
Macrophage-Induced Exacerbation of Nasopharyngeal Inflammatory Lymphocytes in COVID-19 Disease

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
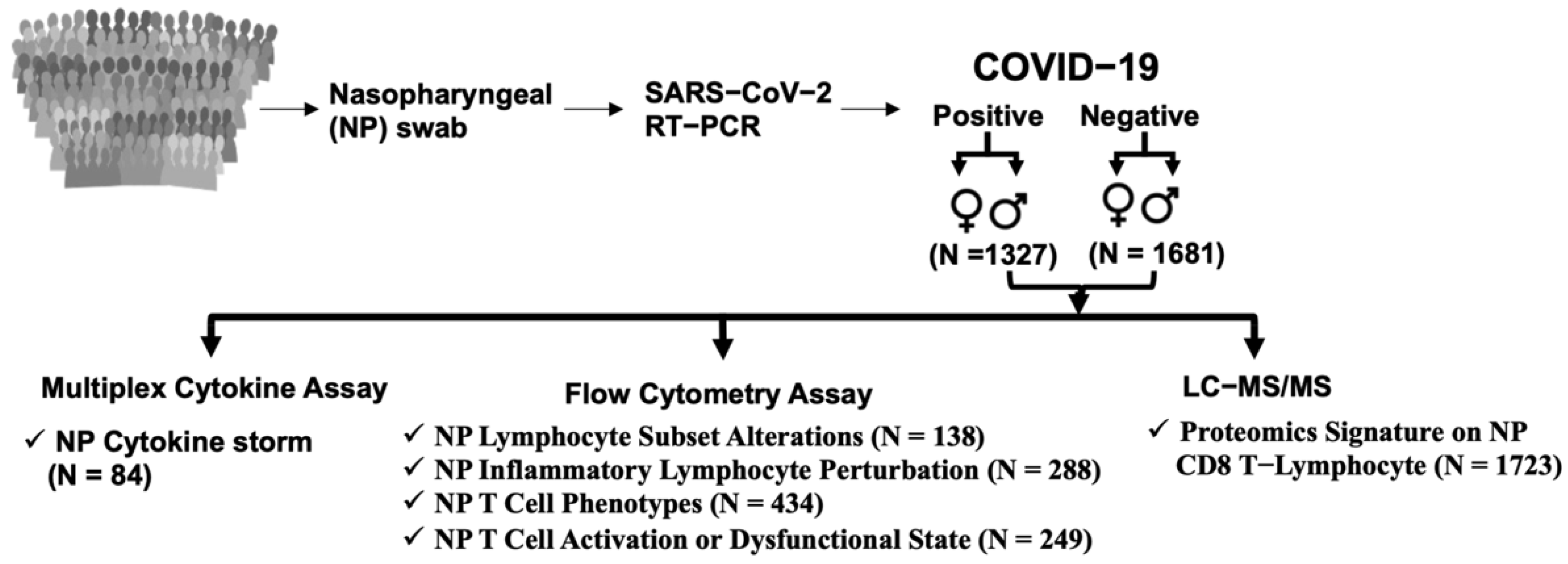
2.1. Subject Details
2.2. Characteristics of Patients
2.3. SARS-CoV-2 RT-PCR Assay
2.4. Sample Collection and Processing
2.5. Multiplex Cytokine Assay
2.6. Flow Cytometric Staining of Nasal Cells
2.7. PBMCs Isolation and Differentiation into Macrophages
2.8. SARS-CoV-2 Spike (S) Protein Induces the Production of Cytokines from Human PBMCs
2.9. Role of Macrophages upon SARS-CoV-2 Infection
2.10. In Vitro Analysis of T-Cell Inflammatory Responses
2.11. The Proliferation of Lymphocyte Subsets in the Presence of Distinct Macrophages Co-Culture
2.12. Identification of SARS-CoV-2-Induced Nasopharyngeal (NP) Surfactome in CD8 T-Cells from Patient NP Samples
2.13. Identification of a Surfactome in CD8 T-Cells in the Presence of SARS-CoV-2 Trimer S Protein-Induced Macrophage Subsets
2.14. Sample Preparation for Surfactome Analysis by Mass Spectrometry
2.15. LC-MS/MS Analysis and Data Processing
2.16. Statistical Analysis
3. Results
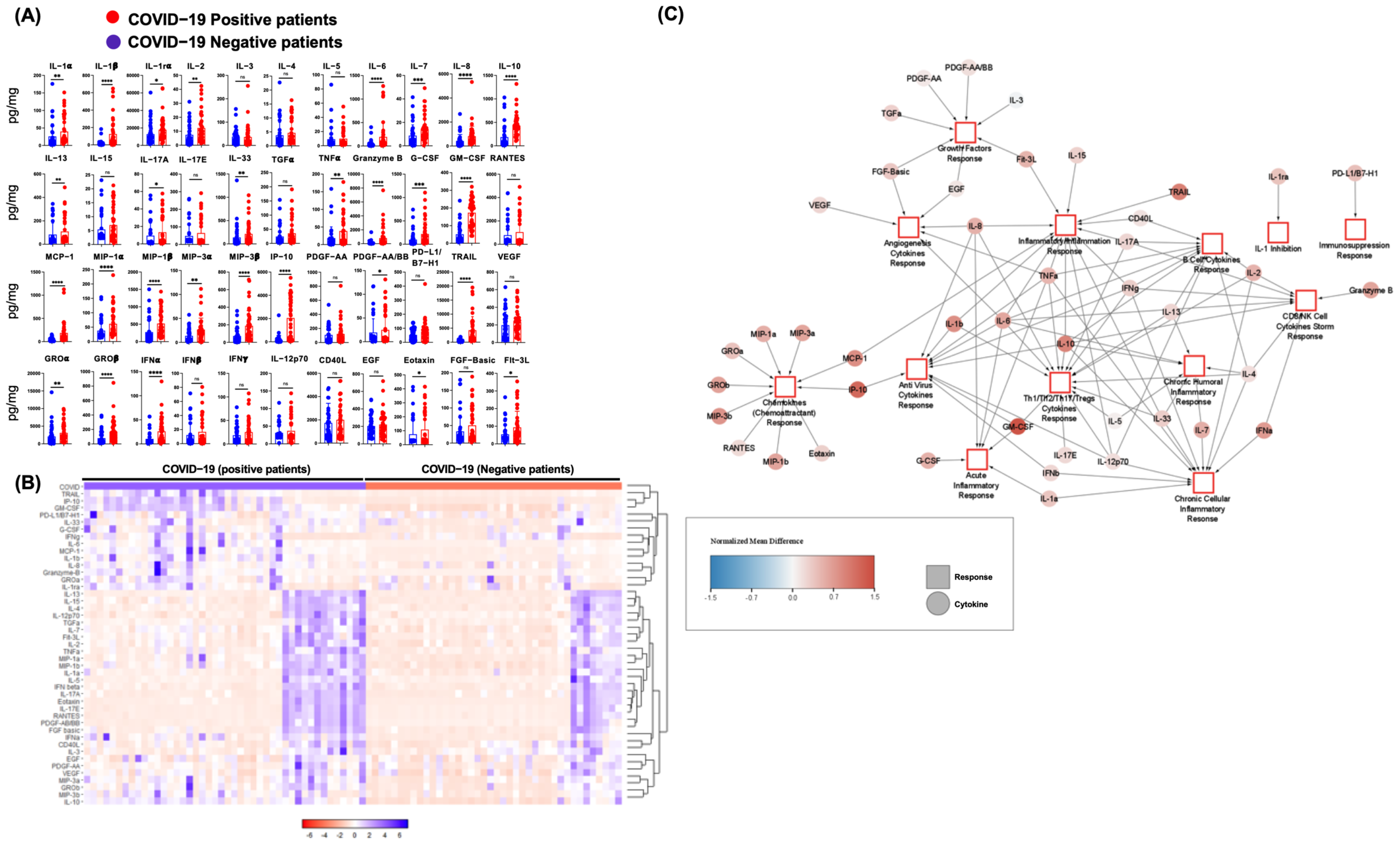
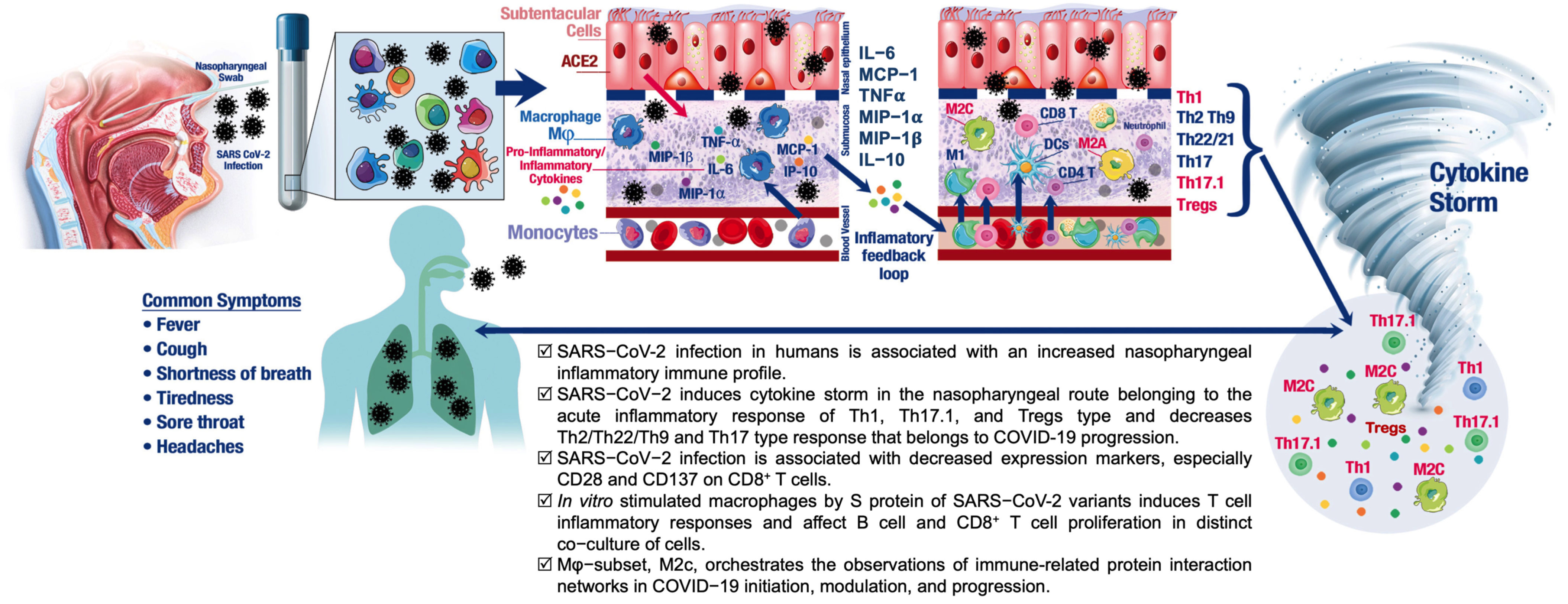
3.1. SARS-CoV-2 Infection in Humans Is Associated with an NP Cytokine Storm
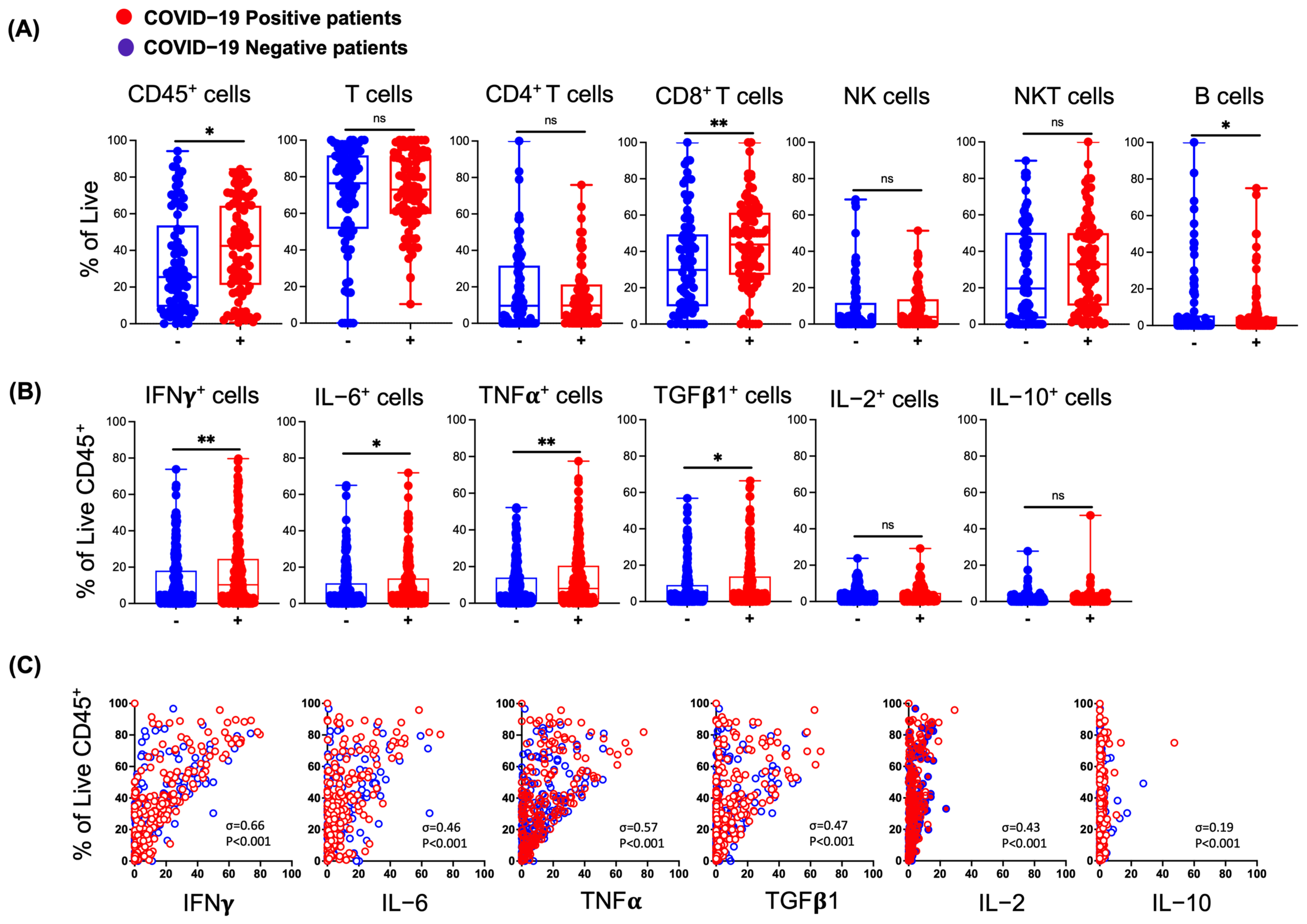
3.2. Effects of SARS-CoV-2 Infection on Lymphocytes in the Nasal Mucosa
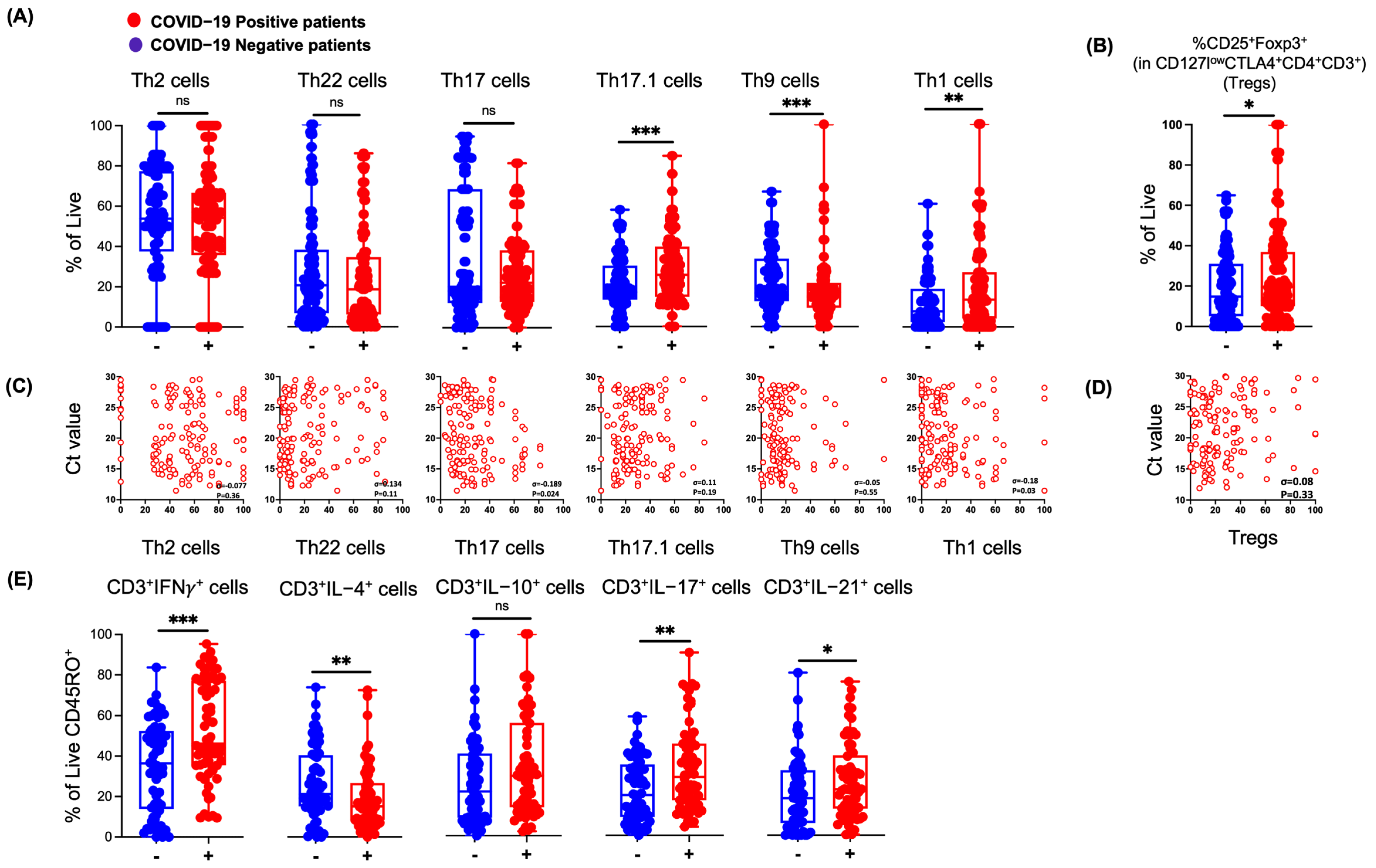
3.3. SARS-CoV-2 Infection in Humans Results in Changes in NP Th Lymphocytes
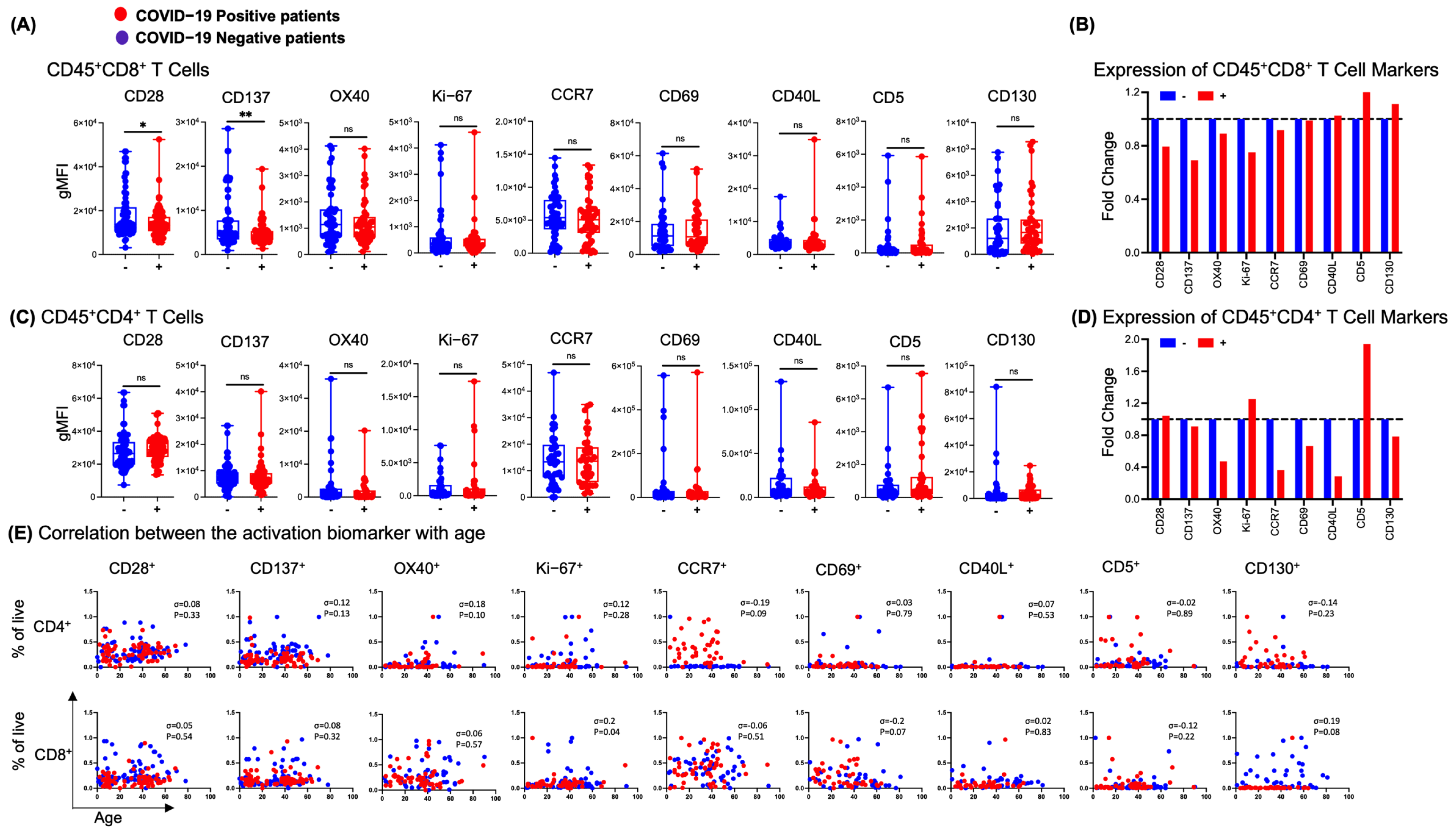
3.4. NP T-Cell Activation or Dysfunctional State Is Associated with SARS-CoV-2 Infection
