1. Introduction
The members of the genus
Mycobacterium are responsible for some of the most difficult-to-diagnose and treat bacterial infections in humans and animals and can be divided into two groups based on growth rate. Rapid growers form visible colonies on solid media often within seven days, while slow growers take longer [
1]. The slow growth rate of clinically relevant mycobacteria makes diagnosing mycobacterial infections via culture a major challenge.
According to the World Health Organization (WHO), an estimated 1.3 million people died in 2022 from tuberculosis (TB), a treatable infectious disease. Between the 2015–2030 period, losses of USD 983 billion are expected if disease control is not improved [
2], with the largest burden placed on low- and middle-income countries (LMICs). TB is caused by the members of the
Mycobacterium tuberculosis complex (MTBC) and there has been a call for new diagnostics to be developed [
3].
Mycobacterium bovis is a member of the MTBC and causes bovine TB (bTB), a zoonotic infection that primarily affects the lungs and is a significant problem to the agricultural industry in the UK and Republic of Ireland. This chronic infectious disease has led to the slaughter of over 25,000 cattle in England in 2021 [
4]. Control efforts cost the taxpayer approximately GBP 70 million and farmers GBP 50 million each year in England [
5].
Johne’s disease (JD) is caused by
Mycobacterium avium subsp.
paratuberculosis (MAP) and is responsible for chronic wasting in infected animals. The disease is endemic in commercial ruminant herds worldwide [
6]. The associated clinical features have been estimated to cost farmers up to GBP 26 per dairy cow per year [
7]; given the tight margins farmers operate within, this is an important economic concern.
Effective diagnostics are the cornerstone of control and, in humans, treatment for these infectious diseases. The welfare and economic importance of TB, bTB and JD underline the need for diagnostics to be affordable, sensitive, specific, user-friendly, rapid and robust, equipment-free and deliverable tools for end-users [
8]. Diagnostics encompassing these criteria are vital to meeting the targets described in the END TB strategy [
9] and the roadmap for zoonotic tuberculosis [
10]. New diagnostics are essential, but it is unfeasible for these to be a “one size fits all” approach. A holistic approach that utilizes current tests and involves all stakeholders, including policy makers, is essential to tackle these diseases.
Tuberculosis, bovine TB and Johne’s disease are all caused by mycobacterial pathogens. The thick and waxy cell wall common to
Mycobacterium makes these bacteria difficult to lyse [
11], preventing access to molecular biomarkers inside the cell such as DNA. This difficulty negatively impacts molecular diagnostic tests such as PCR [
12].
Bacteriophage are viruses that infect bacteria and can have a lytic or lysogenic life cycle. Mycobacteriophage—phage that infect mycobacteria—have been used as molecular tools in the study of mycobacteria, especially their genetics [
13], and have recently been used for therapeutic purposes [
14]. Here, we use mycobacteriophage to lyse mycobacteria and to act as an efficient viable lysis too, bypassing the limitations of traditional lysis methods [
12]. The Actiphage
® assay has been developed to detect pathogenic bacteria from cattle and human blood [
12]. Here, bacteriophage are used as a viable lysing agent and, when coupled with qPCR, can be used to detect viable mycobacteria [
12,
15]. DNA filtration, clean-up and concentration are also required to ensure the sensitive detection of viable cells, which adds to the time, steps and equipment needed to carry out a test and limits the assay to provincial specialists in reference laboratories.
Loop-mediated isothermal amplification (LAMP) is a promising nucleic acid amplification test (NAAT). LAMP can produce up to 10
9 copies of the amplified DNA within an hour and is highly specific. LAMP reactions contain two or three pairs of primers, which recognize up to eight specific locations on the target DNA [
16]. LAMP has been used to detect the signature DNA of many pathogens and can be used simply, without the need for complex equipment, facilitating its application in the field or low-resource settings [
17]. Additionally, LAMP has been reported to be more resistant to inhibition from components in blood compared to conventional qPCR [
18].
Here, we aimed to determine whether LAMP assays could be developed to detect low levels of M. tuberculosis, M. bovis and MAP. Once optimized, we aimed to determine whether LAMP could be used in combination with phage-mediated lysis (termed phage-LAMP) to bring detection closer to care via removing centrifugation, DNA clean and concentrating steps and the need for complex detection equipment.
