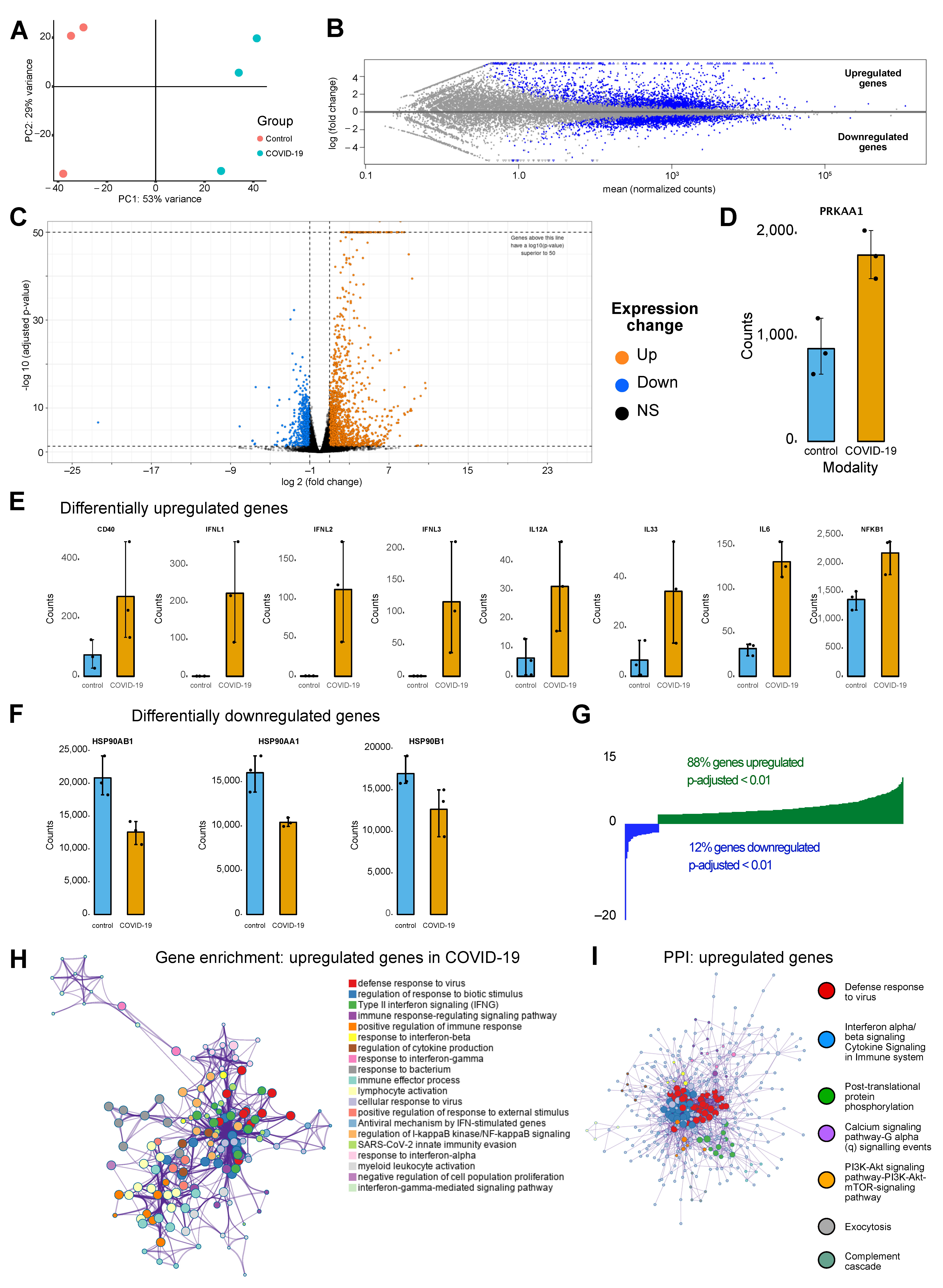
2.1. Transcriptomic Analysis
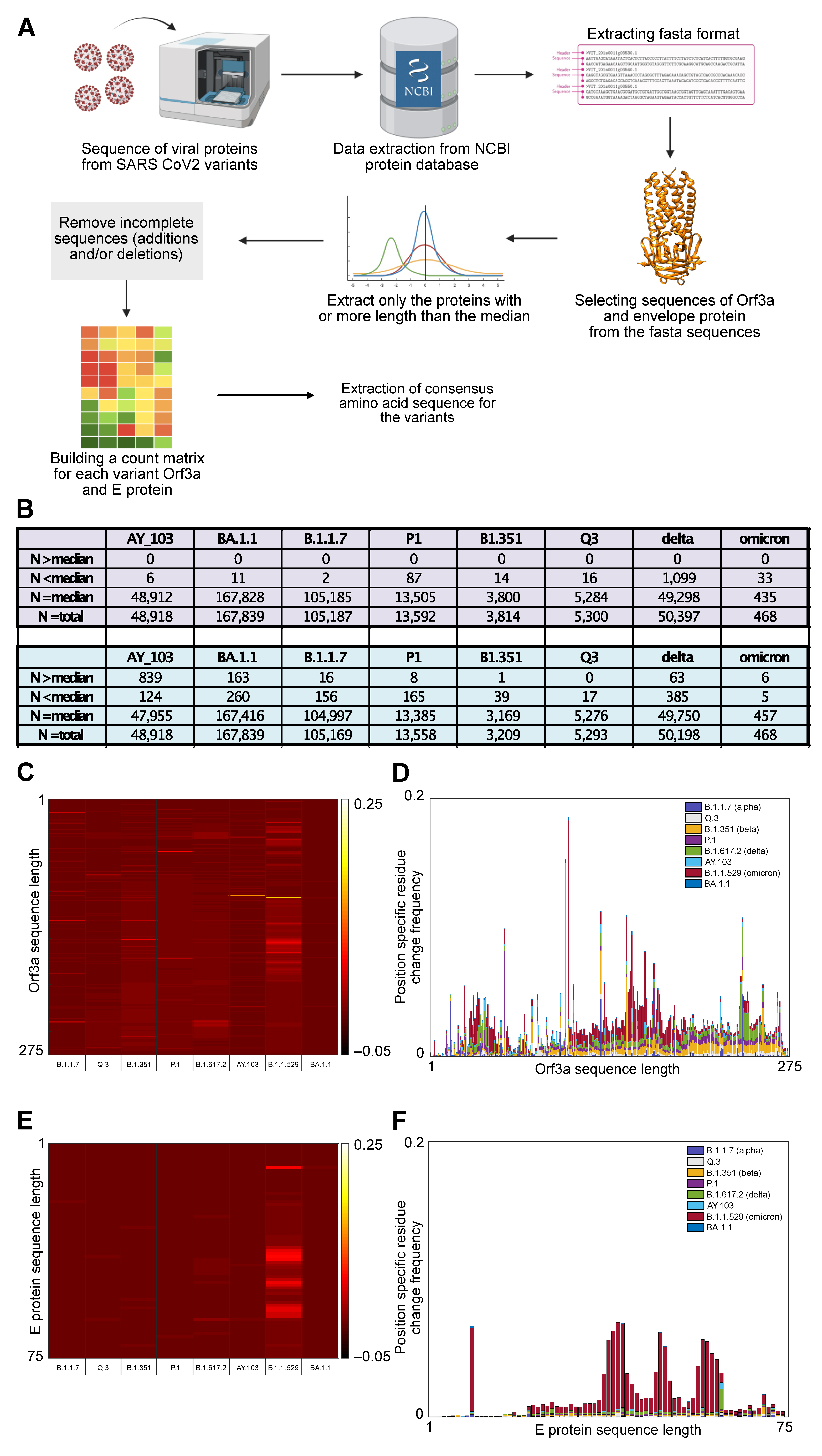
2.2. Position-Specific Amino Acid Residue Frequency Calculation
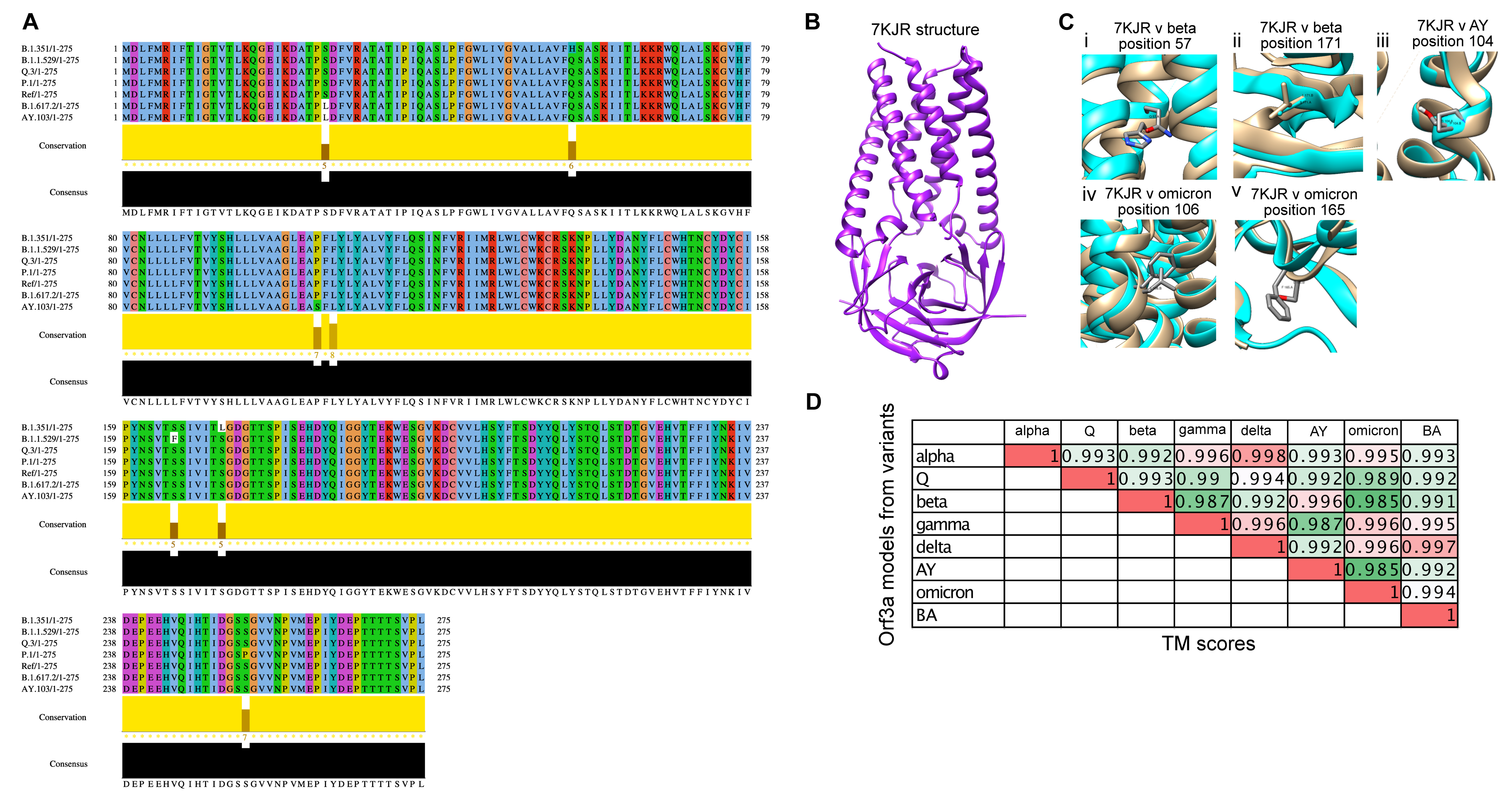
2.3. Multiple Sequence Alignment
2.4. Homology Modelling of the Orf3a Protein and E Protein
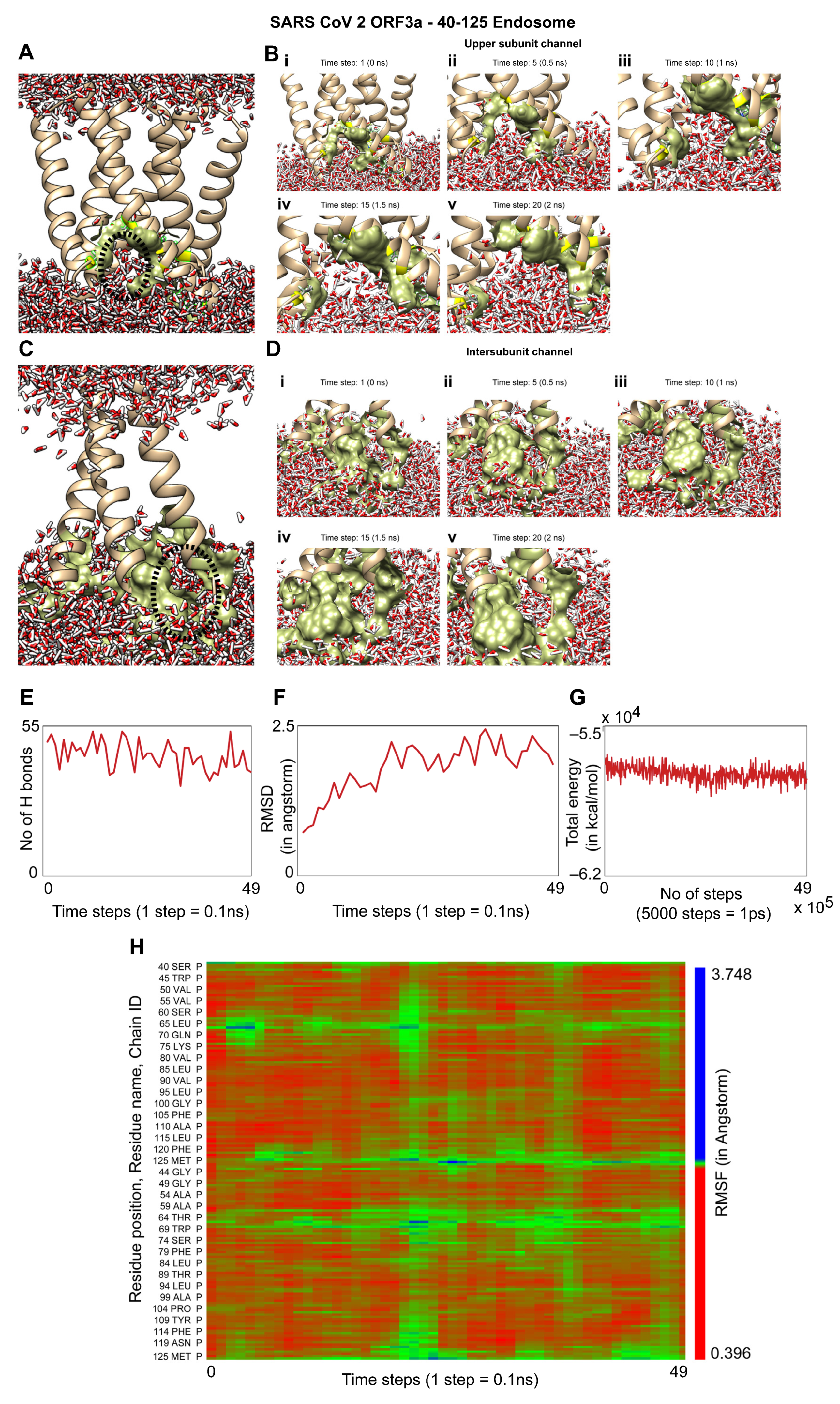
2.5. Generation of the Protein–Membrane System
2.6. Molecular Dynamics Simulations Using NAMD and VMD
2.7. Effect of Point Mutations Using ProtParam and SDM2
2.8. Statistical Analysis
3. Results
3.1. Upregulated Genes in COVID Condition Reflect Cellular Processes Impacted by Ion Channelling Activity
3.2. Intra-Variant Sequence Analysis of SARS-CoV-2 Viroporins, Orf3a and Envelope (E) Protein
3.3. Structural Modelling and Analysis of Representative Mutations in Orf3a across SARS-CoV-2 Variants
3.4. Molecular Dynamics Simulation of the TM Region of Orf3a (40-125) from SARS-CoV-2 in Endosomal Membrane Mimicking System
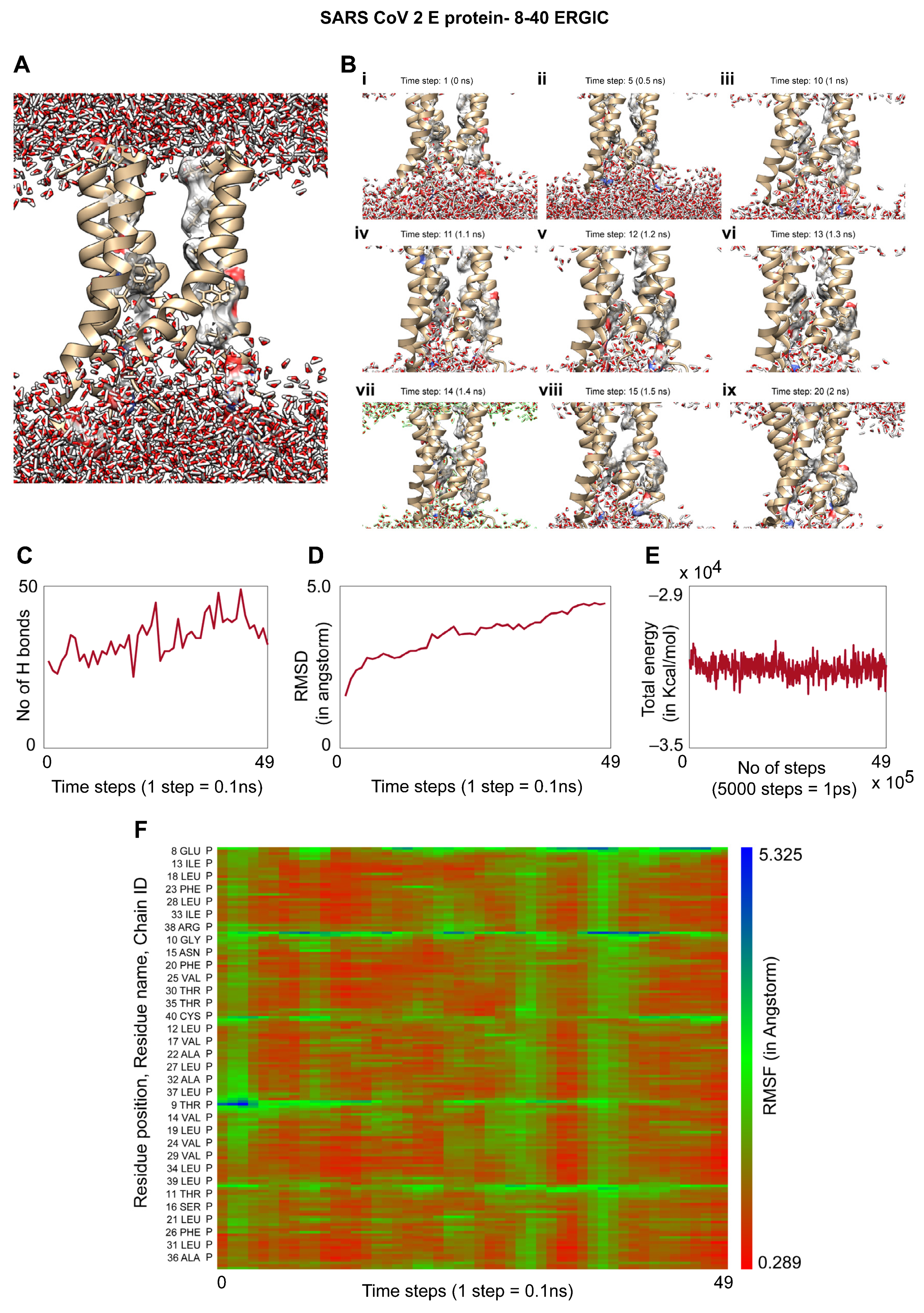