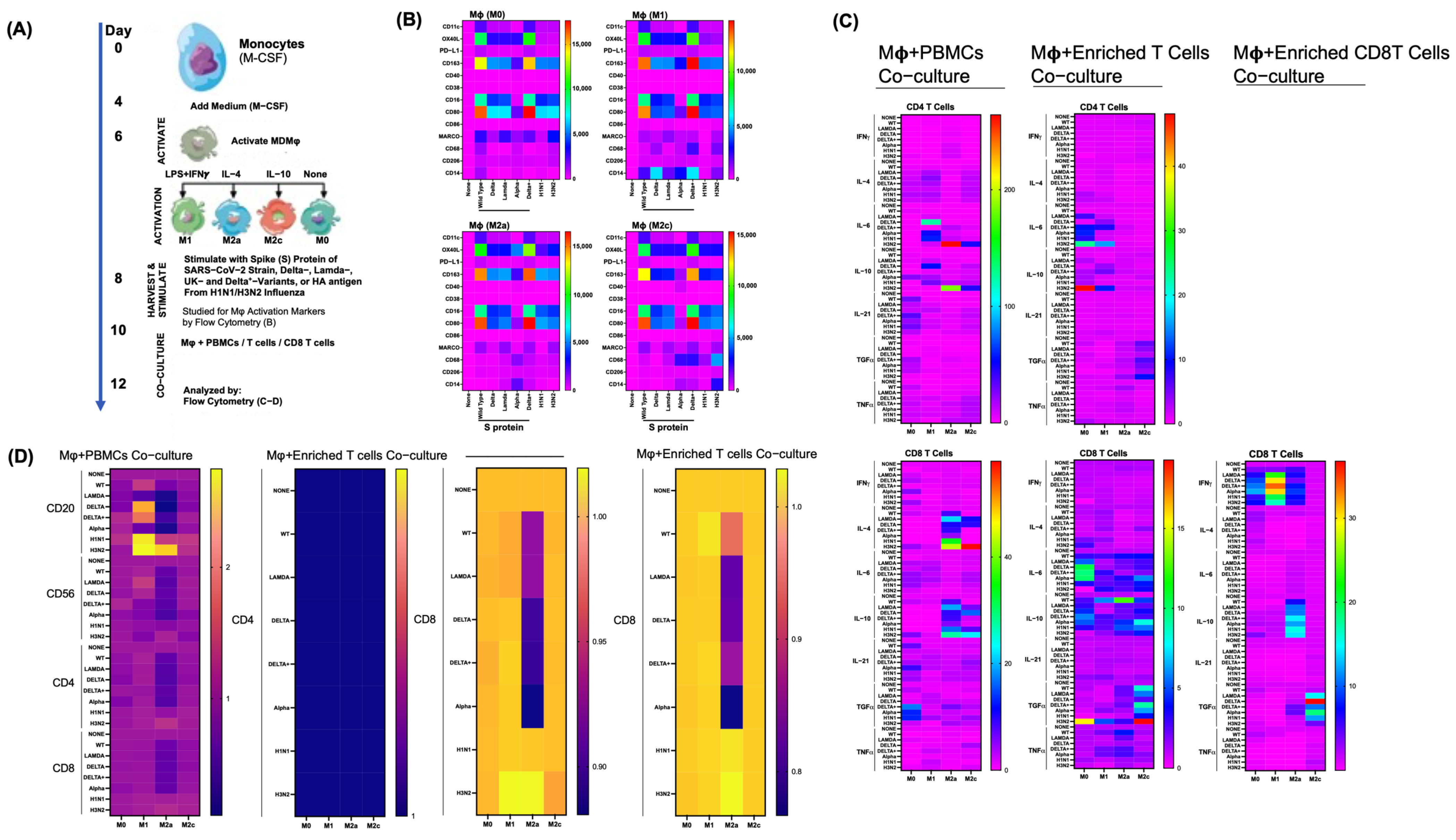
3.5. Differential Effects of Macrophage (M) Subtypes on SARS-CoV-2 Infection
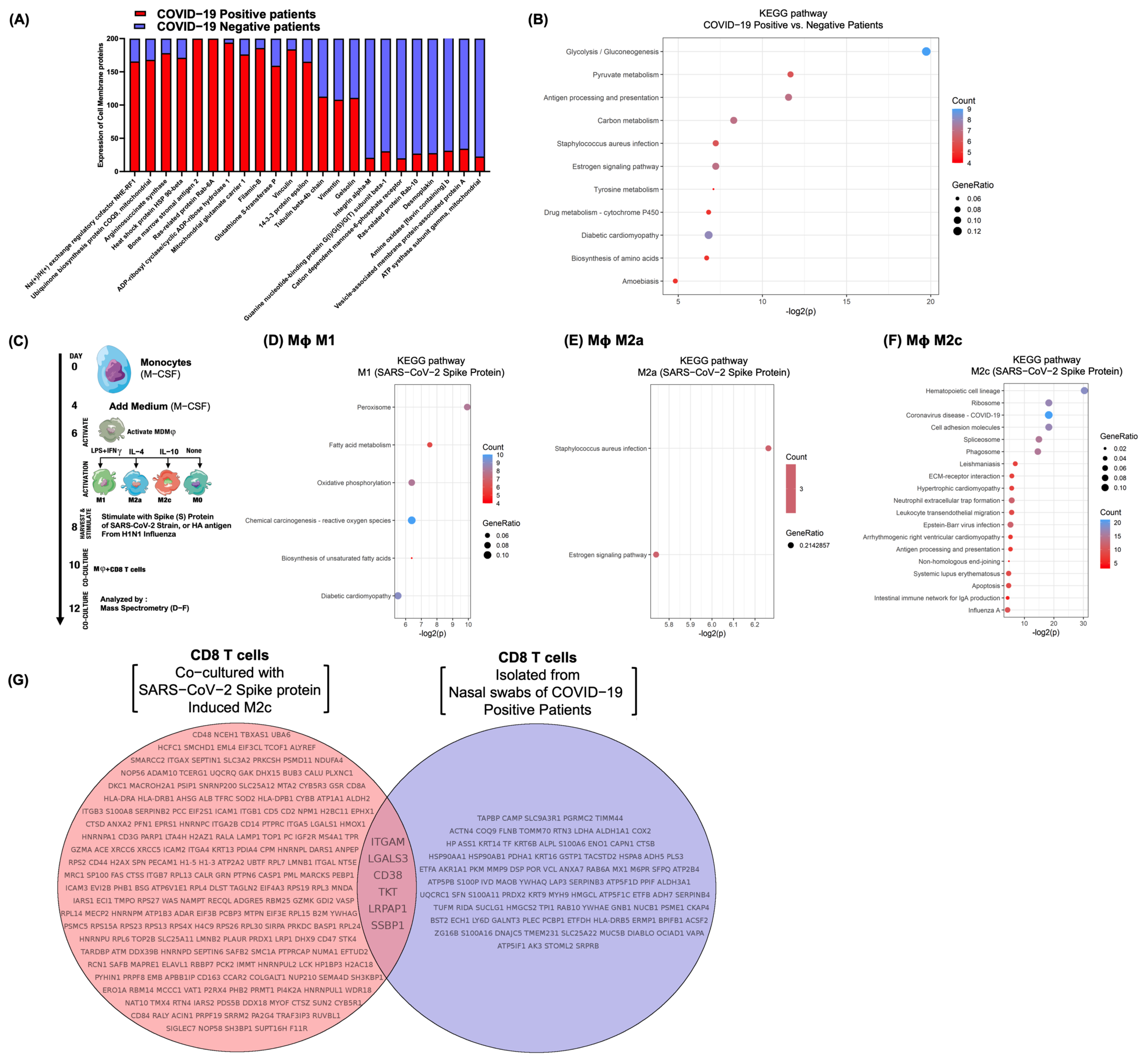
3.6. Differential Surfactome Expression on NP CD8 T-Lymphocytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lai, C.; Shih, T.; Ko, W.; Tang, H.; Hsueh, P. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef]
- Raveendran, A.; Jayadevan, R.; Sashidharan, S. Long COVID: An overview. Diabetes Metab. Syndr. Clin. Res. Rev. 2021, 15, 869–875. [Google Scholar] [CrossRef]
- Merad, M.; Blish, C.; Sallusto, F.; Iwasaki, A. The immunology and immunopathology of COVID-19. Science 2022, 375, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, Y.; Takeuchi, O.; Kato, H.; Kumar, H.; Matsui, K.; Morii, E.; Aozasa, K.; Kawai, T.; Akira, S. Alveolar macrophages are the primary interferon-α producer in pulmonary infection with RNA viruses. Immunity 2007, 27, 240–252. [Google Scholar] [CrossRef] [Green Version]
- Kasuga, Y.; Zhu, B.; Jang, K.; Yoo, J. Innate immune sensing of coronavirus and viral evasion strategies. Exp. Mol. Med. 2021, 53, 723–736. [Google Scholar] [CrossRef]
- Schultze, J.; Aschenbrenner, A. COVID-19 and the human innate immune system. Cell 2021, 184, 1671–1692. [Google Scholar] [CrossRef]
- Yoo, J.; Kato, H.; Fujita, T. Sensing viral invasion by RIG-I like receptors. Curr. Opin. Microbiol. 2014, 20, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Wathelet, M.; Orr, M.; Frieman, M.; Baric, R. Severe acute respiratory syndrome coronavirus evades antiviral signaling: Role of nsp1 and rational design of an attenuated strain. J. Virol. 2007, 81, 11620–11633. [Google Scholar] [CrossRef] [Green Version]