2. Materials and Methods
2.1. Strains, Cultures and Enumeration of Mycobacteria
Mycobacterial strains used in this study were M. smegmatis (MC2155); M. bovis BCG (Pasteur); Mycobacterium avium subsp. paratuberculosis strain (ATCC 19698); and a MAP goat strain (clinical isolate). Mycobacteria were maintained in liquid culture in Middlebrook 7H9 (Sigma–Aldrich, St. Louis, MI, USA) and solid culture on 7H10 (Sigma–Aldrich, USA) agar, both supplemented with OADC (10%; BD, London, UK). Growth media for MAP were also supplemented with mycobactin J (2 μg/μL; IDVet, Montpellier, France). All liquid cultures were incubated at 37 °C without shaking. The bacteriophage used was a lab strain of D29.
When assay specificity was assessed, approximately 1 × 105 cfu/mL of each bacterium was used in each experiment to ensure lack of detection was not due to a poor sensitivity of the assay, and all were lab strains maintained on Brain Heart Infusion media (Oxoid, Hampshire, UK).
To determine the concentration of mycobacteria in liquid culture, the enumeration method was carried out according to Swift et al. [
19]. In brief, serial dilutions of cultured mycobacteria in Middlebrook 7H9 supplemented with OADC and CaCl
2 were mixed with phage D29 (approximately 10
8 pfu/mL) and incubated at 37 °C for 1 h, allowing phage to bind and infect mycobacteria. Extracellular phages were inactivated with virucide (ferrous ammonium sulfate, 10 mM) for 5 min at ambient temperature. The virucide was neutralized via diluting with 5 mL Middlebrook 7H9 supplemented with OADC and CaCl
2 media before plating on 7H10 agar (0.75%
w/
v agar final). The mycobacterial cell number was determined via counting plaques on the
M. smegmatis reporter lawn (recorded as pfu/mL).
2.2. Bacteria Isolation from Blood and Use of Phage-LAMP in Blood
To isolate the bacteria from blood, a truncated processing method based on the MolYsis™ Basic 5 (Molzym GmbH & Co. KG, Bremen, Germany) was used. Briefly, whole blood (up to 5 mL) was mixed with buffer CM (2 mL) and vortexed for 15 s. The sample was left on the bench at ambient temperature for 5 min. Buffer SU (2 mL) and MolDNase (10 µL) were added to the sample and vortexed for 15 s. The sample was incubated on the bench for 15 min at ambient temperature before centrifuging (9500× g; 10 min). The supernatant was removed, and the pellet was resuspended in 7H9 Media plus OADC (1 mL). The sample was then centrifuged (13,000× g; 3 min) before use in the phage assays.
2.3. Actiphage Method
The Actiphage method was carried out according to Swift et al. [
12]. Before the assay, blood cells were depleted using MolYsis™ Basic 5. Briefly, the processed samples were suspended in 100 μL phage D29 (approximately 10
8 pfu/mL) and incubated for 3.5 h at 37 °C. Following incubation, the lysate was centrifuged (13,000×
g; 3 min) in 0.22 µm filter tubes (Spin-X, Costar, London, UK). The filtrate was then cleaned and concentrated (Monarch
® PCR & DNA Cleanup Kit (5 μg), New England Biotech, Ipswich, MA, USA) until the elution step, where 10 µL (double loaded) of molecular grade water (pre-warmed to 55 °C) was used to elute DNA. The eluted DNA was used as template DNA for nucleic acid amplification.
Blood spiking experiments were carried out using fresh, defibrinated sheep blood (Oxoid Ltd., Basingstoke, UK). Blood (3 mL) was spiked with cultures of MAP or M. bovis BCG to a final concentration of 104 cells/mL and serially diluted to approximately 100 cells/mL. Blood cells were lysed using MolYsis™ Basic 5. Lysed blood was then processed using the Actiphage method or phage-LAMP assay.