3.5. Molecular Dynamics of the TM Region of SARS-CoV-2 E Protein in an ERGIC Membrane Mimic Shows Water Movement
3.6. Effects of Mutations on Different Regions of the E Protein and Orf3a
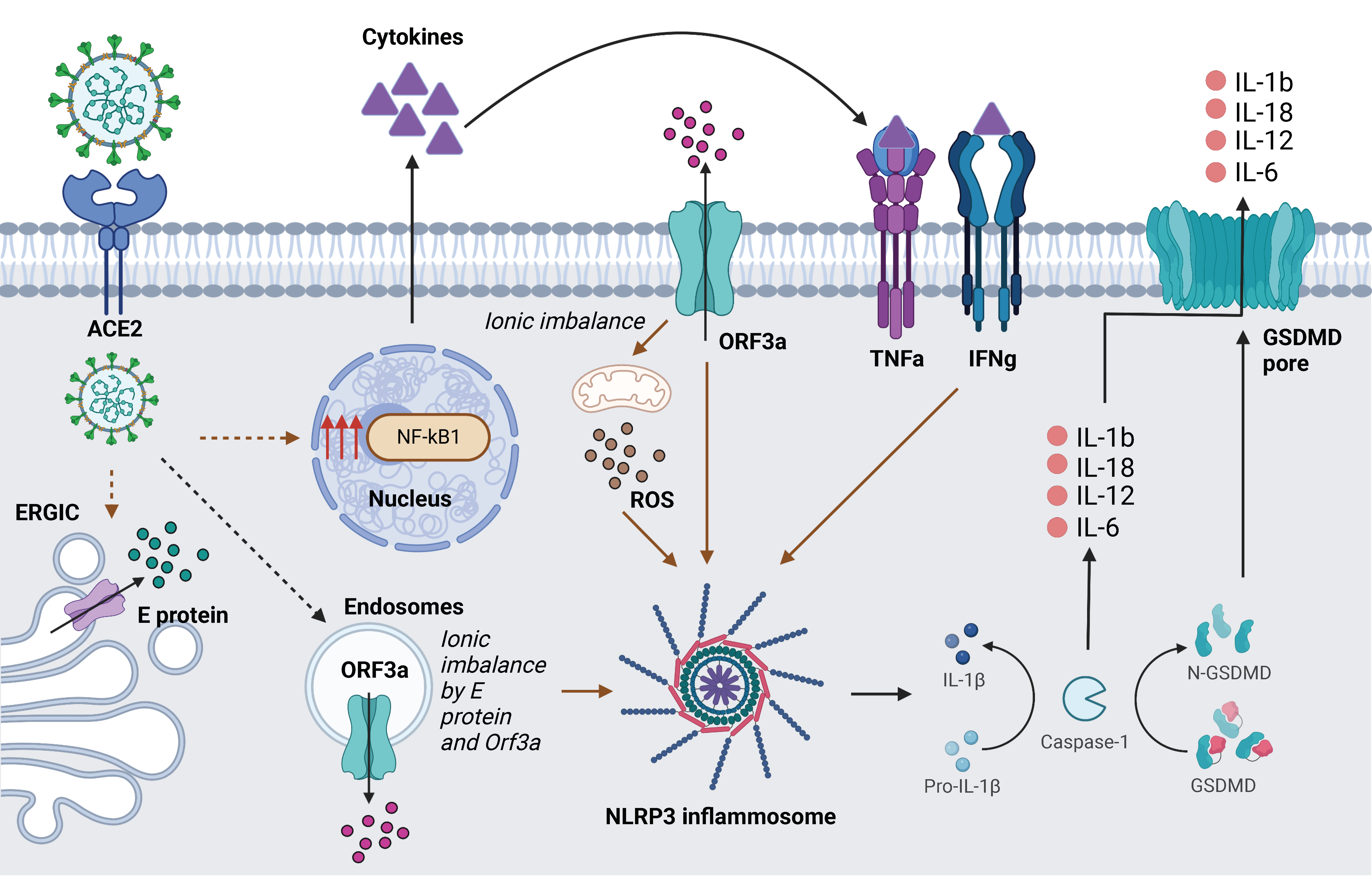
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Ballie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: An interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Haas, E.J.; Angulo, F.J.; McLaughlin, J.M.; Anis, E.; Singer, S.R.; Khan, F.; Brooks, N.; Smaja, M.; Mircus, G.; Pan, K.; et al. Impact and effectiveness of mRNA BNT162b2 vaccine against SARS-CoV-2 infections and COVID-19 cases, hospitalisations, and deaths following a nationwide vaccination campaign in Israel: An observational study using national surveillance data. Lancet 2021, 397, 1819–1829. [Google Scholar] [CrossRef]
- Sadoff, J.; Gray, G.; Vandebosch, A.; Cárdenas, V.; Shukarev, G.; Grinsztejn, B.; Goepfert, P.A.; Truyers, C.; Fennema, H.; Spiessens, B.; et al. Safety and Efficacy of Single-Dose Ad26.COV2.S Vaccine against COVID-19. N. Engl. J. Med. 2021, 384, 2187–2201. [Google Scholar] [CrossRef]