- Ratia, K.; Saikatendu, K.; Santarsiero, B.; Barretto, N.; Baker, S.; Stevens, R.; Mesecar, A. Severe acute respiratory syndrome coronavirus papain-like protease: Structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. USA 2006, 103, 5717–5722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sladkova, T.; Kostolansky, F. The role of cytokines in the immune response to influenza A virus infection. Acta Virol. 2006, 50, 151. [Google Scholar] [PubMed]
- Prokunina-Olsson, L.; Alphonse, N.; Dickenson, R.; Durbin, J.; Glenn, J.; Hartmann, R.; Kotenko, S.; Lazear, H.; O’Brien, T.; Odendall, C.; et al. COVID-19 and emerging viral infections: The case for interferon lambda. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [Green Version]
- Primorac, D.; Vrdoljak, K.; Brlek, P.; Pavelić, E.; Molnar, V.; Matišić, V.; Ivkošić, I.; Parčina, M. Adaptive Immune Responses and Immunity to SARS-CoV-2. Front. Immunol. 2022, 13, 848582. [Google Scholar] [CrossRef]
- Hue, S.; Beldi-Ferchiou, A.; Bendib, I.; Surenaud, M.; Fourati, S.; Frapard, T.; Rivoal, S.; Razazi, K.; Carteaux, G.; Delfau-Larue, M.; et al. Uncontrolled innate and impaired adaptive immune responses in patients with COVID-19 acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2020, 202, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Lyudovyk, O.; Kim, J.; Qualls, D.; Hwee, M.; Lin, Y.; Boutemine, S.; Elhanati, Y.; Solovyov, A.; Douglas, M.; Chen, E.; et al. Impaired humoral immunity is associated with prolonged COVID-19 despite robust CD8 T-cell responses. Cancer Cell 2022, 40, 738–753.e5. [Google Scholar] [CrossRef] [PubMed]
