
Microbiome Dynamics in Samia cynthia ricini: Impact of Growth Stage and Dietary Variations


Abstract
:1. Introduction
2. Materials and Method
2.1. Sample Collection, Rearing, and Growth Characteristics Observation
2.2. DNA Extraction and Sequencing
2.3. Sequence Retrieval, Quality Control, and Processing
2.4. Compositional Analyses
2.5. Functional Prediction
3. Results
3.1. Silkworm Rearing and Growth Observations
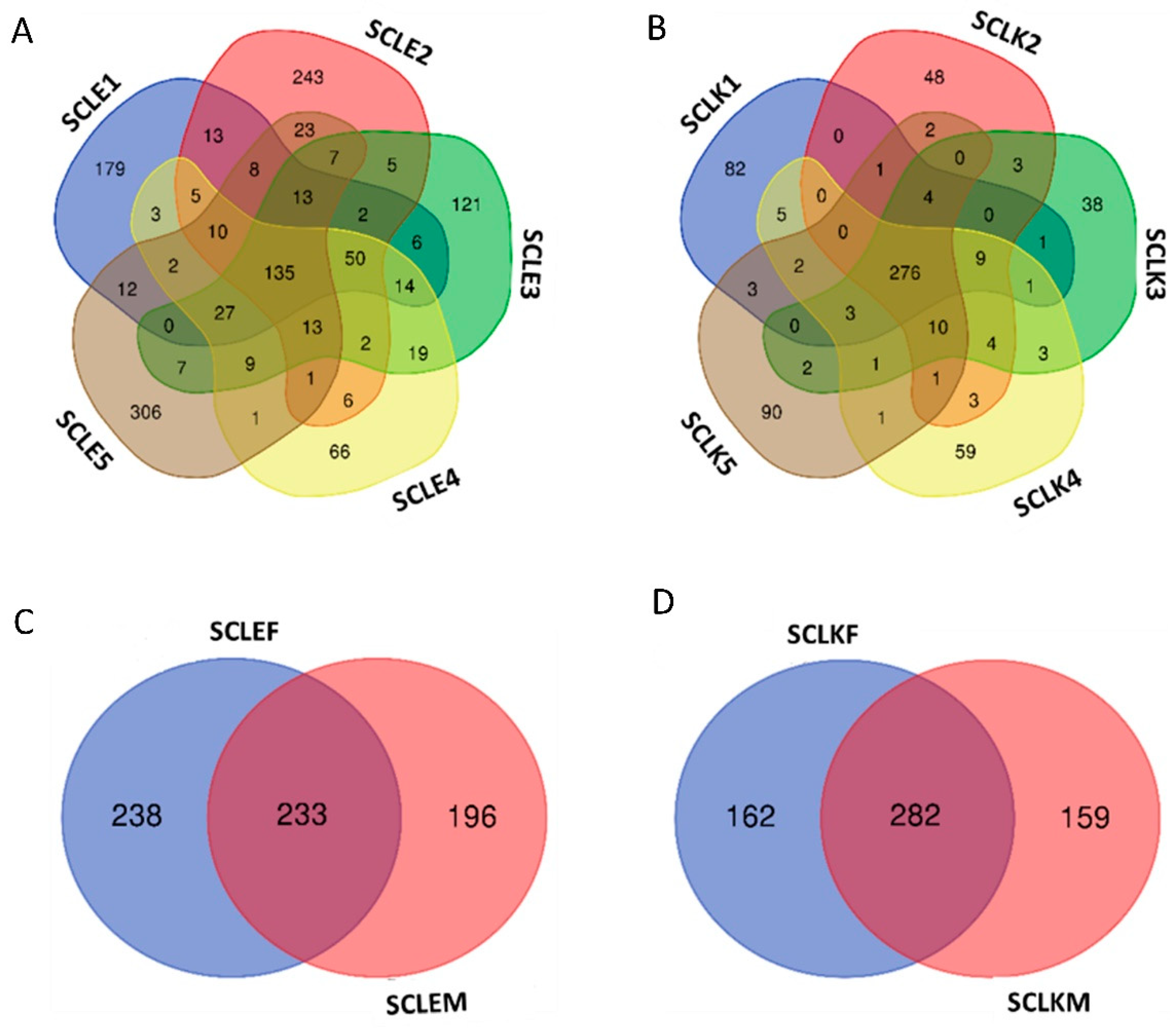
3.2. Sequence Processing, Quality Control, and Bioinformatic Analyses
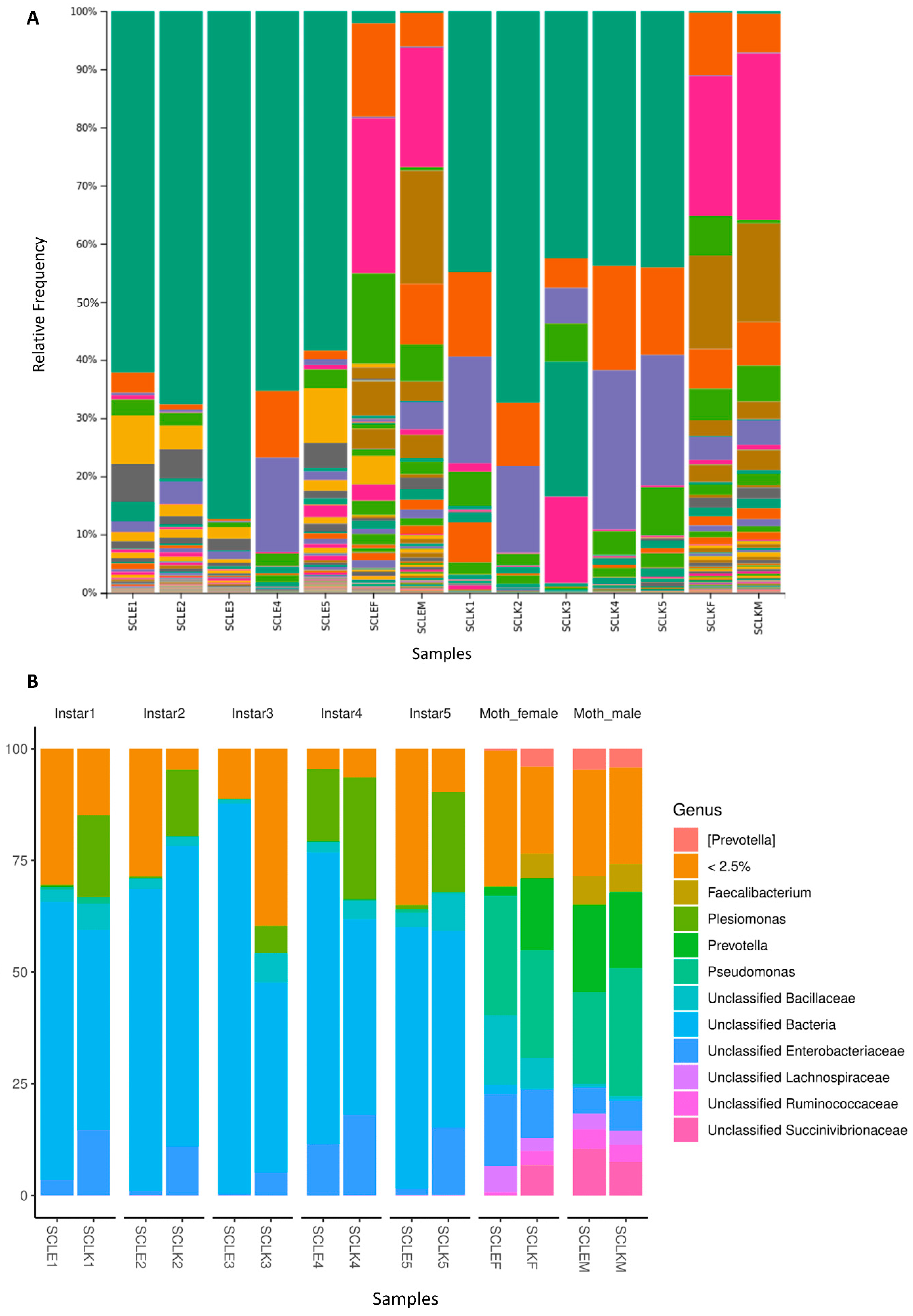
3.3. Microbiome Composition
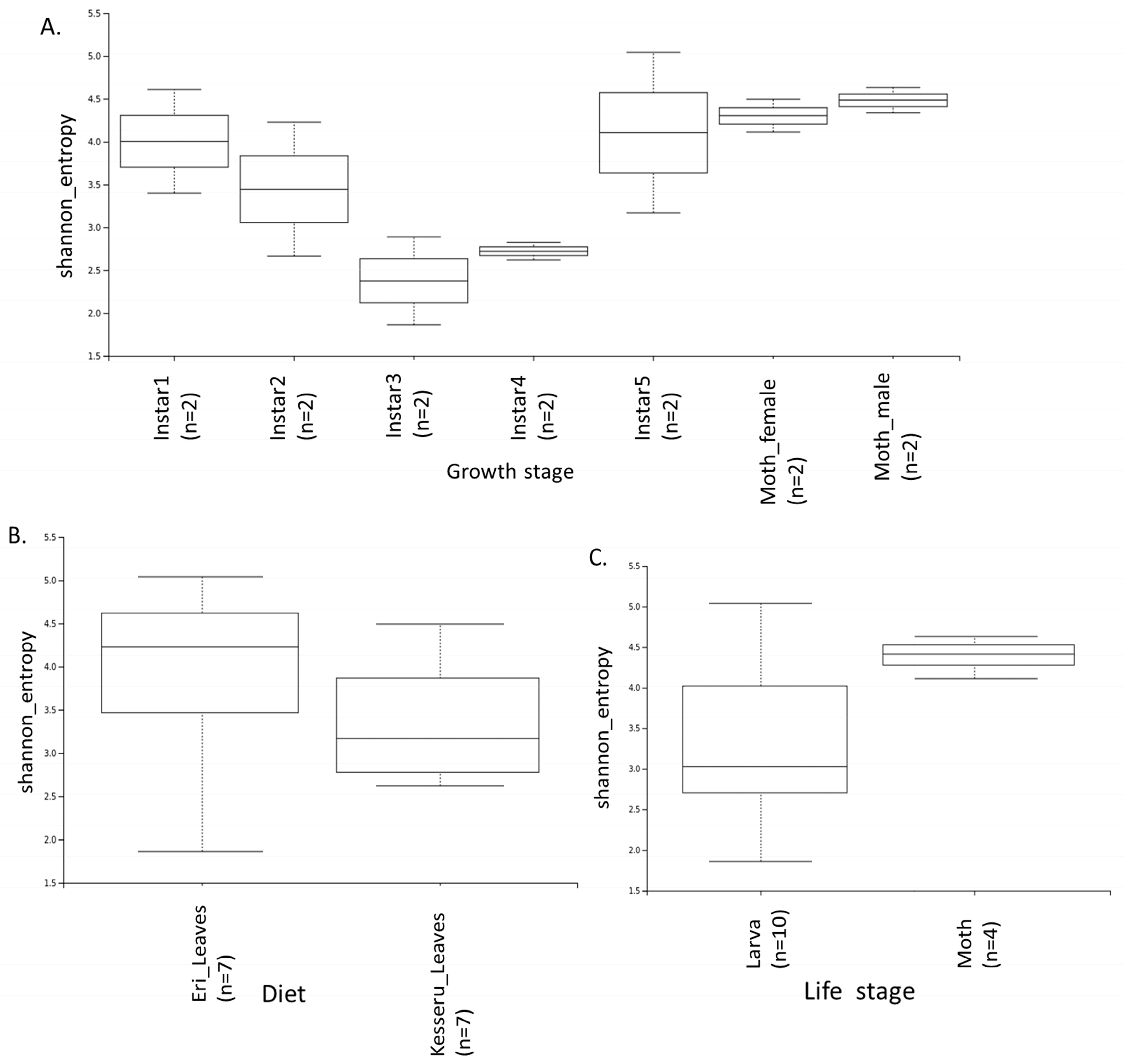
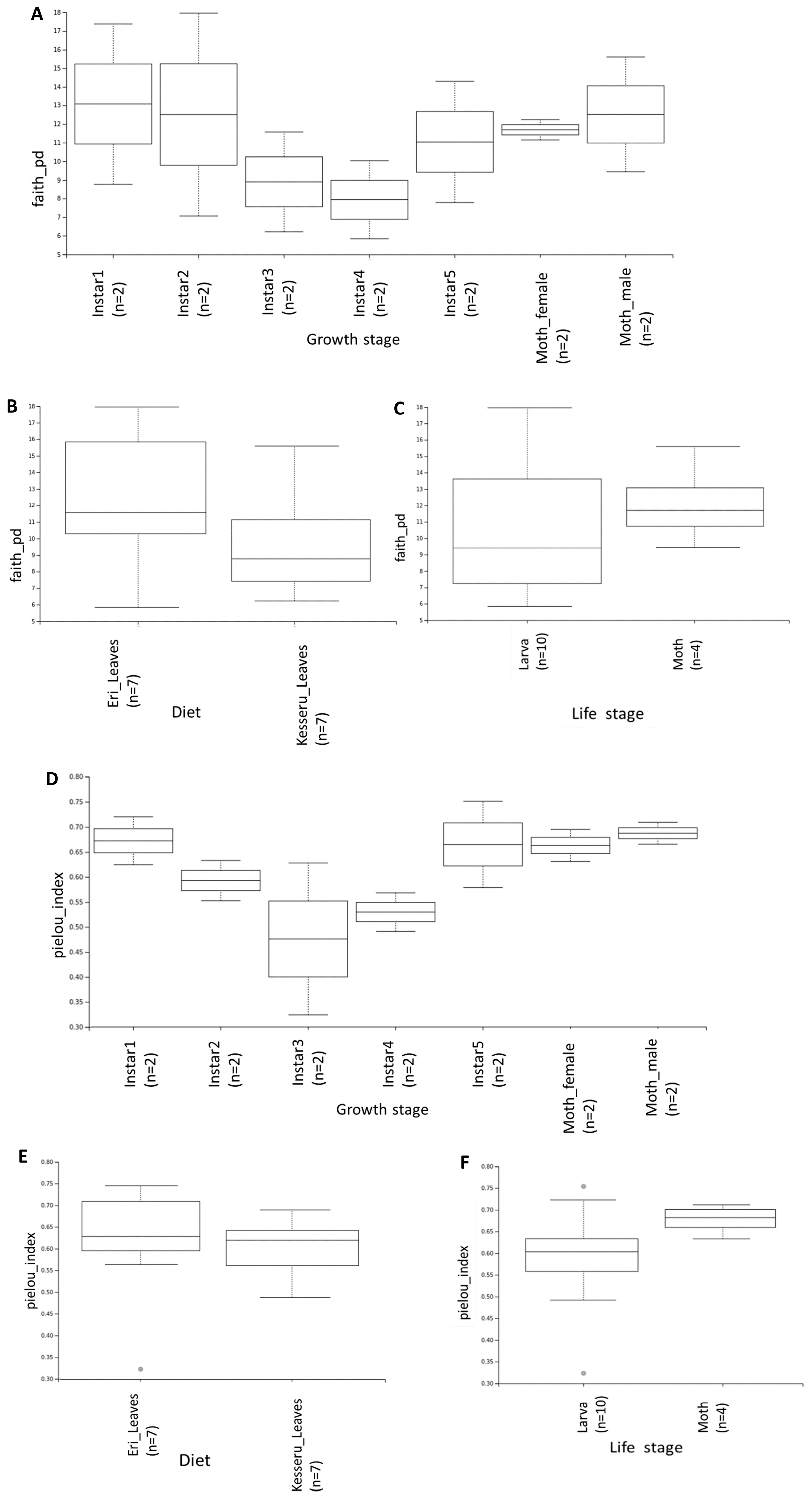
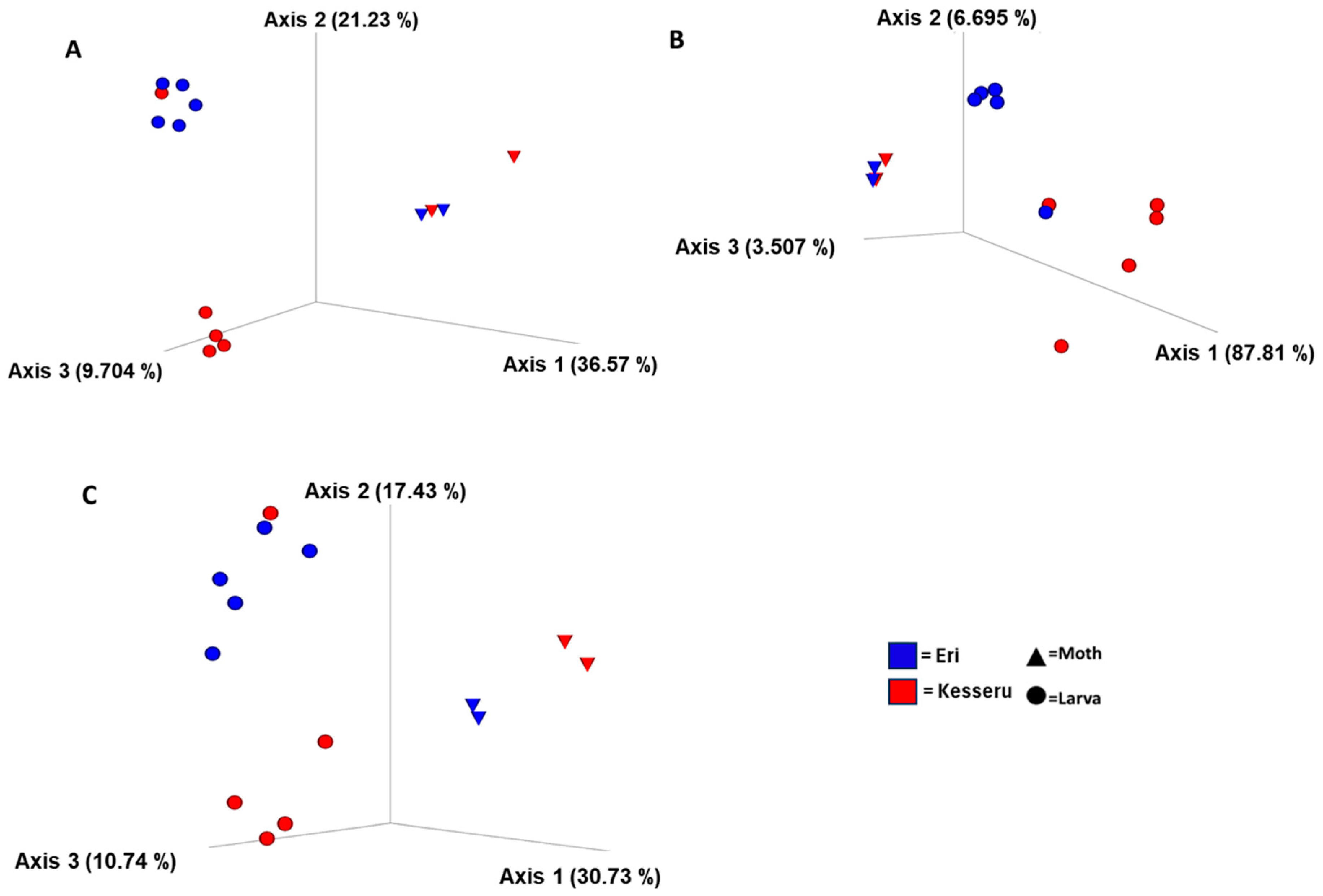
3.4. Diversity Analyses
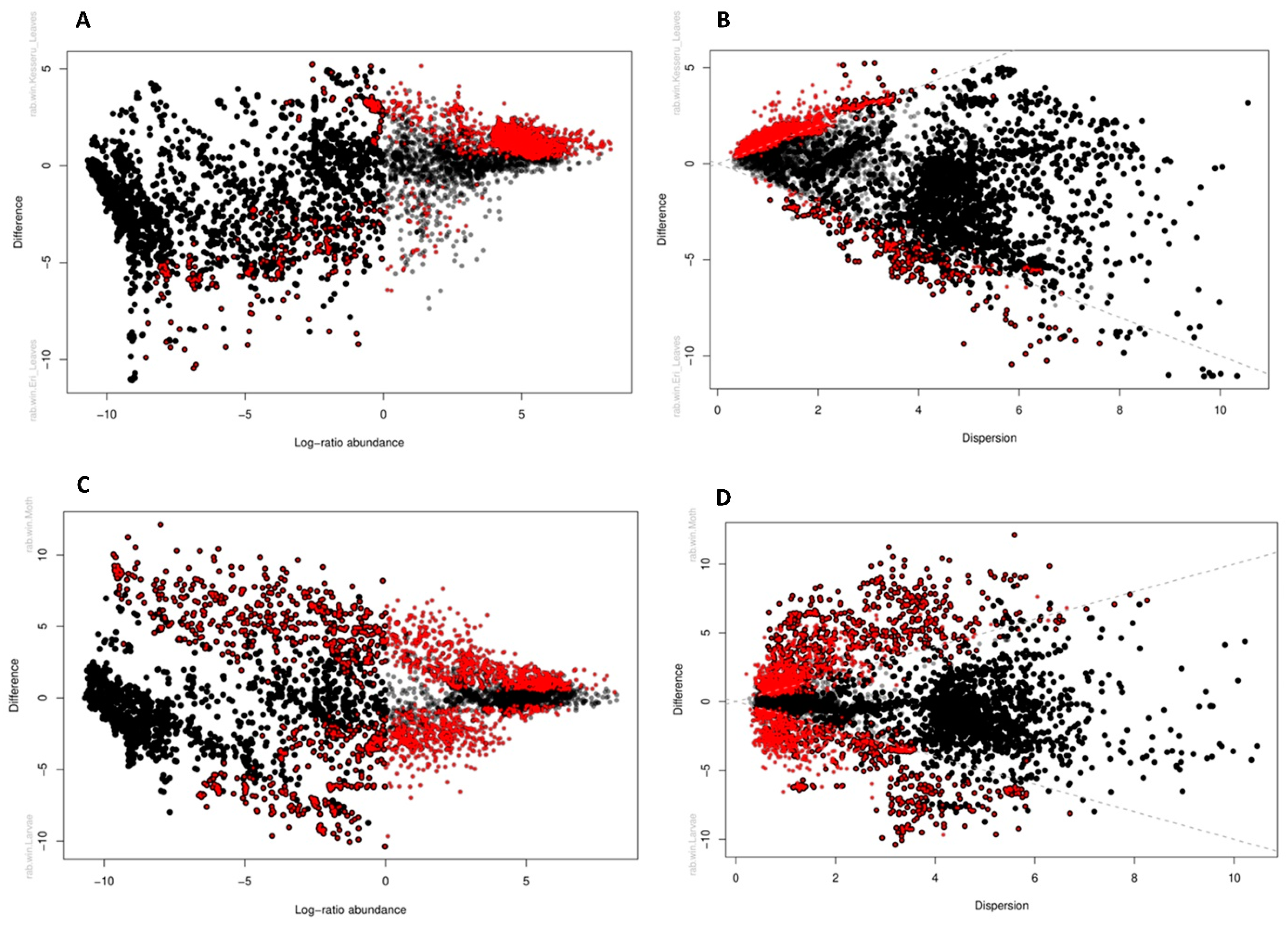
3.5. Differential Abundances
3.6. Predictive Functional Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HCN | Hydrogen cyanide |
| Dfls | Disease-free layings |
| PHEC | Phenol/chloroform |
| PBS | Phosphate-buffered solution |
| SDS | Sodium dodecyl sulphate |
| DNA | Deoxyribonucleic acid |
| QIIME2 | Quantitative insights into microbial ecology 2 |
| DADA | Divisive amplicon de-noising algorithm |
| ASV | Amplicon sequence variants |
| OTU | Operational taxonomic unit |
| PCoA | Principal coordinates analysis |
| NMDS | Non-metric multidimensional scaling |
| DA | Differential abundance |
| ANCOM-BC | Analysis of compositions of microbiomes with bias correction |
| MEGAN | MEtaGenome analyser |
| CLR | Centred-log ration |
| FDRs | False discovery rates |
| PICRUSt2 | Phylogenetic investigation of communities by reconstruction of unobserved states |
| KO | Kegg-Orthology |
| KEGG | Kyoto encyclopedia of genes and genomes |
| Faith-PD | Faith’s phylogenetic diversity |
| PERMANOVA | Permutational multivariate analysis of variance |
| PWY | Formaldehyde oxidation/detoxification |
| RNA | Ribonucleic acid |
| GABA | Gamma-aminobutyric acid |
| MAGs | Metagenome-assembled genomes |
| HGTs | Horizontal gene transfers |
| ARGs | Antibiotic resistance genes |
References
