
Translation Enhancement by a Short Nucleotide Insertion at 5′UTR: Application to an In Vitro Cell-Free System and a Photosynthetic Bacterium

Abstract
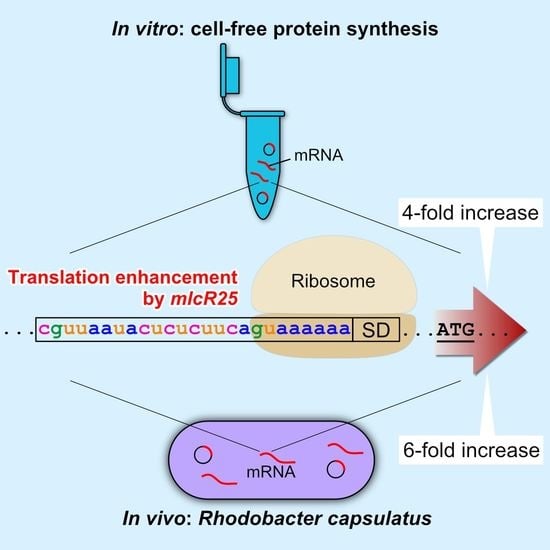
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids
2.3. In Vitro Transcription
2.4. In Vitro Translation
2.5. Western Blotting
2.6. RNA Purification and Real-Time PCR
2.7. β-Galactosidase Assay
2.8. Statistical Analysis
3. Results
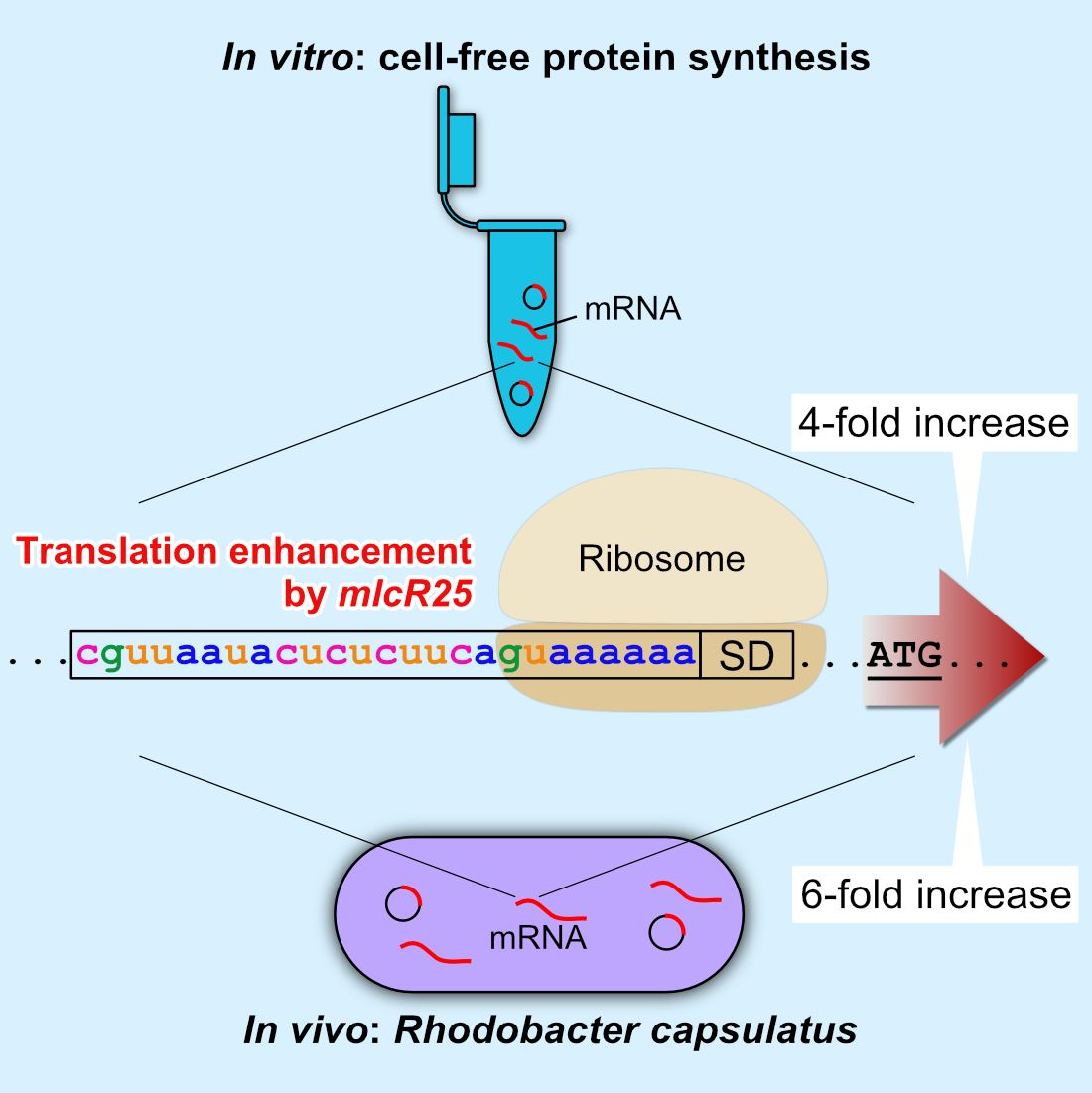
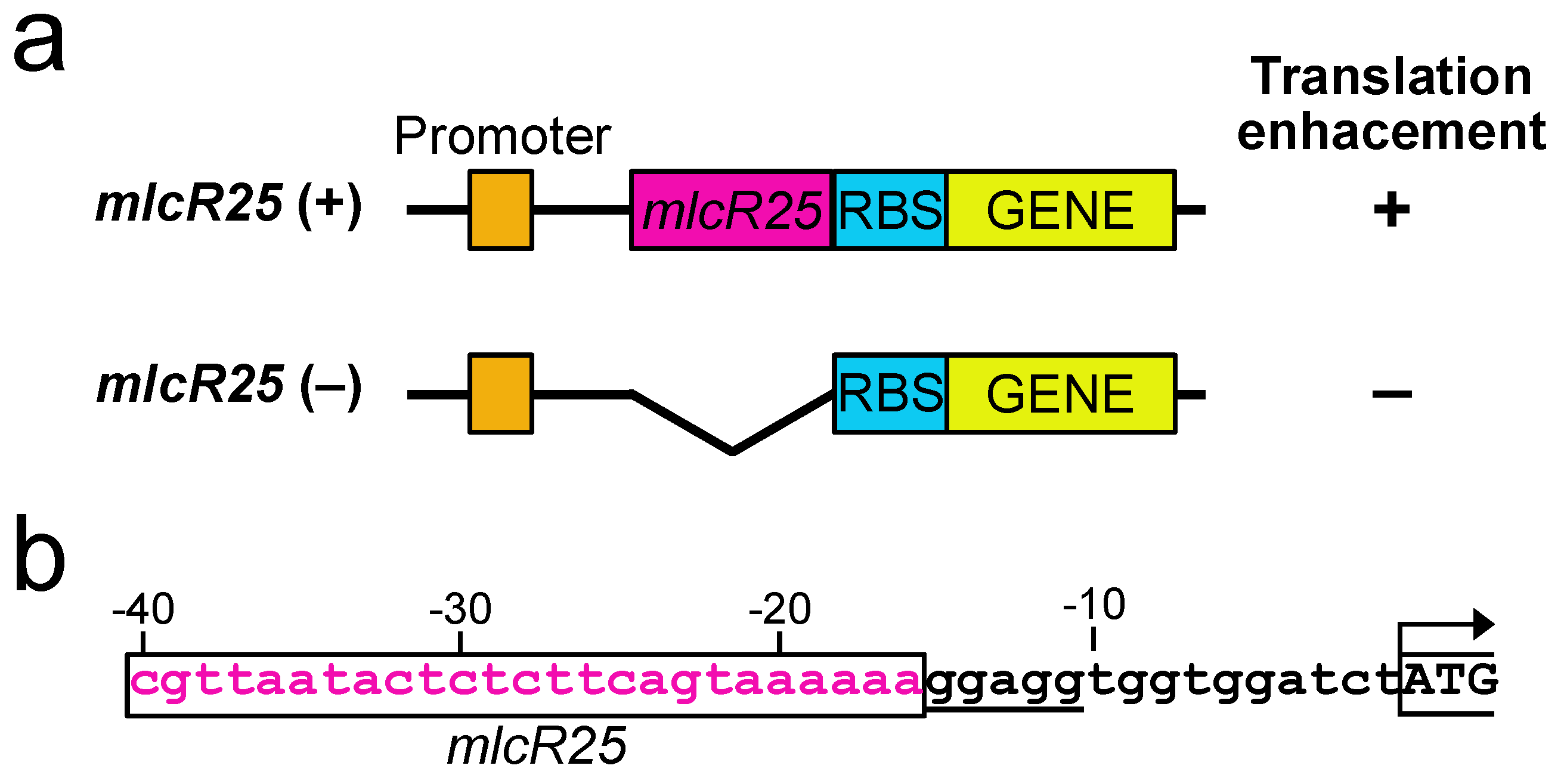
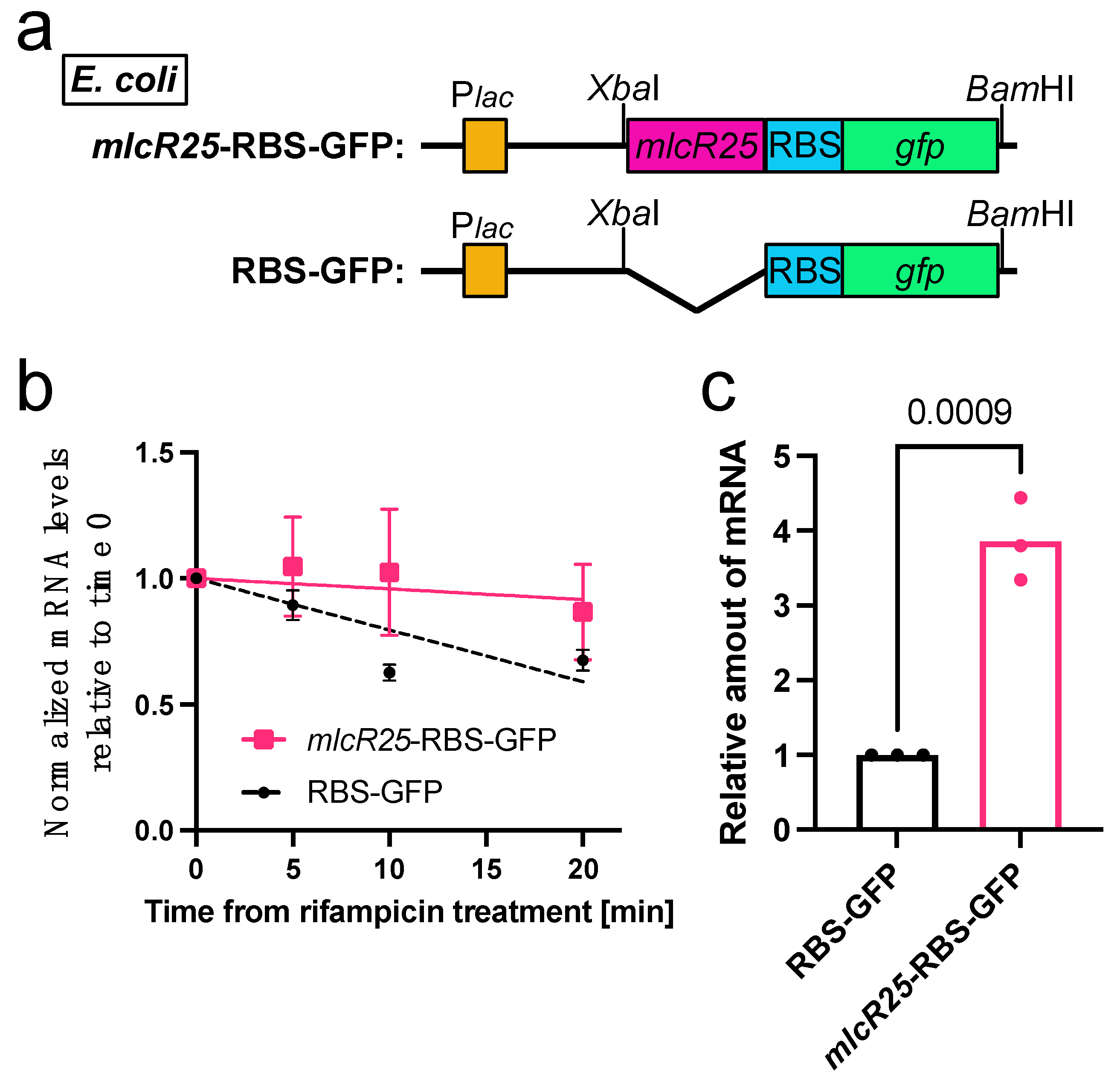
3.1. Effect of mlcR25 on mRNA Longevity in E. coli
3.2. mlcR25 Confers Enhanced Translational Efficiency on mRNA
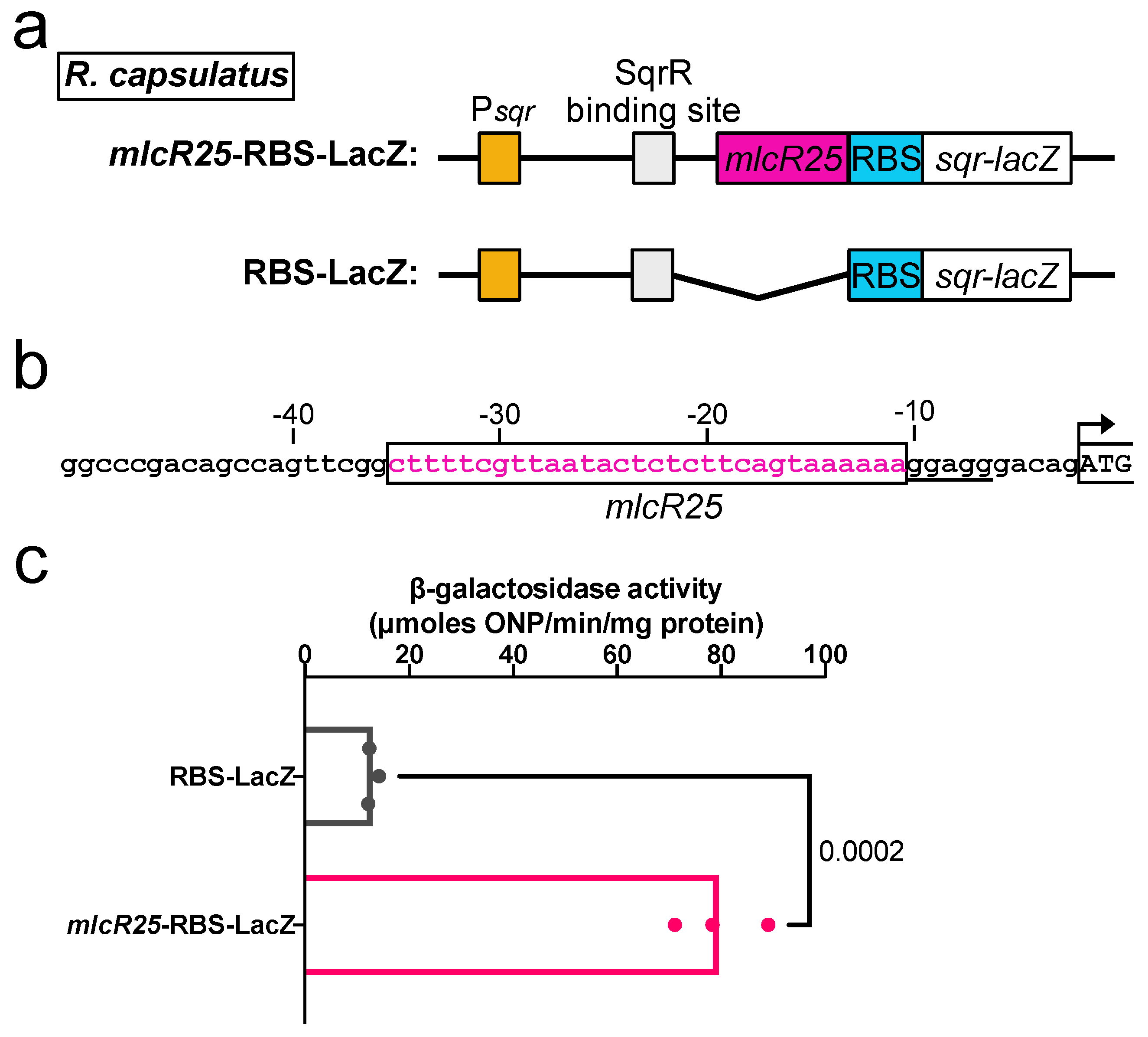
3.3. Application to a Photosynthetic Bacterium
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shine, J.; Dalgarno, L. The 3’ Terminal Sequence of Escherichia coli 16S Ribosomal RNA: Complementarity to Nonsense Triplets and Ribosome Binding Sites. Proc. Natl. Acad. Sci. USA 1974, 71, 1342–1346. [Google Scholar] [CrossRef]
- Boël, G.; Letso, R.; Neely, H.; Price, W.N.; Wong, K.H.; Su, M.; Luff, J.D.; Valecha, M.; Everett, J.K.; Acton, T.B.; et al. Codon Influence on Protein Expression in E. coli Correlates with mRNA Levels. Nature 2016, 529, 358–363. [Google Scholar] [CrossRef] [Green Version]
- Belasco, J.G. mRNA Degradation in Prokaryotic Cells: An Overview; Woodhead Publishing Limited: Sawston, UK, 1993. [Google Scholar]
- Carrier, T.A.; Keasling, J.D. Controlling Messenger RNA Stability in Bacteria: Strategies for Engineering Gene Expression. Biotechnol. Prog. 1997, 13, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Carrier, T.A.; Keasling, J.D. Engineering mRNA Stability in E. coli by the Addition of Synthetic Hairpins Using a 5′ Cassette System. Biotechnol. Bioeng. 1997, 55, 577–580. [Google Scholar] [CrossRef]