- Xia, S.; Duan, K.; Zhang, Y.; Zhao, D.; Zhang, H.; Xie, Z.; Li, X.; Peng, C.; Zhang, Y.; Zhang, W.; et al. Effect of an Inactivated Vaccine Against SARS-CoV-2 on Safety and Immunogenicity Outcomes: Interim Analysis of 2 Randomized Clinical Trials. JAMA 2020, 324, 951. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zeng, G.; Pan, H.; Li, C.; Hu, Y.; Chu, K.; Han, W.; Chen, Z.; Tang, R.; Yin, W.; et al. Safety, tolerability, and immunogenicity of an inactivated SARS-CoV-2 vaccine in healthy adults aged 18–59 years: A randomised, double-blind, placebo-controlled, phase 1/2 clinical trial. Lancet Infect. Dis. 2021, 21, 181–192. [Google Scholar] [CrossRef]
- Asghari, A.; Naseri, M.; Safari, H.; Saboory, E.; Parsamanesh, N. The Novel Insight of SARS-CoV-2 Molecular Biology and Pathogenesis and Therapeutic Options. DNA Cell Biol. 2020, 39, 1741–1753. [Google Scholar] [CrossRef] [PubMed]
- Cevik, M.; Kuppalli, K.; Kindrachuk, J.; Peiris, M. Virology, transmission, and pathogenesis of SARS-CoV-2. BMJ 2020, 371, m3862. [Google Scholar] [CrossRef]
- Redondo, N.; Zaldívar-López, S.; Garrido, J.J.; Montoya, M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front Immunol. 2021, 12, 708264. [Google Scholar] [CrossRef] [PubMed]
- Silvas, J.A.; Vasquez, D.M.; Park, J.G.; Chiem, K.; Allué-Guardia, A.; Garcia-Vilanova, A.; Platt, R.N.; Miorin, L.; Kehrer, T.; Cupic, A.; et al. Contribution of SARS-CoV-2 Accessory Proteins to Viral Pathogenicity in K18 Human ACE2 Transgenic Mice. J. Virol. 2021, 95, e0040221. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ. 2020, 98, 495–504. [Google Scholar] [CrossRef]
- Sarkar, M.; Saha, S. Structural insight into the role of novel SARS-CoV-2 E protein: A potential target for vaccine development and other therapeutic strategies. PLoS ONE 2020, 15, e0237300. [Google Scholar] [CrossRef]