- Adamo, S.; Chevrier, S.; Cervia, C.; Zurbuchen, Y.; Raeber, M.; Yang, L.; Sivapatham, S.; Jacobs, A.; Baechli, E.; Rudiger, A.; et al. Profound dysregulation of T cell homeostasis and function in patients with severe COVID-19. Allergy 2021, 76, 2866–2881. [Google Scholar] [CrossRef]
- Gallo, O.; Locatello, L.; Mazzoni, A.; Novelli, L.; Annunziato, F. The central role of the nasal microenvironment in the transmission, modulation, and clinical progression of SARS-CoV-2 infection. Mucosal Immunol. 2021, 14, 305–316. [Google Scholar] [CrossRef]
- Hellings, P.; Steelant, B. Epithelial barriers in allergy and asthma. J. Allergy Clin. Immunol. 2020, 145, 1499–1509. [Google Scholar] [CrossRef]
- McAuley, J.; Corcilius, L.; Tan, H.; Payne, R.; McGuckin, M.; Brown, L. The cell surface mucin MUC1 limits the severity of influenza A virus infection. Mucosal Immunol. 2017, 10, 1581–1593. [Google Scholar] [CrossRef] [Green Version]
- Linden, S.; Sutton, P.; Karlsson, N.; Korolik, V.; McGuckin, M. Mucins in the mucosal barrier to infection. Mucosal Immunol. 2008, 1, 183–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, S.; Colonna, M. Innate lymphoid cells in mucosal immunity. Front. Immunol. 2019, 10, 861. [Google Scholar] [CrossRef] [Green Version]
- Roukens, A.; Pothast, C.; König, M.; Huisman, W.; Dalebout, T.; Tak, T.; Azimi, S.; Kruize, Y.; Hagedoorn, R.; Zlei, M.; et al. Prolonged activation of nasal immune cell populations and development of tissue-resident SARS-CoV-2-specific CD8+ T cell responses following COVID-19. Nat. Immunol. 2022, 23, 23–32. [Google Scholar] [CrossRef]