- Carrier, T.A.; Keasling, J.D. Library of Synthetic 5’ Secondary Structures To Manipulate mRNA Stability in Escherichia coli. Biotechnol. Prog. 1999, 15, 58–64. [Google Scholar] [CrossRef]
- Radhakrishnan, A.; Green, R. Connections Underlying Translation and mRNA Stability. J. Mol. Biol. 2016, 428, 3558–3564. [Google Scholar] [CrossRef] [PubMed]
- Grunberg-Manago, M. Messenger RNA Stability and Its Role in Control of Gene Expression in Bacteria and Phages. Annu. Rev. Genet. 1999, 33, 193–227. [Google Scholar] [CrossRef]
- Kudla, G.; Murray, A.W.; Tollervey, D.; Plotkin, J.B. Coding-Sequence Determinants of Gene Expression in Escherichia coli. Science 2009, 324, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Plotkin, J.B.; Kudla, G. Synonymous but Not the Same: The Causes and Consequences of Codon Bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Hanson, G.; Coller, J. Translation and Protein Quality Control: Codon Optimality, Bias and Usage in Translation and mRNA Decay. Nat. Rev. Mol. Cell. Biol. 2018, 19, 20–30. [Google Scholar] [CrossRef]
- Kondo, T.; Yumura, S. Strategies for Enhancing Gene Expression in Escherichia coli. Appl. Microbiol. Biotechnol. 2020, 104, 3825–3834. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Yumura, S. Translation Enhancement by a Dictyostelium Gene Sequence in Escherichia coli. Appl. Microbiol. Biotechnol. 2019, 103, 3501–3510. [Google Scholar] [CrossRef] [PubMed]
- Olins, P.O.; Rangwala, S.H. A Novel Sequence Element Derived from Bacteriophage T7 mRNA Acts as an Enhancer of Translation of the LacZ Gene in Escherichia coli. J. Biol. Chem. 1989, 264, 16973–16976. [Google Scholar] [CrossRef]
- Jang, S.H.; Cha, J.W.; Han, N.S.; Jeong, K.J. Development of Bicistronic Expression System for the Enhanced and Reliable Production of Recombinant Proteins in Leuconostoc Citreum. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Gao, X.; Zhao, Z.; Li, A.; Wang, Y.; Yang, Y.; Liu, X.; Bai, Z. Enhanced Production of Recombinant Proteins in Corynebacterium Glutamicum by Constructing a Bicistronic Gene Expression System. Microb. Cell. Fact. 2020, 19, 113. [Google Scholar] [CrossRef]