- Gilbert, S.F.; Sapp, J.; Tauber, A.I. A symbiotic view of life: We have never been individuals. Q. Rev. Biol. 2012, 87, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Groussin, M.; Mazel, F.; Alm, E.J. Co-evolution and Co-speciation of Host-Gut Bacteria Systems. Cell Host Microbe 2020, 28, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Nair, S. Dynamics of Insect–Microbiome Interaction Influence Host and Microbial Symbiont. Front. Microbiol. 2020, 11, 1357. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, Y.; Hosokawa, T.; Nikoh, N.; Meng, X.-Y.; Kamagata, Y.; Fukatsu, T. Host-symbiont co-speciation and reductive genome evolution in gut symbiotic bacteria of acanthosomatid stinkbugs. BMC Biol. 2009, 7, 2. [Google Scholar] [CrossRef]
- Rolff, J.; Johnston, P.R.; Reynolds, S. Complete metamorphosis of insects. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20190063. [Google Scholar] [CrossRef]
- Wilbur, H.M. Complex life cycles. Annu. Rev. Ecol. Syst. 1980, 11, 67–93. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, F.; Lu, X. Diversity and Functional Roles of the Gut Microbiota in Lepidopteran Insects. Microorganisms 2022, 10, 1234. [Google Scholar] [CrossRef]
- Voirol, L.R.P.; Frago, E.; Kaltenpoth, M.; Hilker, M.; Fatouros, N.E. Bacterial symbionts in lepidoptera: Their diversity, transmission, and impact on the host. Front. Microbiol. 2018, 9, 556. [Google Scholar]
- Engel, P.; Moran, N.A. The gut microbiota of insects—Diversity in structure and function. FEMS Microbiol. Rev. 2013, 37, 699–735. [Google Scholar] [CrossRef]
- Hasan, N.; Yang, H. Factors affecting the composition of the gut microbiota, and its modulation. PeerJ 2019, 7, e7502. [Google Scholar] [CrossRef]
- Douglas, A.E. Multiorganismal Insects: Diversity and Function of Resident Microorganisms. Annu. Rev. Entomol. 2015, 60, 17. [Google Scholar] [CrossRef] [PubMed]
- Mason, C.J.; Clair, A.S.; Peiffer, M.; Gomez, E.; Jones, A.G.; Felton, G.W.; Hoover, K. Diet influences proliferation and stability of gut bacterial populations in herbivorous lepidopteran larvae. PLoS ONE 2020, 15, e0229848. [Google Scholar] [CrossRef]
- Chen, B.; Teh, B.-S.; Sun, C.; Hu, S.; Lu, X.; Boland, W.; Shao, Y. Biodiversity and Activity of the Gut Microbiota across the Life History of the Insect Herbivore Spodoptera littoralis. Sci. Rep. 2016, 6, 29505. [Google Scholar] [CrossRef] [PubMed]