- Tomazic, P.; Darnhofer, B.; Birner-Gruenberger, R. Nasal mucus proteome and its involvement in allergic rhinitis. Expert Rev. Proteom. 2020, 17, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Ayass, M.; Cao, W.; Zhang, J.; Dai, J.; Zhu, K.; Tripathi, T.; Griko, N.; Pashkov, V.; Abi-Mosleh, L. Noninvasive nasopharyngeal proteomics of COVID-19 patient identify abnormalities related to complement and coagulation cascade and mucosal immune system. PLoS ONE 2022, 17, e0274228. [Google Scholar] [CrossRef]
- Ssemaganda, A.; Nguyen, H.; Nuhu, F.; Jahan, N.; Card, C.; Kiazyk, S.; Severini, G.; Keynan, Y.; Su, R.; Ji, H.; et al. Expansion of cytotoxic tissue-resident CD8+ T cells and CCR6+ CD161+ CD4+ T cells in the nasal mucosa following mRNA COVID-19 vaccination. Nat. Commun. 2022, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Jiang, J.; Li, J.; Zhou, C.; Chen, Y.; Lu, H.; He, F. The Characteristics of Microbiome and Cytokines in Healthy Implants and Peri-Implantitis of the Same Individuals. J. Clin. Med. 2022, 11, 5817. [Google Scholar] [CrossRef]
- Jochems, S.; Piddock, K.; Rylance, J.; Adler, H.; Carniel, B.; Collins, A.; Gritzfeld, J.; Hancock, C.; Hill, H.; Reine, J.; et al. Novel analysis of immune cells from nasal microbiopsy demonstrates reliable, reproducible data for immune populations, and superior cytokine detection compared to nasal wash. PLoS ONE 2017, 12, e0169805. [Google Scholar] [CrossRef]