2.4. Phage-LAMP Assay
The phage-LAMP assay was performed via first isolating bacteria from blood (
Section 2.3). The pellet was resuspended in 100 μL phage D29 and incubated for 3.5 h at 37 °C. Following incubation, the lysate was centrifuged (13,000×
g; 3 min) in Rapid tubes. Then, 5 μL was used as template in a LAMP 25 μL reaction consisting of 1× WarmStart
® LAMP master mix (New England Biotech, USA), 1× LAMP Fluorescent Dye (New England Biotech, USA) and 1× primer master mix. The IS6110, IS900 and RD4 master mixes contained 0.5% DMSO, and the RD4 master mix contained 55 μM guanidine hydrochloride.
2.5. NAAT Optimization and Testing
Three separate LAMP assays were optimized, targeting the
Mycobacterium tuberculosis complex group of mycobacteria (IS6110),
M. bovis (RD4) and MAP (IS900). Briefly, a master mix was prepared and each 25 μL reaction consisted of 1× WarmStart
® LAMP master mix (New England Biotech, USA), 1× LAMP Fluorescent Dye (New England Biotech, USA) and 1× primer master mix. A matrix of effects was investigated to optimize each LAMP assay. This included determining the optimal reaction temperature using a temperature gradient; (60–67.5 °C) optimizing the loop forward and loop reverse primer concentrations (0.8, 1.4 and 2.4 μM); determining the effect of primer sets with phosphorothioated FIP (F1c) and BIP (B1c) regions in the presence of urea (3.6 μM; Thermo Fisher Scientific, Waltham, MA, USA); and determining the effect of LAMP enhancers, guanidine hydrochloride (40 µM; Thermo Fisher Scientific) and DMSO (2%; Thermo Fisher Scientific). LAMP primer sequences and modifications are available in
Table S1.
2.6. Analytical Sensitivity and Specificity Testing
The limit of detection was determined via enumerating the mycobacteria and serially diluting to a concentration of 100 cells/mL in either 7H9(+) media or spiked sheep blood (Oxoid Ltd., UK). These dilutions were processed with phage, and 5 μL of non-purified lysate was used as a template in either the PCR assays or MTBC, MAP and M. bovis LAMP assays. When the effect of the clean and concentrate step was examined, this process was repeated in 7H9(+), except at the end of the Actiphage method, the clean and concentration step was not included. A negative no-template control was carried out for every run.
A range of non-target bacteria were tested in each LAMP assay to assess specificity. For each bacterial species, one colony was harvested and subjected to crude DNA extraction (boiling at 95 °C for 10 min). Then 5 μL of DNA was used as the template and tested in each phage-LAMP assay.
2.7. LAMP Product Visualisaion
Fluorescence was measured in real-time and melt-curve analysis was performed using the on-board Genie® II software (OptiGene Limited, Horsham, UK). For color reactions, 0.5 μL of ×1000 SYBR green 1 (Sigma-Aldrich, St. Louis, MI, USA) was added to the tube cap before amplification and shaken after amplification. Relative color was quantified via measuring the hue value in the Hue Value Saturation (HSV) model using Color Grab™ (Loomatix Ltd., Haifa, Isreal); images for analysis were captured using a Moto G5 Android phone (Motorola, Schaumburg, IL, USA). Amplification was then confirmed with gel electrophoresis using 10 μL product on a 2% TAE gel containing 0.01% GelRed (Sigma-Aldrich, St. Louis, MI, USA) followed by UV-transillumination.
2.8. PCR Amplification
For PCR reactions, DNA samples from the phage lysates (5 μL) were used as template for amplification of signature sequences. Members of the MTBC were identified using IS6110 qPCR [
20] and for MAP an IS900 specific qPCR method was used [
21]. Cycling conditions were 1 cycle of 95 °C for 5 min, followed by 40 cycles of 95 °C for 30 s, followed by 62 °C (MAP) or 65 °C (MTBC) for 30 s. Fluorescence reading was captured at the annealing step. After cycling was complete, melt curve analysis was carried out. All qPCR reactions were carried out using QuantiFast
® SYBR green master mix (Qiagen, Hilden, Germany).