- Li, D.; Fu, G.; Tu, R.; Jin, Z.; Zhang, D. High-Efficiency Expression and Secretion of Human FGF21 in Bacillus subtilis by Intercalation of a Mini-Cistron Cassette and Combinatorial Optimization of Cell Regulatory Components. Microb. Cell. Fact. 2019, 18, 17. [Google Scholar] [CrossRef] [Green Version]
- Schoner, B.E.; Belagaje, R.M.; Schoner, R.G. Translation of a Synthetic Two-Cistron mRNA in Escherichia coli. Proc. Natl. Acad. Sci. USA 1986, 83, 8506–8510. [Google Scholar] [CrossRef]
- Mutalik, V.K.; Guimaraes, J.C.; Cambray, G.; Lam, C.; Christoffersen, M.J.; Mai, Q.-A.; Tran, A.B.; Paull, M.; Keasling, J.D.; Arkin, A.P.; et al. Precise and Reliable Gene Expression via Standard Transcription and Translation Initiation Elements. Nat. Methods 2013, 10, 354–360. [Google Scholar] [CrossRef]
- Ishida, H.; Hata, Y.; Kawato, A.; Abe, Y. Improvement of the GlaB Promoter Expressed in Solid-State Fermentation (SSF) of Aspergillus Oryzae. Biosci. Biotechnol. Biochem. 2006, 70, 1181–1187. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Wu, Y.; Zheng, Z.; Chen, N.; Luo, X.; Tang, H.; Keasling, J.D. A Synthetic Promoter System for Well-Controlled Protein Expression with Different Carbon Sources in Saccharomyces cerevisiae. Microb. Cell. Fact. 2021, 20, 202. [Google Scholar] [CrossRef]
- Shimizu, Y.; Inoue, A.; Tomari, Y.; Suzuki, T.; Yokogawa, T.; Nishikawa, K.; Ueda, T. Cell-Free Translation Reconstituted with Purified Components. Nat. Biotechnol. 