- Hedley, B.; Cheng, G.; Keeney, M.; Kern, W.; Padurean, A.; Luider, J.; Chin-Yee, I.; Lowes, L.; Rohrbach, J.; Ortega, R.; et al. A multicenter study evaluation of the ClearLLab 10C panels. Cytom. Part B Clin. Cytom. 2021, 100, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Lurier, E.; Dalton, D.; Dampier, W.; Raman, P.; Nassiri, S.; Ferraro, N.; Rajagopalan, R.; Sarmady, M.; Spiller, K. Transcriptome analysis of IL-10-stimulated (M2c) macrophages by next-generation sequencing. Immunobiology 2017, 222, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Vuorela, A.; Freitag, T.; Leskinen, K.; Pessa, H.; Härkönen, T.; Stracenski, I.; Kirjavainen, T.; Olsen, P.; Saarenpää-Heikkilä, O.; Ilonen, J.; et al. Enhanced influenza A H1N1 T cell epitope recognition and cross-reactivity to protein-O-mannosyltransferase 1 in Pandemrix-associated narcolepsy type 1. Nat. Commun. 2021, 12, 2283. [Google Scholar] [CrossRef]
- Bispo, E.; Silva-Carvalho, A.; Sousa, M.; Neves, F.; Carvalho, J.; Arganaraz, E.; Saldanha-Araujo, F. Differential peripheral blood mononuclear cell reactivity against SARS-CoV-2 proteins in naive and previously infected subjects following COVID-19 vaccination. Clin. Immunol. Commun. 2022, 2, 172–176. [Google Scholar] [CrossRef]
- Bausch-Fluck, D.; Hofmann, A.; Bock, T.; Frei, A.; Cerciello, F.; Jacobs, A.; Moest, H.; Omasits, U.; Gundry, R.; Yoon, C.; et al. A mass spectrometric-derived cell surface protein atlas. PLoS ONE 2015, 10, e0121314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Doyle, W.; Skoner, D.; Gentile, D. Nasal cytokines as mediators of illness during the common cold. Curr. Allergy Asthma