- Jing, T.Z.; Qi, F.H.; Wang, Z.Y. Most dominant roles of insect gut bacteria: Digestion, detoxification, or essential nutrient provision? Microbiome 2020, 8, 38. [Google Scholar] [CrossRef]
- Ma, Q.; Cui, Y.; Chu, X.; Li, G.; Yang, M.; Wang, R.; Liang, G.; Wu, S.; Tigabu, M.; Zhang, F.; et al. Gut Bacterial Communities of Lymantria xylina and Their Associations with Host Development and Diet. Microorganisms 2021, 9, 1860. [Google Scholar] [CrossRef]
- Pandiarajan, J.; Krishnan, M. Comparative bacterial survey in the gut of lepidopteran insects with different bionetwork. Microbiology 2018, 87, 103–115. [Google Scholar] [CrossRef]
- Brinkmann, N.; Martens, R.; Tebbe, C.C. Origin and diversity of metabolically active gut bacteria from laboratory-bred larvae of Manduca sexta (Sphingidae, Lepidoptera, Insecta). Appl. Environ. Microbiol. 2008, 74, 7189–7196. [Google Scholar] [CrossRef]
- Henry, L.P.; Bruijning, M.; Forsberg, S.K.G.; Ayroles, J.F. The microbiome extends host evolutionary potential. Nat. Commun. 2021, 12, 5141. [Google Scholar] [CrossRef]
- Engel, P.; Moran, N.A. Functional and evolutionary insights into the simple yet specific gut microbiota of the honey bee from metagenomic analysis. Gut Microbes 2013, 4, 60. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1. [Google Scholar] [CrossRef]
- Lei, W.T.; Huang, K.Y.; Jhong, J.H.; Chen, C.H.; Weng, S.L. Metagenomic analysis of the gut microbiome composition associated with vitamin D supplementation in Taiwanese infants. Sci. Rep. 2021, 11, 2856. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Li, A.; Jiang, J.; Zhou, L.; Yu, Z.; Lu, H.; Xie, H.; Chen, X.; Shao, L.; Zhang, R.; et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 2019, 68, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Veziant, J.; Villéger, R.; Barnich, N.; Bonnet, M. Gut Microbiota as Potential Biomarker and/or Therapeutic Target to Improve the Management of Cancer: Focus on Colibactin-Producing Escherichia coli in Colorectal Cancer. Cancers 2021, 13, 2215. [Google Scholar] [CrossRef]