2.9. Statistical Analysis
LAMP fluorescence was analyzed using Genie®Explorer V 2.0.7.11 (OptiGene Limited, Horsham, UK). Statistical analysis including Pearson’s correlation and Cohen’s kappa was performed using GraphPad Prism V 9.0.0 (GraphPad Software, La Jolla, CA, USA). qPCR results were analyzed using Bio-Rad CFX Maestro V 2.3 (Bio-Rad Laboratories, Hercules, CA, USA).
2.10. Samples and Ethics
Human clinical samples were obtained opportunistically as part of wider TB study where no patient was HIV positive. This study was approved by the Research Ethics Committee (REC) for East Midlands—Nottingham 1, Nottingham, UK (REC 15/EM/0109). The study using cattle blood was approved by the Royal Veterinary College’s Clinical Research Ethical Review Board, (URN 2020 2009-2).
3. Results
3.1. Optimization of LAMP Assays
The performance of the assay is influenced by some factors such as temperature, the time of exposure, loop primer sequence and the composition of the NAAT master mix. Different incubation temperatures ranging from 60 °C to 67.5 °C were tested, with intervals of 1.25 °C. The MTBC (IS6110), MAP (IS900) and M. bovis (RD4) assays performed better at higher temperatures, with the fastest times to detection (15:00, 23:45 and 22:45 min) at 67.5 °C, 66.25 °C and 65 °C, respectively. Performance was improved in the IS6110 assay after changing the loop primer concentrations to 0.8 μM and including 0.5% DMSO; this improved the limit of detection (LOD) by 1 log. In the RD4 assay, the inclusion of 55 μM guanidine hydrochloride improved the time-of-detection (TOD) by 10 min 10 s. The phosphorothioation of primers theoretically lowers amplification temperatures via improving the self-folding efficiency of terminal hairpins. F1c and B1c primers were phosphorothioated and used in the primer master mix with 3.6 μM urea. This prevented detection when fluorescence was measured in real-time, and when the product was run on a 2% TAE gel, no evidence of amplification was seen. Once optimized, the MTBC, MAP and M. bovis assays could each detect 100 cells/mL, and cut-off times were determined as 45, 45 and 80 min, respectively.
3.2. LAMP Assay Read-Out Methods
LAMP assays can be read using several methods. Here, we examined two endpoints: a real-time fluorescent dye and a colorimetric intercalating DNA dye. Amplification was confirmed with gel electrophoresis. Each method could detect down to 1 cell (
Table 1). Only the real-time fluorescent read-out method gave semi-quantitative results (based on time to detection) compared to the colorimetric assay.
3.3. Analytical LAMP Specificity
The optimized LAMP assays were tested with DNA from non-target bacterial species to determine analytical specificity. The results in
Table 2 show there was no non-target amplification in the MTBC, MAP or
M. bovis assays.
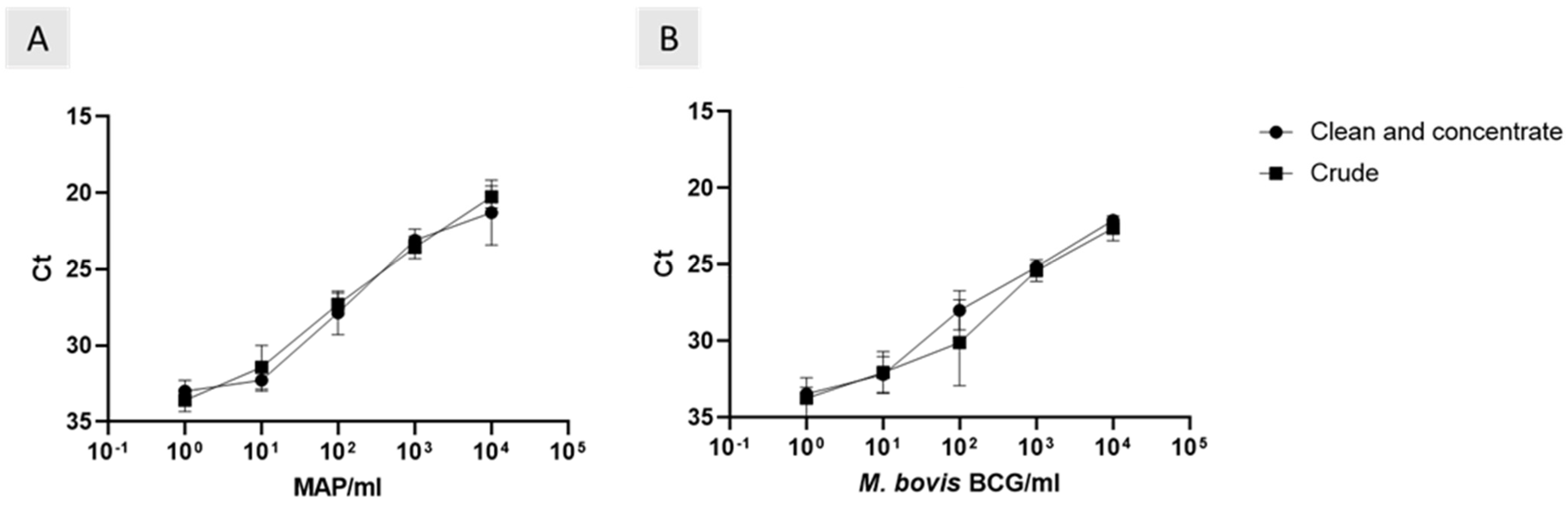
3.4. Effect of Removing DNA Clean and Concentrate on Phage-LAMP Assay