- Guruprasad, L. Human SARS CoV-2 spike protein mutations. Proteins 2021, 89, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Sasi, S.; Pillai, S.G.; Nag, A.; Shukla, D.; Singhal, R.; Phalke, S.; Velu, G.S.K. SARS-CoV-2 Mutations and Their Impact on Diagnostics, Therapeutics and Vaccines. Front. Med. 2022, 9, 815389. [Google Scholar] [CrossRef]
- Jackson, C.B.; Zhang, L.; Farzan, M.; Choe, H. Functional importance of the D614G mutation in the SARS-CoV-2 spike protein. Biochem. Biophys. Res. Commun. 2021, 538, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xing, N.; Meng, K.; Fu, B.; Xue, W.; Dong, P.; Tang, W.; Xiao, Y.; Liu, G.; Luo, H.; et al. Nucleocapsid mutations R203K/G204R increase the infectivity, fitness, and virulence of SARS-CoV-2. Cell Host Microbe 2021, 29, 1788–1801.e6. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Zhou, Y.; Lokugamage, K.G.; Vu, M.N.; Bopp, N.; Crocquet-Valdes, P.A.; Kalveram, B.; Schindewolf, C.; Liu, Y.; Scharton, D.; et al. Nucleocapsid Mutations in SARS-CoV-2 Augment Replication and Pathogenesis. Microbiology. 2021. Available online: http://biorxiv.org/lookup/doi/10.1101/2021.10.14.464390 (accessed on 3 May 2022).