- Allaband, C.; McDonald, D.; Vázquez-Baeza, Y.; Minich, J.J.; Tripathi, A.; Brenner, D.A.; Loomba, R.; Smarr, L.; Sandborn, W.J.; Schnabl, B.; et al. Microbiome 101: Studying, Analyzing, and Interpreting Gut Microbiome Data for Clinicians. Clin. Gastroenterol. Hepatol. 2019, 17, 218. [Google Scholar] [CrossRef]
- Hammer, T.J.; Bowers, M.D. Gut microbes may facilitate insect herbivory of chemically defended plants. Oecologia 2015, 179, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Show, B.K.; Banerjee, S.; Banerjee, A.; Ghosh Thakur, R.; Hazra, A.K.; Mandal, N.C.; Ross, A.B.; Balachandran, S.; Chaudhury, S. Insect gut bacteria: A promising tool for enhanced biogas production. Rev. Environ. Sci. Biotechnol. 2022, 21, 1–25. [Google Scholar] [CrossRef]
- Francoeur, C.B.; Khadempour, L.; Moreira-Soto, R.D.; Gotting, K.; Book, A.J.; Pinto-Tomás, A.A.; Keefover-Ring, K.; Currie, C.R. Bacteria contribute to plant secondary compound degradation in a generalist herbivore system. mBio 2020, 11, e02146-20. [Google Scholar] [CrossRef]
- Kohl, K.D.; Denise Dearing, M. The woodrat gut microbiota as an experimental system for understanding microbial metabolism of dietary toxins. Front. Microbiol. 2016, 7, 1165. [Google Scholar] [CrossRef]
- Krishnan, M.; Bharathiraja, C.; Pandiarajan, J.; Prasanna, V.A.; Rajendhran, J.; Gunasekaran, P. Insect gut microbiome—An unexploited reserve for biotechnological application. Asian Pac. J. Trop. Biomed. 2014, 4, S16. [Google Scholar] [CrossRef]
- Kedir Shifa, K.S.; Emana Getu, E.G.; Waktole Sori, W.S. Rearing Performance of Eri-Silkworm (Samia cynthia ricini Boisduval) (Lepidoptera: Saturniidae) Fed with Different Castor (Ricinus communis L.) Genotypes. J. Entomol. 2013, 11, 25–33. [Google Scholar] [CrossRef]
- Renuka, G.; Shamitha, G. Studies On The Economic Traits Of Eri Silkworm, Samia Cynthia Ricini, In Relation To Seasonal Variations. Int. J. Adv. Res. 2014, 2, 315–322. [Google Scholar]
- Sharma, P.; Chandra Kalita, J. A comparative study on six strains of eri silk worm (samia ricini; donovan) based on morphological traits. Glob. J. Bio-Sci. Biotechnol. 2013, 2, 506–511. [Google Scholar]
- Ahmed, S.A.; Singh, N.I.; Sarkar, C.R. Role of forest biodiversity in conservation of non-mulberry (vanya) silk in India. Munis Entomol. Zool. 2015, 10, 342–357. [Google Scholar]