The Actiphage method required the use of a DNA clean and concentration step before carrying out qPCR. Here, we compared the effect of removing the clean and concentrate step on the phage assays. There was a strong positive correlation between the ct values of crude and clean and concentrated
M. bovis BCG samples (
Figure 1A; r
2 = 0.96, 99% CI = 0.5 to 0.99). The trend was consistent with MAP samples (
Figure 1B; r
2 = 0.99, 99% CI = 0.69 to 0.99). As well as this, removing the clean and concentrate step did not affect the analytical sensitivities (
Figure 1).
3.5. Endpoint Detection Comparison; qPCR vs. LAMP
After the clean and concentrate step was removed from the phage-LAMP assay, the effect of changing the endpoint from qPCR to LAMP was assessed. When the MTBC LOD of the two methods were compared, there was a perfect correlation (κ = 1, 95% CI 1 to 1); this trend was consistent with MAP samples. There was no impact on LOD when changing to the LAMP endpoint.
3.6. Testing Phage-LAMP with Artificially Inoculated Blood
To evaluate the assay performance in clinically relevant sample types, mycobacteria were artificially inoculated into blood and processed using the optimized phage-LAMP assays. The MTBC, RD4 and MAP assays were each able to detect 10
1 cells/mL in blood. In all LAMP assays, the effect of blood was noticeable as the TOD increased and the LOD reduced by a log. Because of the increase in TOD, assay cut-off times were increased by 15 min. When used with blood samples, the MTBC, MAP and RD4 assays used cut-off times of 60, 60 and 100 min, respectively (
Table 3). In this experiment, the colorimetric readout was attempted; however, no positive results were observed.
3.7. Clinical Proof-of-Concept in Animal and Human Samples
As the phage-LAMP assay was capable of detecting clinically relevant levels of mycobacteria in artificially inoculated blood samples, as a proof-of-concept, clinical samples were tested for MAP and MTBC using the phage-LAMP assays.
Initially, blood samples were obtained from two herds with known Johne’s disease problems. Thirty blood samples from cattle that had their Johne’s disease status determined via routine blood ELISA were tested using the phage-LAMP assay. Eight cattle were considered positive for Johne’s based on a red ELISA result, ten were considered at risk based on amber ELISA results and twelve were considered negative based on green ELISA results. The MAP phage-LAMP assay detected viable MAP in 10 samples. Five out of eight ELISA red cattle were positive, five out of ten ELISA amber were positive and no ELISA green cattle were positive for MAP (
Table 4).
To demonstrate that the phage-LAMP assay could detect
M. tuberculosis in clinical human blood samples, a small proof-of-concept experiment was carried out on the blood of ten recent TB contacts that were positive using an interferon-gamma release assay (IGRA) and were recently exposed to
M. tuberculosis. The MTBC phage-LAMP assay detected viable mycobacteria in 70% (7/10) of patients (
Table 5).