2001, 19, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Yumura, S. An Improved Molecular Tool for Screening Bacterial Colonies Using GFP Expression Enhanced by a Dictyostelium Sequence. Biotechniques 2020, 68, 91–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Inoue, K.; Sakurai, H.; Nagashima, K.V.P. Effects of the Deletion of Hup Genes Encoding the Uptake Hydrogenase on the Activity of Hydrogen Production in the Purple Photosynthetic Bacterium Rubrivivax gelatinosus IL144. J. Gen. Appl. Microbiol. 2017, 63, 274–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, P.F.; Wall, J.D.; Gest, H. Characterization of Rhodopseudomonas capsulata. Arch Microbiol. 1975, 105, 207–216. [Google Scholar] [CrossRef]
- Minton, N.P. Improved Plasmid Vectors for the Isolation of Translational Lac Gene Fusions. Gene 1984, 31, 269–273. [Google Scholar] [CrossRef]
- Fellay, R.; Frey, J.; Krisch, H. Interposon Mutagenesis of Soil and Water Bacteria: A Family of DNA Fragments Designed for in Vitro Insertional Mutagenesis of Gram-Negative Bacteria. Gene 1987, 52, 147–154. [Google Scholar] [CrossRef]
- Young, D.A.; Bauer, C.E.; Williams, J.C.; Marrs, B.L. Gentic Evidence for Superoperonal Organization of Genes for Photosynthesis Pigments and Pigment-Binding Proteins in Rhodobacter capsulatus. Mol. Gen. Genet. 1989, 218, 1–12. [Google Scholar] [CrossRef]
- Sganga, M.W.; Bauer, C.E. Regulatory Factors Controlling Photosynthetic Reaction Center and Light-Harvesting Gene Expression in Rhodobacter capsulatus. Cell 1992, 68, 945–954. [Google Scholar] [CrossRef]
- Shimizu, T.; Shen, J.; Fang, M.; Zhang, Y.; Hori, K.; Trinidad, J.C.; Bauer, C.E.; Giedroc, D.P.; Masuda, S. Sulfide-Responsive Transcriptional Repressor SqrR Functions as a Master Regulator of Sulfide-Dependent Photosynthesis. Proc. Natl. Acad. Sci. USA 2017, 114, 2355–2360. [Google Scholar] [CrossRef]