- Velayudhan, K.; Balachandran, N.; RadhaKrishnan, S.; Singh, B.K.; Jayaprakash, P. Biodiversity in Eri Silkworm Samia Ricini (Donovan) Genetic Resources and its Conservation. J. Aquat. Biol. Fish. 2014, 2, 817–824. [Google Scholar]
- Government of Assam, India. Handloom Textiles & Sericulture. Host Plants of Eri Silkworm. Available online: https://hts.assam.gov.in/portlet-innerpage/host-plants-of-eri-silkworm (accessed on 15 July 2020).
- Churchandpur, Manipur. Improved Varieties of Silkworm Food Plants. Available online: https://silks.csb.gov.in/churchandpur/improved-varieties-of-silkworm-food-plants/ (accessed on 10 August 2020).
- Zhang, N.; He, J.; Shen, X.; Sun, C.; Muhammad, A.; Shao, Y. Contribution of sample processing to gut microbiome analysis in the model Lepidoptera, silkworm Bombyx mori. Comput. Struct. Biotechnol. J. 2021, 19, 4658–4668. [Google Scholar] [CrossRef]
- Keen, J.N.; Austin, M.K.; Huang, L.S.; Messing, S.; Wyatt, J.D. Efficacy of Soaking in 70% Isopropyl Alcohol on Aerobic Bacterial Decontamination of Surgical Instruments and Gloves for Serial Mouse Laparotomies. J. Am. Assoc. Lab. Anim. Sci. 2010, 49, 832. [Google Scholar]
- Gerasimidis, K.; Bertz, M.; Quince, C.; Brunner, K.; Bruce, A.; Combet, E.; Calus, S.; Loman, N.; Ijaz, U.Z. The effect of DNA extraction methodology on gut microbiota research applications. BMC Res. Notes 2016, 9, 365. [Google Scholar] [CrossRef]
- García-Alegría, A.M.; Anduro-Corona, I.; Pérez-Martínez, C.J.; Corella-Madueño, M.A.G.; Rascón-Durán, M.L.; Astiazaran-Garcia, H. Quantification of DNA through the nanodrop spectrophotometer: Methodological validation using standard reference material and sprague dawley rat and human DNA. Int. J. Anal. Chem. 2020, 2020, 8896738. [Google Scholar] [CrossRef]
- Olson, N.D.; Morrow, J.B. DNA extract characterization process for microbial detection methods development and validation. BMC Res. Notes 2012, 5, 668. [Google Scholar] [CrossRef]
- Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 20 October 2021).
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Draw Venn Diagram. Available online: https://bioinformatics.psb.ugent.be/webtools/Venn/ (accessed on 12 February 2022).