- Ilmjärv, S.; Abdul, F.; Acosta-Gutiérrez, S.; Estarellas, C.; Galdadas, I.; Casimir, M.; Alessandrini, M.; Gervasio, F.L.; Krause, K.H. Concurrent mutations in RNA-dependent RNA polymerase and spike protein emerged as the epidemiologically most successful SARS-CoV-2 variant. Sci. Rep. 2021, 11, 13705. [Google Scholar] [CrossRef] [PubMed]
- Eskier, D.; Karakülah, G.; Suner, A.; Oktay, Y. RdRp mutations are associated with SARS-CoV-2 genome evolution. PeerJ 2020, 8, e9587. [Google Scholar] [CrossRef] [PubMed]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [PubMed]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.; Verdiá-Báguena, C.; Castaño-Rodriguez, C.; Aguilella, V.; Enjuanes, L. Relevance of Viroporin Ion Channel Activity on Viral Replication and Pathogenesis. Viruses 2015, 7, 3552–3573. [Google Scholar] [CrossRef]
- Li, J.Y.; Liao, C.H.; Wang, Q.; Tan, Y.J.; Luo, R.; Qiu, Y.; Ge, X.Y. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020, 286, 198074. [Google Scholar] [CrossRef] [PubMed]
- Mandala, V.S.; McKay, M.J.; Shcherbakov, A.A.; Dregni, A.J.; Kolocouris, A.; Hong, M. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nat. Struct. Mol. Biol. 2020, 27, 1202–1208. [Google Scholar] [CrossRef]
- Mariano, G.; Farthing, R.J.; Lale-Farjat, S.L.M.; Bergeron, J.R.C. Structural Characterization of SARS-CoV-2: Where We Are, and Where We Need to Be. Front. Mol. Biosci. 2020, 7, 605236. [Google Scholar] [CrossRef] [PubMed]
- Singh Tomar, P.P.; Arkin, I.T. SARS-CoV-2 E protein is a potential ion channel that can be inhibited by Gliclazide and Memantine. Biochem. Biophys. Res. Commun. 2020, 530, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Kern, D.M.; Sorum, B.; Mali, S.S.; Hoel, C.M.; Sridharan, S.; Remis, J.P.; Toso, D.B.; Kotecha, A.; Bautista, D.M.; Brohawn, S.G. Cryo-EM structure of SARS-CoV-2 ORF3a in lipid nanodiscs. Nat. Struct. Mol. Biol. 2021, 28, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Fong, P. SARS-CoV-2: Pushing the E(nvelope). J. Physiol. 2021, 599, 2785–2786. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cruz-cosme, R.; Zhuang, M.W.; Liu, D.; Liu, Y.; Teng, S.; Wang, P.H.; Tang, Q. A systemic and molecular study of subcellular localization of SARS-CoV-2 proteins. Signal Transduct. Target. Ther. 2020, 5, 269. [Google Scholar] [CrossRef] [PubMed]
- Miao, G.; Zhao, H.; Li, Y.; Ji, M.; Chen, Y.; Shi, Y.; Bi, Y.; Wang, P.; Zhang, H. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell 2021, 56, 427–442.e5. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Ng, Y.; Tam, J.; Liu, D. Human Coronaviruses: A Review of Virus–Host Interactions. Diseases 2016, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Garcia, D.; Bekdash, R.; Abbott, G.W.; Yazawa, M.; Harrison, N.L. The envelope protein of SARS-CoV-2 increases intra-Golgi pH and forms a cation channel that is regulated by pH. J. Physiol. 2021, 599, 2851–2868. [Google Scholar] [CrossRef]
- Fan, E.; Beitler, J.R.; Brochard, L.; Calfee, C.S.; Ferguson, N.D.; Slutsky, A.S.; Brodie, D. COVID-19-associated acute respiratory distress syndrome: Is a different approach to management warranted? Lancet Respir. Med. 2020, 8, 816–821. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Akinyemi, I.A.; Chitre, S.A.; Loeb, J.C.; Lednicky, J.A.; McIntosh, M.T.; Bhaduri-McIntosh, S. SARS-CoV-2 viroporin encoded by ORF3a triggers the NLRP3 inflammatory pathway. Virology 2022, 568, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Shah, A. Novel Coronavirus-Induced NLRP3 Inflammasome Activation: A Potential Drug Target in the Treatment of COVID-19. Front. Immunol. 2020, 11, 1021. [Google Scholar] [CrossRef] [PubMed]
- Azkur, A.K.; Akdis, M.; Azkur, D.; Sokolowska, M.; Veen, W.; Brüggen, M.; O’Mahony, L.; Gao, Y.; Nadeau, K.; Akdis, C.A. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy 2020, 75, 1564–1581. [Google Scholar] [CrossRef]
- Shah, V.K.; Firmal, P.; Alam, A.; Ganguly, D.; Chattopadhyay, S. Overview of Immune Response During SARS-CoV-2 Infection: Lessons From the Past. Front. Immunol. 2020, 11, 1949. [Google Scholar] [CrossRef]
- García, L.F. Immune Response, Inflammation, and the Clinical Spectrum of COVID-19. Front. Immunol. 2020, 11, 1441. [Google Scholar] [CrossRef]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Katsura, H.; Sontake, V.; Tata, A.; Kobayashi, Y.; Edwards, C.E.; Heaton, B.E.; Konkimalla, A.; Asakura, T.; Mikami, Y.; Fritch, E.J.; et al. Human Lung Stem Cell-Based Alveolospheres Provide Insights into SARS-CoV-2-Mediated Interferon Responses and Pneumocyte Dysfunction. Cell Stem. Cell 2020, 27, 890–904.e8. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Baxevanis, A.D. Current Protocols in Bioinformatics; John Wiley and Sons: New York, NY, USA, 2004; Available online: http://onlinelibrary.wiley.com/book/10.1002/0471250953 (accessed on 26 September 2021).