- Shimizu, Y.; Kanamori, T.; Ueda, T. Protein Synthesis by Pure Translation Systems. Methods 2005, 36, 299–304. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, K.; Zhou, L.; Lim, Q.; Zou, R.; Stephanopoulos, G.; Too, H.P. Novel Reference Genes for Quantifying Transcriptional Responses of Escherichia coli to Protein Overexpression by Quantitative PCR. BMC Mol. Biol. 2011, 12, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komarova, A.V.; Tchufistova, L.S.; Dreyfus, M.; Boni, I.V. AU-Rich Sequences within 5’ Untranslated Leaders Enhance Translation and Stabilize mRNA in Escherichia coli. J. Bacteriol. 2005, 187, 1344–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umezawa, H.; Mizuno, S.; Yamazaki, H.; Nitta, K. Inhibition of DNA-Dependent RNA Synthesis by Rifamycins. J. Antibiot. (Tokyo) 1968, 21, 234–236. [Google Scholar] [CrossRef] [Green Version]
- Kanamori, T.; Fujino, Y.; Ueda, T. PURE Ribosome Display and Its Application in Antibody Technology. Biochim. Biophys. Acta. Proteins Proteom. 2014, 1844, 1925–1932. [Google Scholar] [CrossRef]
- Endo, Y.; Takai, K.; Ueda, T. Cell-Free Protein Production; Endo, Y., Takai, K., Ueda, T., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2010; Volume 607, ISBN 978-1-60327-330-5. [Google Scholar]
- Nagumo, Y.; Fujiwara, K.; Horisawa, K.; Yanagawa, H.; Doi, N. PURE mRNA Display for in Vitro Selection of Single-Chain Antibodies. J. Biochem. 2016, 159, 519–526. [Google Scholar] [CrossRef]
- Katzen, F.; Fletcher, J.E.; Yang, J.P.; Kang, D.; Peterson, T.C.; Cappuccio, J.A.; Blanchette, C.D.; Sulchek, T.; Chromy, B.A.; Hoeprich, P.D.; et al. Insertion of Membrane Proteins into Discoidal Membranes Using a Cell-Free Protein Expression Approach. J. Proteome Res. 2008, 7, 3535–3542. [Google Scholar] [CrossRef]
- Forster, A.C.; Church, G.M. Towards Synthesis of a Minimal Cell. Mol. Syst. Biol. 2006, 2. [Google Scholar] [CrossRef] [Green Version]
- Matsubayashi, H.; Ueda, T. Purified Cell-Free Systems as Standard Parts for Synthetic Biology. Curr. Opin. Chem. Biol. 2014, 22, 158–162. [Google Scholar] [CrossRef]