- RStudio. Open Source & Professional Software for Data Science Teams—RStudio. Available online: https://www.rstudio.com/ (accessed on 15 November 2022).
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef]
- Konopiński, M.K. Shannon diversity index: A call to replace the original Shannon’s formula with unbiased estimator in the population genetics studies. PeerJ 2020, 8, e9391. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228. [Google Scholar] [CrossRef]
- Davenport, J.M. New approxzmations to theexact distribution of the kruskal-wallis test statistic. Commun. Stat. Theory Methods 1976, 5, 1335–1348. [Google Scholar]
- Kelly, B.J.; Gross, R.; Bittinger, K.; Sherrill-Mix, S.; Lewis, J.D.; Collman, R.G.; Bushman, F.D.; Li, H. Power and sample-size estimation for microbiome studies using pairwise distances and PERMANOVA. Bioinformatics 2015, 31, 2461–2468. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Lin, H.; das Peddada, S. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020, 11, 3514. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Peschel, S.; Müller, C.L.; von Mutius, E.; Boulesteix, A.L.; Depner, M. NetCoMi: Network construction and comparison for microbiome data in R. Brief. Bioinform. 2021, 22, bbaa290. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Fernandes, A.D.; Macklaim, J.M.; Linn, T.G.; Reid, G.; Gloor, G.B. ANOVA-Like Differential Expression (ALDEx) Analysis for Mixed Population RNA-Seq. PLoS ONE 2013, 8, e67019. [Google Scholar] [CrossRef]
- Welch, B.L. The Significance of the Difference Between Two Means When the Population Variances Are Unequal. Biometrika 1938, 29, 350–362. [Google Scholar] [CrossRef]
- Altman, D.G.; Bland, J.M. Measurement in Medicine: The Analysis of Method Comparison Studies. Statistician 1983, 32, 307. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes—A 2019 update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Effect of Differential Feeding on Eri Silk Worm (Samia Ricini Donovan) and Its Pupal Protein Concentration. Available online: https://www.researchgate.net/publication/319178276_Effect_of_Differential_Feeding_on_Eri_Silk_Worm_Samia_Ricini_Donovan_and_its_Pupal_Protein_Concentration (accessed on 16 December 2021).