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B., III; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Im, W. Automated Builder and Database of Protein/Membrane Complexes for Molecular Dynamics Simulations. PLoS ONE 2007, 2, e880. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.R.; Pan, H.; Wu, D.; Nekouzadeh, A.; Decker, K.F.; Cui, J.; Baker, N.A.; Sept, D.; Rudy, Y. A multiscale model linking ion-channel molecular dynamics and electrostatics to the cardiac action potential. Proc. Natl. Acad. Sci. USA 2009, 106, 11102–11106. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Pandurangan, A.P.; Ochoa-Montaño, B.; Ascher, D.B.; Blundell, T.L. SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017, 45, W229–W235. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Föller, M. Regulation of ion channels and transporters by AMP-activated kinase (AMPK). Channels 2014, 8, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, M.; Amri Maleh, P.; Bagherzadeh, M.; Moulana, Z.; Sepidarkish, M.; Rezanejad, M.; Mirzakhani, M.; Ebrahimpour, S.; Ghorbani, H.; Ahmadnia, Z.; et al. Linkage of Lambda Interferons in Protection Against Severe COVID-19. J. Interferon Cytokine Res. 2021, 41, 149–152. [Google Scholar] [CrossRef]
- Hariharan, A.; Hakeem, A.R.; Radhakrishnan, S.; Reddy, M.S.; Rela, M. The Role and Therapeutic Potential of NF-kappa-B Pathway in Severe COVID-19 Patients. Inflammopharmacology 2021, 29, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Wilfong, E.M.; Croze, R.; Fang, X.; Schwede, M.; Niemi, E.; López, G.Y.; Lee, J.W.; Nakamura, M.C.; Matthay, M.A. Proinflammatory cytokines and ARDS pulmonary edema fluid induce CD40 on human mesenchymal stromal cells—A potential mechanism for immune modulation. PLoS ONE 2020, 15, e0240319. [Google Scholar]
- Notz, Q.; Schmalzing, M.; Wedekink, F.; Schlesinger, T.; Gernert, M.; Herrmann, J.; Sorger, L.; Weismann, D.; Schmid, B.; Sitter, M.; et al. Pro- and Anti-Inflammatory Responses in Severe COVID-19-Induced Acute Respiratory Distress Syndrome—An Observational Pilot Study. Front. Immunol. 2020, 11, 581338. [Google Scholar] [CrossRef]
- Lin, S.; Wu, H.; Wang, C.; Xiao, Z.; Xu, F. Regulatory T Cells and Acute Lung Injury: Cytokines, Uncontrolled Inflammation, and Therapeutic Implications. Front. Immunol. 2018, 9, 1545. [Google Scholar] [CrossRef]
- Kanzawa, N.; Nishigaki, K.; Hayashi, T.; Ishii, Y.; Furukawa, S.; Niiro, A.; Yasui, F.; Kohara, M.; Morita, K.; Matsushima, K.; et al. Augmentation of chemokine production by severe acute respiratory syndrome coronavirus 3a/X1 and 7a/X4 proteins through NF-κB activation. FEBS Lett. 2006, 580, 6807–6812. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeño, J.M.; Fernandez-Delgado, R.; Fett, C.; Castaño-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Andersen, M.N.; Rasmussen, H.B. AMPK: A regulator of ion channels. Commun. Integr. Biol. 2012, 5, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Barrese, V.; Stott, J.B.; Figueiredo, H.B.; Aubdool, A.A.; Hobbs, A.J.; Jepps, T.A.; McNeish, A.J.; Greenwood, I.A. Angiotensin II Promotes K V 7.4 Channels Degradation Through Reduced Interaction With HSP90 (Heat Shock Protein 90). Hypertension 2018, 71, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef]
- Gonzalez, M.E.; Carrasco, L. Viroporins. FEBS Lett. 2003, 552, 28–34. [Google Scholar] [CrossRef]
- McClenaghan, C.; Hanson, A.; Lee, S.J.; Nichols, C.G. Coronavirus Proteins as Ion Channels: Current and Potential Research. Front. Immunol. 2020, 11, 573339. [Google Scholar] [CrossRef]
- Rahbar, M.R.; Jahangiri, A.; Khalili, S.; Zarei, M.; Mehrabani-Zeinabad, K.; Khalesi, B.; Pourzardosht, N.; Hessami, A.; Nezafat, N.; Sadraei, S.; et al. Hotspots for mutations in the SARS-CoV-2 spike glycoprotein: A correspondence analysis. Sci. Rep. 2021, 11, 23622. [Google Scholar] [CrossRef] [PubMed]
- Schrörs, B.; Riesgo-Ferreiro, P.; Sorn, P.; Gudimella, R.; Bukur, T.; Rösler, T.; Löwer, M.; Sahin, U. Large-scale analysis of SARS-CoV-2 spike-glycoprotein mutants demonstrates the need for continuous screening of virus isolates. PLoS ONE 2021, 16, e0249254. [Google Scholar]