- Niwa, T.; Kanamori, T.; Ueda, T.; Taguchi, H. Global Analysis of Chaperone Effects Using a Reconstituted Cell-Free Translation System. Proc. Natl. Acad. Sci. USA 2012, 109, 8937–8942. [Google Scholar] [CrossRef]
- Lavickova, B.; Maerkl, S.J. A Simple, Robust, and Low-Cost Method to Produce the PURE Cell-Free System. ACS Synth. Biol. 2019, 8, 455–462. [Google Scholar] [CrossRef]
- Roy, A.; Shukla, A.K.; Haase, W.; Michel, H. Employing Rhodobacter sphaeroides to Functionally Express and Purify Human G Protein-Coupled Receptors. Biol. Chem. 2008, 389, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Erbakan, M.; Curtis, B.S.; Nixon, B.T.; Kumar, M.; Curtis, W.R. Advancing Rhodobacter sphaeroides as a Platform for Expression of Functional Membrane Proteins. Protein. Expr. Purif. 2015, 115, 109–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laible, P.D.; Scott, H.N.; Henry, L.; Hanson, D.K. Towards Higher-Throughput Membrane Protein Production for Structural Genomics Initiatives. J. Struct. Funct. Genom. 2004, 5, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Kappler, U.; McEwan, A.G. A System for the Heterologous Expression of Complex Redox Proteins in Rhodobacter capsulatus: Characterisation of Recombinant Sulphite:Cytochrome c Oxidoreductase from Starkeya Novella. FEBS Lett. 2002, 529, 208–214. [Google Scholar] [CrossRef] [Green Version]
- Katzke, N.; Arvani, S.; Bergmann, R.; Circolone, F.; Markert, A.; Svensson, V.; Jaeger, K.-E.; Heck, A.; Drepper, T. A Novel T7 RNA Polymerase Dependent Expression System for High-Level Protein Production in the Phototrophic Bacterium Rhodobacter capsulatus. Protein. Expr. Purif. 2010, 69, 137–146. [Google Scholar] [CrossRef]
- Cullen, P.J.; Kaufman, C.K.; Bowman, W.C.; Kranz, R.G. Characterization of the Rhodobacter capsulatus Housekeeping RNA Polymerase. J. Biol. Chem. 1997, 272, 27266–27273. [Google Scholar] [CrossRef] [Green Version]