4. Discussion
Bacteriophage technology shows great potential in aiding the diagnosis of mycobacterial infections. However, these assays need to be practical, affordable and as close to care as possible. The previous iterations of phage-based assays, such as the Actiphage method, although extremely sensitive, rely on multiple centrifugation and DNA clean-up steps as well as the use of qPCR, which limits its use to centralized laboratories [
12]. Here, we describe the combined use of phage-mediated DNA release and LAMP as a tool to bring phage-based diagnostics closer to care.
4.1. LAMP Optimization
Several LAMP assays have been described for the detection of
M. tuberculosis complex,
M. bovis and MAP [
22,
23,
24]. Their development, however, was not carried out in the context of a phage-based assay and thus required optimization to maximize the sensitivity and ensure they were compatible with bacteriophage.
Initially, the temperature at which the LAMP assay was carried out was optimized. The temperature at which LAMP assays operate is crucial to ensure efficient enzyme activity and DNA amplification. Using a temperature gradient, the MTBC (IS6110), MAP (IS900) and M. bovis (RD4) assays performed better at higher temperatures than those reported, with the fastest time to detection at 67.5 °C, 66.25 °C and 65 °C, respectively.
LAMP assays can also be improved using enhancers such as urea and primer phosphorothioation. Cai et al. [
25] showed urea and the phosphorothioation of primers could lower amplification temperature; however, this effect was not observed in our experiments, possibly owing to the longer amplification time used [
25]. Alternatively, the high GC content of the MTBC and MAP F1c and B1c primers may influence the stability of the foldback hairpin structures.
Including DMSO improved both the time to detection and LOD. Because LAMP is still in its infancy of development, less is known about chemical effects on amplification in comparison to PCR. Nevertheless, DMSO affects PCR via making it more heat-labile, which destabilizes the DNA structure and lowers denaturation temperature [
26]. We theorize that the same mode of action is true for LAMP. Why DMSO improved the time to detection is unknown, but it may be a by-product of this effect.
Guanidine hydrochloride has been reported to improve the time to detection [
27]. We found that it acted as an inhibitor in MTBC and MAP assays but improved time to detection in the RD4 assay without affecting analytical sensitivity or specificity. Speculated to improve base pairing between primers and target sequences, the increased length of RD4 FIP and BIP primers (in comparison to MTBC and MAP) may explain this effect.
4.2. Experimental Phage-LAMP Evaluation
The observed LOD of the optimized LAMP assays was 10
0 cells/ mL. The use of lower LF/LR primer concentrations and DMSO in the IS6110 LAMP assay improved the LOD compared to the original reporting, which did not examine the effect of DMSO [
23]. The LOD and TOD in the RD4 assay were improved via changing LF/LR primer concentration and including DMSO and guanidine hydrochloride. These factors were also not examined by the original authors [
24]. These improvements in TOD and LOD suggest that optimization steps involving primer concentrations and common LAMP enhancers should be considered when applying LAMP assays to detect very low levels of bacterial DNA from clinical samples.
As LAMP assays are thought to be less prone to inhibitors than PCR [
18], the DNA clean-up that is included in the Actiphage method was removed. There was no difference between the TOD or LOD of lysates processed with or without the clean and concentration step. This is likely because this experiment was processed in media. When comparing LAMP and phage-LAMP assays in media and blood, there was a clear increase in TOD and reduction in LOD. This effect was also observed during the development of the Actiphage method [
12]. This suggests that some components of blood may inhibit the LAMP assay, similar to PCR [
28], but this is less frequently associated with LAMP reactions. Though LAMP is tolerant to inhibition [
18], it is not impervious to inhibition.
Different LAMP readouts were explored, and a real-time readout method was adopted. When used in media, the sensitivity of both the real-time and colorimetric outputs was equivalent; however, when used in spiked blood, the colorimetric readout failed to produce a color change in any sample. Because the clean and concentration step was removed, the lysate at the end of the Actiphage method was suspended with red blood cell precipitate. This likely obscured any color change, creating a negative result.