- Manzoor, R.; Igarashi, M.; Takada, A. Influenza A Virus M2 Protein: Roles from Ingress to Egress. Ijms 2017, 18, 2649. [Google Scholar] [CrossRef]
- Jones, S. Death by viroporin. Nat. Rev. Microbiol. 2007, 5, 907. [Google Scholar] [CrossRef]
- Zamzami, N.; Kroemer, G. The mitochondrion in apoptosis: How Pandora’s box opens. Nat. Rev. Mol. Cell Biol. 2001, 2, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Farag, N.S.; Breitinger, U.; El-Azizi, M.; Breitinger, H.G. The p7 viroporin of the hepatitis C virus contributes to liver inflammation by stimulating production of Interleukin-1β. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewski, R.; Matalon, S.; Collawn, J.F. Ion channels of the lung and their role in disease pathogenesis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L859–L872. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, X. Acute respiratory failure in COVID-19: Is it “typical” ARDS? Crit. Care 2020, 24, 198. [Google Scholar] [CrossRef] [PubMed]
- Torres Acosta, M.A.; Singer, B.D. Pathogenesis of COVID-19-induced ARDS: Implications for an ageing population. Eur. Respir. J. 2020, 56, 2002049. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Orf3a | E protein | |
|---|---|---|
| Lipids | Intracellular compartments | |
| Endosome | ERGIC | |
| POPC | 120 | 94 |
| POPE | 44 | 42 |
| PSM | 60 | Not present |
| Cholesterol | 120 | 28 |
| bGMP | 28 | Not present |
| WT | 8.57 | 0 | 1.128 |
| Channel forming | |||
| E8A | 8.99 | 0 | 1.199 |
| N15A | 8.57 | 1.74 | 1.199 |
| F26A | 8.57 | 0.97 | 1.115 |
| Membrane anchoring part | |||
| L37A | 8.57 | −0.36 | 1.101 |
| R38A | 7.69 | 0 | 1.212 |
| WT | 5.55 | 0 | 0.275 |
| Initial neck constriction | |||
| F43A | 5.55 | −0.02 | 0.271 |
| L46A | 5.55 | 0.15 | 0.267 |
| I47A | 5.55 | 0.5 | 0.265 |
| V50A | 5.55 | 0.84 | 0.266 |
| Q57A | 5.55 | 0.39 | 0.294 |
| S58A | 5.55 | 1.49 | 0.284 |
| C81A | 5.55 | −0.78 | 0.272 |
| N82A | 5.55 | 1.49 | 0.294 |
| L85A | 5.55 | 0.15 | 0.267 |
| L95A | 5.55 | −2.68 | 0.267 |
| L96A | 5.55 | 0.15 | 0.267 |
| F105A | 5.55 | −0.02 | 0.271 |
| Y109A | 5.55 | −1.07 | 0.286 |
| Q116A | 5.55 | 0.39 | 0.294 |
| Upper tunnel | |||
| K61A | 5.4 | 0.11 | 0.295 |
| K75A | 5.4 | 0.31 | 0.295 |
| H78A | 5.43 | 0.61 | 0.293 |
| F79A | 5.55 | 0.97 | 0.271 |
| I123A | 5.55 | 0.5 | 0.265 |
| L127A | 5.55 | 0.15 | 0.267 |
| L139A | 5.55 | 0.15 | 0.267 |
| D142A | 5.71 | −0.21 | 0.294 |
| Lower tunnel | |||
| L65A | 5.55 | −0.4 | 0.267 |
| K66A | 5.4 | 0.18 | 0.295 |
| L71A | 5.55 | −3.22 | 0.267 |
| Y141A | 5.55 | −0.51 | 0.286 |
| N144A | 5.55 | 0.03 | 0.294 |
| N161A | 5.55 | 0.03 | 0.294 |
| Y189A | 5.55 | −0.94 | 0.286 |
| Lipid interacting residues | |||
| I62A | 5.55 | −0.08 | 0.265 |
| T64A | 5.55 | 0.6 | 0.284 |
| K66A | 5.4 | 0.18 | 0.295 |
| I118A | 5.55 | 0.5 | 0.265 |
| R122A | 5.4 | 0.41 | 0.297 |
| R126A | 5.4 | −1.71 | 0.297 |
| D142A | 5.71 | −0.21 | 0.294 |
| N144A | 5.55 | 0.03 | 0.294 |
| F207A | 5.55 | −0.02 | 0.271 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarkar, M.; Etheimer, P.; Hannothiaux, V.; Saha, S. SARS-CoV-2 Viroporins: A Multi-Omics Insight from Nucleotides to Amino Acids. Appl. Microbiol. 2022, 2, 572-593. https://doi.org/10.3390/applmicrobiol2030045
Sarkar M, Etheimer P, Hannothiaux V, Saha S. SARS-CoV-2 Viroporins: A Multi-Omics Insight from Nucleotides to Amino Acids. Applied Microbiology. 2022; 2(3):572-593. https://doi.org/10.3390/applmicrobiol2030045
Chicago/Turabian StyleSarkar, Manish, Paul Etheimer, Victor Hannothiaux, and Soham Saha. 2022. "SARS-CoV-2 Viroporins: A Multi-Omics Insight from Nucleotides to Amino Acids" Applied Microbiology 2, no. 3: 572-593. https://doi.org/10.3390/applmicrobiol2030045
APA StyleSarkar, M., Etheimer, P., Hannothiaux, V., & Saha, S. (2022). SARS-CoV-2 Viroporins: A Multi-Omics Insight from Nucleotides to Amino Acids. Applied Microbiology, 2(3), 572-593. https://doi.org/10.3390/applmicrobiol2030045