- Naylor, G.W.; Addlesee, H.A.; Gibson, L.C.D.; Hunter, C.N. The Photosynthesis Gene Cluster of Rhodobacter sphaeroides. Photosynth Res. 1999, 62, 121–139. [Google Scholar] [CrossRef]
- Alberti, M.; Burke, D.H.; Hearst, J.E. Structure and Sequence of the Photosynthesis Gene Cluster. In Anoxygenic Photosynthetic Bacteria; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1995; pp. 1083–1106. [Google Scholar]
- Nakagawa, S.; Niimura, Y.; Miura, K.; Gojobori, T. Dynamic Evolution of Translation Initiation Mechanisms in Prokaryotes. Proc. Natl. Acad. Sci. USA 2010, 107, 6382–6387. [Google Scholar] [CrossRef]
- Friedman, D.I.; Pollara, B.; Gray, E.D. Structural Studies of the Ribosomes of Rhodopseudomonas spheroides. J. Mol. Biol. 1966, 22, 53–66. [Google Scholar] [CrossRef]
- Robinson, A.; Sykes, J. A Comparison of the Unfolding and Dissociation of the Large Ribosome Subunits from Rhodopseudomonas spheroides N.C.I.B. 8253 and Escherichia coli M.R.E. 600. Biochem. J. 1973, 133, 739–747. [Google Scholar] [CrossRef]
- Duval, M.; Korepanov, A.; Fuchsbauer, O.; Fechter, P.; Haller, A.; Fabbretti, A.; Choulier, L.; Micura, R.; Klaholz, B.P.; Romby, P.; et al. Escherichia coli Ribosomal Protein S1 Unfolds Structured mRNAs onto the Ribosome for Active Translation Initiation. PLoS Biol. 2013, 11, e1001731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunel, C.; Romby, P.; Sacerdot, C.; De Smit, M.; Graffe, M.; Dondon, J.; Van Duin, J.; Ehresmann, B.; Ehresmann, C.; Springer, M. Stabilised Secondary Structure at a Ribosomal Binding Site Enhances Translational Repression in E. coli. J. Mol. Biol. 1995, 253, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Emory, S.A.; Bouvet, P.; Belasco, J.G. A 5’-Terminal Stem-Loop Structure Can Stabilize mRNA in Escherichia coli. Genes Dev. 1992, 6, 135–148. [Google Scholar] [CrossRef] [Green Version]