- Baruah, M.; Bari, P. Studies on Larval Weight and Shell Ratio of Eri Silkworm (Philosamia ricini) on Castor, Kesseru and Treated Kesseru by Foliar Spray. Int. J. Comput. Appl. Eng. Sci. 2012, 2, 133–137. [Google Scholar]
- Canani, R.B.; De Filippis, F.; Nocerino, R.; Laiola, M.; Paparo, L.; Calignano, A.; De Caro, C.; Coretti, L.; Chiariotti, L.; Gilbert, J.A.; et al. Specific signatures of the gut microbiota and increased levels of butyrate in children treated with fermented cow’s milk containing heat-killed Lactobacillus paracasei CBA L74. Appl. Environ. Microbiol. 2017, 83, e01206-17. [Google Scholar]
- Kaakoush, N.O. Insights into the role of Erysipelotrichaceae in the human host. Front. Cell. Infect. Microbiol. 2015, 5, 84. [Google Scholar] [CrossRef]
- Jiang, W.; Wu, N.; Wang, X.; Chi, Y.; Zhang, Y.; Qiu, X.; Hu, Y.; Li, J.; Liu, Y. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci. Rep. 2015, 5, 8096. [Google Scholar] [CrossRef]
- Ng, S.H.; Stat, M.; Bunce, M.; Simmons, L.W. The influence of diet and environment on the gut microbial community of field crickets. Ecol. Evol. 2018, 8, 4704. [Google Scholar] [CrossRef]
- van Teijlingen, N.H.; Helgers, L.C.; Zijlstra-Willems, E.M.; van Hamme, J.L.; Ribeiro, C.M.; Strijbis, K.; Geijtenbeek, T.B. Vaginal dysbiosis associated-bacteria Megasphaera elsdenii and Prevotella timonensis induce immune activation via dendritic cells. J. Reprod. Immunol. 2020, 138, 103085. [Google Scholar] [CrossRef]
- Iljazovic, A.; Roy, U.; Gálvez, E.J.C.; Lesker, T.R.; Zhao, B.; Gronow, A.; Amend, L.; Will, S.E.; Hofmann, J.D.; Pils, M.C.; et al. Perturbation of the gut microbiome by Prevotella spp. enhances host susceptibility to mucosal inflammation. Mucosal Immunol. 2020, 14, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Tainchum, K.; Dupont, C.; Chareonviriyaphap, T.; Jumas-Bilak, E.; Bangs, M.J.; Manguin, S. Bacterial Microbiome in Wild-Caught Anopheles Mosquitoes in Western Thailand. Front. Microbiol. 2020, 11, 965. [Google Scholar] [CrossRef] [PubMed]
- Zamakhchari, M.; Wei, G.; Dewhirst, F.; Lee, J.; Schuppan, D.; Oppenheim, F.G.; Helmerhorst, E.J. Identification of Rothia Bacteria as Gluten-Degrading Natural Colonizers of the Upper Gastro-Intestinal Tract. PLoS ONE 2011, 6, e24455. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Jin, B.-J.; Bi, Q.-F.; Li, K.-J.; Sun, C.-L.; Lin, X.-Y.; Zhu, Y.-G. Variations of earthworm gut bacterial community composition and metabolic functions in coastal upland soil along a 700-year reclamation chronosequence. Sci. Total Environ. 2022, 804, 149994. [Google Scholar] [CrossRef]
- Sapountzis, P.; Zhukova, M.; Shik, J.Z.; Schiott, M.; Boomsma, J.J. Reconstructing the functions of endosymbiotic mollicutes in fungus-growing ants. Elife 2018, 7, e39209. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, S.; Jiang, Q.; Bai, Y.; Shen, G.; Li, S.; Ding, W. Using community analysis to explore bacterial indicators for disease suppression of tobacco bacterial wilt. Sci. Rep. 2016, 6, 36773. [Google Scholar] [CrossRef]
- Fijan, S. Microorganisms with Claimed Probiotic Properties: An Overview of Recent Literature. Int. J. Environ. Res. Public Health 2014, 11, 4745. [Google Scholar] [CrossRef]
- Zuko, Y.; Maeda, K. The effect of intestinal Bifidobacterium on the output of Bombyx mori. Peer J. Prepr. 2018. [Google Scholar] [CrossRef]
- Soliman, S. Micro-Organisms Supplementation to Mulberry Silkworm, Bombyx mori L. Egypt. Acad. J. Biol. Sci. Entomol. 2017, 10, 57–64. [Google Scholar]
- Nishida, S.; Ono, Y.; Sekimizu, K. Lactic acid bacteria activating innate immunity improve survival in bacterial infection model of silkworm. Drug Discov. Ther. 2016, 10, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Fujita, Y.; Ren, Q.; Ma, M.; Dong, C.; Hashimoto, K. Bifidobacterium in the gut microbiota confer resilience to chronic social defeat stress in mice. Sci. Rep. 2017, 7, 45942. [Google Scholar] [CrossRef] [PubMed]
- Socała, K.; Doboszewska, U.; Szopa, A.; Serefko, A.; Włodarczyk, M.; Zielińska, A.; Poleszak, E.; Fichna, J.; Wlaź, P. The role of microbiota-gut-brain axis in neuropsychiatric and neurological disorders. Pharmacol. Res. 2021, 172, 105840. [Google Scholar] [CrossRef] [PubMed]
- Maltz, R.M.; Keirsey, J.; Kim, S.C.; Mackos, A.R.; Gharaibeh, R.Z.; Moore, C.C.; Xu, J.; Somogyi, A.; Bailey, M.T. Social stress affects colonic inflammation, the gut microbiome, and short chain fatty acid levels and receptors. J. Pediatr. Gastroenterol. Nutr. 2019, 68, 533. [Google Scholar] [CrossRef]