Rep. 2005, 5, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.; Nair, S.; Revai, K.; Grady, J.; Chonmaitree, T. Nasopharyngeal acute phase cytokines in viral upper respiratory infection: Impact on acute otitis media in children. Pediatr. Infect. Dis. J. 2009, 28, 1002. [Google Scholar] [CrossRef] [Green Version]
- Bian, J.; Nie, W.; Zang, Y.; Fang, Z.; Xiu, Q.; Xu, X. Clinical aspects and cytokine response in adults with seasonal influenza infection. Int. J. Clin. Exp. Med. 2014, 7, 5593. [Google Scholar]
- Lopez, S.; Shaikh, N.; Johnson, M.; Liu, H.; Martin, J.; Williams, J. Viral Coinfection and Nasal Cytokines in Children With Clinically Diagnosed Acute Sinusitis. Front. Pediatr. 2022, 9, 1563. [Google Scholar] [CrossRef]
- Lee, N.; Wong, C.; Chan, P.; Chan, M.; Wong, R.; Lun, S.; Ngai, K.; Lui, G.; Wong, B.; Lee, S.; et al. Cytokine response patterns in severe pandemic 2009 H1N1 and seasonal influenza among hospitalized adults. PLoS ONE 2011, 6, e26050. [Google Scholar] [CrossRef]
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020, 53, 25–32. [Google Scholar] [CrossRef]
- Bandopadhyay, P.; D’Rozario, R.; Lahiri, A.; Sarif, J.; Ray, Y.; Paul, S.; Roy, R.; Maiti, R.; Chaudhuri, K.; Bagchi, S.; et al. Nature and dimensions of systemic hyperinflammation and its attenuation by convalescent plasma in severe COVID-19. J. Infect. Dis. 2021, 224, 565–574. [Google Scholar] [CrossRef]
- Takahashi, T.; Ellingson, M.; Wong, P.; Israelow, B.; Lucas, C.; Klein, J.; Silva, J.; Mao, T.; Oh, J.; Tokuyama, M.; et al. Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature 2020, 588, 315–320. [Google Scholar] [CrossRef]