The specificity of the assay comes from two aspects. First, the D29 phage only infects and lyses mycobacterial hosts; therefore, DNA from non-mycobacteria should only be present in low amounts. Second, we show the LAMP assays only amplify the targeted insertion sequences. The original reporting of the MAP LAMP assay [
20] did not examine specificity; here it is demonstrated that MAP LAMP is specific. Furthermore, the range of bacterial species for which MTBC LAMP is known to not produce false positives has been expanded from those originally reported [
22].
4.3. Use of Phage-LAMP in Clinical Samples
The phage-LAMP assay was then applied to two sets of blood samples, one from cattle and another from humans. This was to determine whether viable mycobacteria could be detected using the assay. The first set came from cattle on two herds with a prevalence of Johne’s disease of 15% based on serial ELISA testing. The results showed that the phage-LAMP assay detected MAP in 5/8 ELISA red cattle, 5/10 ELISA amber cattle, but no viable MAP was detected in the ELISA green cattle. These results suggest that the phage-LAMP assay can detect viable MAP in animals suspected to have Johne’s based on ELISA. As serum ELISAs do not have 100% sensitivity and specificity [
29], further work would be required to determine the clinical sensitivity and specificity of the phage-LAMP assay in Johne’s infected herds with a range of prevalences, longitudinally.
The phage-LAMP assay was used on a small set of opportunistically collected human blood samples as part of a wider TB study. Only samples that came from IGRA-positive patients were tested and our results showed that 7/10 patients had viable mycobacteria in their blood as determined by the phage-LAMP assay. In the same way as the Johne’s samples, IGRAs are unable to differentiate remote or recent infection, and IGRAs do not determine which patients have or will go on to have an active TB infection [
29,
30]. Therefore, further samples would need to be collected along with further diagnostic data to determine the sensitivity and specificity of phage-LAMP. However, these proof-of-concept data suggest the assay is capable of detecting MTBC organisms in clinical samples.
It should also be mentioned that immunoreactivity assays of mycobacterial infections are a poor gold standard for current metabolically active infections for both TB and Johne’s disease that are detected with phage-based assays [
31]. Determining the performance of these novel assays using the immunological benchmarks is therefore challenging to interpret. In the future, for the evaluation of this method, alternative outcomes will be sought, including additional clinical information on the patient/animal samples as well as having an appropriate negative control group for infected vs. non-infected.
4.4. Future Perspectives and Moving Closer to the Point-of-Care
The WHO has called for new molecular diagnostics that can be used close to the point-of-care from samples other than sputum [
32]. Our phage-LAMP assay has the potential to meet this call, and further work will be carried out to evaluate this potential.
The speed of detection using phage-LAMP is limited by the 3.5 h incubation during the phage-mediated lysis step. This is due to the D29 replication cycle, which is seemingly unavoidable. The 5.5 h time to detection of phage-LAMP does not preclude its application in district-level labs and still allows results to be relayed within the day.
The cost of phage-LAMP is estimated at USD 12.45 per test (
Table S2). This is above the WHO’s recommended USD 5 per test for a human TB test. The commercialization of the technology is expected to lower costs. This price does not include infrastructure costs.
Using LAMP as an endpoint instead of qPCR has several advantages. Time to detection is faster, it does not need a constant power source, equipment is cheaper and portable, and no clean and concentration step is needed. This represents a move closer to the point-of-care that is more applicable in low- and middle-income countries.
One aspect not explored in this study, or the original reporting of RD4 LAMP [
23], is the effect of BCG vaccination on specificity. In the UK, cattle are not vaccinated; however, a CattleBCG vaccine has recently been developed by the APHA [
33] and is currently under field testing [
34]. The persistence of the attenuated strain may create false positives. This should be examined in future work.
Logically, the next stage of development is to adapt the method into a true point-of-care NAAT assay. Certain requirements currently limit this advancement. The requirement of a centrifuge forces the assay to be in a centralized location. The need for a cold chain during the transport of LAMP reagents will increase the costs. A true point-of-care test should produce results during the initial clinic visit [
8]; thus, the time to detection needs to be shortened.
Further to this, we will need to explore how novel diagnostic tests such as this fit into the testing protocols, and how further testing can be supported by policy makers, clinicians and other stakeholders, which will be crucial to successfully tackling these diseases.


 ,
, 

{kind=link}