- Dance, A. The search for microbial dark matter. Nature 2020, 582, 301–303. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Diet | Growth Stage |
|---|---|---|
| SCLE1 | Eri Leaves | Instar 1 |
| SCLE2 | Eri Leaves | Instar 2 |
| SCLE3 | Eri Leaves | Instar 3 |
| SCLE4 | Eri Leaves | Instar 4 |
| SCLE5 | Eri Leaves | Instar 5 |
| SCLEF | Eri Leaves | Moth Female |
| SCLEM | Eri Leaves | Moth Male |
| SCLK1 | Kesseru Leaves | Instar 1 |
| SCLK2 | Kesseru Leaves | Instar 2 |
| SCLK3 | Kesseru Leaves | Instar 3 |
| SCLK4 | Kesseru Leaves | Instar 4 |
| SCLK5 | Kesseru Leaves | Instar 5 |
| SCLKF | Kesseru Leaves | Moth Female |
| SCLKM | Kesseru Leaves | Moth Male |
| Sample ID | Input | Filtered | Percentage of Input Passed Filter | De-noised | Merged | Percentage of Input Merged | Non-Chimeric | Percentage of Input Non-Chimeric |
|---|---|---|---|---|---|---|---|---|
| SCLE1 | 274,989 | 245,925 | 89.43 | 234,527 | 212,445 | 77.26 | 176,241 | 64.09 |
| SCLE2 | 280,585 | 228,509 | 81.44 | 221,840 | 213,177 | 75.98 | 193,690 | 69.03 |
| SCLE3 | 229,700 | 170,838 | 74.37 | 165,618 | 160,699 | 69.96 | 143,542 | 62.49 |
| SCLE4 | 149,293 | 103,102 | 69.06 | 101,425 | 97,566 | 65.35 | 78,317 | 52.46 |
| SCLE5 | 246,061 | 207,266 | 84.23 | 203,619 | 193,794 | 78.76 | 184,845 | 75.12 |
| SCLEF | 113,215 | 85,623 | 75.63 | 81,183 | 66,811 | 59.01 | 42,276 | 37.34 |
| SCLEM | 330,290 | 126,527 | 38.31 | 119,896 | 89,418 | 27.07 | 32,703 | 9.9 |
| SCLK1 | 167,516 | 120,398 | 71.87 | 117,746 | 108,255 | 64.62 | 68,185 | 40.7 |
| SCLK2 | 152,280 | 111,113 | 72.97 | 109,436 | 104,636 | 68.71 | 80,647 | 52.96 |
| SCLK3 | 159,959 | 111,203 | 69.52 | 109,329 | 101,584 | 63.51 | 60,491 | 37.82 |
| SCLK4 | 168,904 | 96,377 | 57.06 | 93,810 | 86,834 | 51.41 | 44,061 | 26.09 |
| SCLK5 | 162,808 | 102,294 | 62.83 | 99,241 | 91,310 | 56.08 | 53,879 | 33.09 |
| SCLKF | 477,230 | 200,533 | 42.02 | 192,900 | 152,748 | 32.01 | 60,634 | 12.71 |
| SCLKM | 361,644 | 154,512 | 42.72 | 147,553 | 114,555 | 31.68 | 46,117 | 12.75 |
| Metric | Unfiltered_Table | Filtered_Table |
|---|---|---|
| Number of Samples | 14 | 14 |
| Number of features | 6341 | 1139 |
| Total Frequency | 1,265,628 | 980,296 |
| Feature ID | Taxonomy |
|---|---|
| 656881 | k__Bacteria; p__Proteobacteria; c__Gammaproteobacteria; o__Enterobacteriales; f__Enterobacteriaceae; g__; s__ |
| 289174 | k__Bacteria; p__Proteobacteria; c__Gammaproteobacteria; o__Enterobacteriales; f__Enterobacteriaceae; g__Plesiomonas; s__shigelloides |
| 4438565 | k__Bacteria; p__Proteobacteria; c__Gammaproteobacteria; o__Pseudomonadales; f__Pseudomonadaceae; g__Pseudomonas; s__ |
| 4406763 | k__Bacteria; p__Firmicutes; c__Bacilli; o__Bacillales; f__Bacillaceae; g__; s__ |
| 749805 | k__Bacteria; p__Proteobacteria; c__Betaproteobacteria |
| 686593 | k__Bacteria; p__Proteobacteria; c__Alphaproteobacteria; o__Rhizobiales; f__Rhizobiaceae; g__; s__ |
| 945326 | k__Bacteria; p__Firmicutes; c__Bacilli; o__Bacillales; f__Bacillaceae; g__; s__ |
| 73760 | k__Bacteria; p__Proteobacteria; c__Betaproteobacteria; o__MKC10; f__; g__; s__ |
| 126133 | k__Bacteria; p__Proteobacteria; c__Gammaproteobacteria; o__Pseudomonadales; f__Moraxellaceae; g__Acinetobacter; s__ |
| 516809 | k__Bacteria; p__Proteobacteria; c__Gammaproteobacteria; o__Xanthomonadales; f__Xanthomonadaceae; g__Stenotrophomonas; s__ |
| 212688 | k__Bacteria; p__Proteobacteria; c__Alphaproteobacteria; o__Caulobacterales; f__Caulobacteraceae; g__; s__ |
| Group | p-Value | Test Statistic |
|---|---|---|
| Growth stages | 0.167 | 1.271346 |
| Diet | 0.13 | 1.44053 |
| Life stages (Larva vs. Moth) | 0.001 | 4.94633 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bharali, B.; Basumatary, P.; Bora, U. Microbiome Dynamics in Samia cynthia ricini: Impact of Growth Stage and Dietary Variations. Appl. Microbiol. 2025, 5, 40. https://doi.org/10.3390/applmicrobiol5020040
Bharali B, Basumatary P, Bora U. Microbiome Dynamics in Samia cynthia ricini: Impact of Growth Stage and Dietary Variations. Applied Microbiology. 2025; 5(2):40. https://doi.org/10.3390/applmicrobiol5020040
Chicago/Turabian StyleBharali, Biju, Pulakeswar Basumatary, and Utpal Bora. 2025. "Microbiome Dynamics in Samia cynthia ricini: Impact of Growth Stage and Dietary Variations" Applied Microbiology 5, no. 2: 40. https://doi.org/10.3390/applmicrobiol5020040
APA StyleBharali, B., Basumatary, P., & Bora, U. (2025). Microbiome Dynamics in Samia cynthia ricini: Impact of Growth Stage and Dietary Variations. Applied Microbiology, 5(2), 40. https://doi.org/10.3390/applmicrobiol5020040