- Arnold, T.E.; Yu, J.; Belasco, J.G. mRNA Stabilization by the OmpA 5’ Untranslated Region: Two Protective Elements Hinder Distinct Pathways for mRNA Degradation. RNA 1998, 4, 319–330. [Google Scholar] [PubMed]
- Chen, L.H.; Emory, S.A.; Bricker, A.L.; Bouvet, P.; Belasco, J.G. Structure and Function of a Bacterial mRNA Stabilizer: Analysis of the 5’ Untranslated Region of OmpA mRNA. J. Bacteriol. 1991, 173, 4578–4586. [Google Scholar] [CrossRef] [Green Version]
- Irastortza-Olaziregi, M.; Amster-Choder, O. Coupled Transcription-Translation in Prokaryotes: An Old Couple With New Surprises. Front. Microbiol. 2020, 11, 624830. [Google Scholar] [CrossRef]
- Petersen, C. Translation and mRNA Stability in Bacteria: A Complex Relationship. In Control of Messenger RNA Stability; Academic Press: Waltham, MA, USA, 1993; pp. 117–145. [Google Scholar]
- Iost, I.; Dreyfus, M. The Stability of Escherichia coli LacZ mRNA Depends upon the Simultaneity of Its Synthesis and Translation. EMBO J. 1995, 14, 3252–3261. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kondo, T.; Shimizu, T. Translation Enhancement by a Short Nucleotide Insertion at 5′UTR: Application to an In Vitro Cell-Free System and a Photosynthetic Bacterium. Appl. Microbiol. 2023, 3, 687-697. https://doi.org/10.3390/applmicrobiol3030047
Kondo T, Shimizu T. Translation Enhancement by a Short Nucleotide Insertion at 5′UTR: Application to an In Vitro Cell-Free System and a Photosynthetic Bacterium. Applied Microbiology. 2023; 3(3):687-697. https://doi.org/10.3390/applmicrobiol3030047
Chicago/Turabian StyleKondo, Tomo, and Takayuki Shimizu. 2023. "Translation Enhancement by a Short Nucleotide Insertion at 5′UTR: Application to an In Vitro Cell-Free System and a Photosynthetic Bacterium" Applied Microbiology 3, no. 3: 687-697. https://doi.org/10.3390/applmicrobiol3030047