- Doerre, A.; Doblhammer, G. The influence of gender on COVID-19 infections and mortality in Germany: Insights from age-and gender-specific modeling of contact rates, infections, and deaths in the early phase of the pandemic. PLoS ONE 2022, 17, e0268119. [Google Scholar] [CrossRef]
- Jin, J.; Bai, P.; He, W.; Wu, F.; Liu, X.; Han, D.; Liu, S.; Yang, J. Gender differences in patients with COVID-19: Focus on severity and mortality. Front. Public Health 2020, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Ketteler, M.; Johnson, R.; Lindholm, B.; Pecoits-Filho, R.; Riella, M.; Heimbürger, O.; Cederholm, T.; Girndt, M. IL-10, IL-6, and TNF-α: Central factors in the altered cytokine network of uremia—The good, the bad, and the ugly. Kidney Int. 2005, 67, 1216–1233. [Google Scholar] [CrossRef] [Green Version]
- Zhong, W.; Jiang, Y.; Ma, H.; Wu, J.; Jiang, Z.; Zhao, L. Elevated levels of CCR6+ T helper 22 cells correlate with skin and renal impairment in systemic lupus erythematosus. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Galván-Peña, S.; Leon, J.; Chowdhary, K.; Michelson, D.; Vijaykumar, B.; Yang, L.; Magnuson, A.; Chen, F.; Manickas-Hill, Z.; Piechocka-Trocha, A.; et al. Profound Treg perturbations correlate with COVID-19 severity. Proc. Natl. Acad. Sci. USA 2021, 118, e2111315118. [Google Scholar] [CrossRef]
- Borthwick, N.; Lowdell, M.; Salmon, M.; Akbar, A. Loss of CD28 expression on CD8+ T cells is induced by IL-2 receptor γ chain signalling cytokines and type I IFN, and increases susceptibility to activation-induced apoptosis. Int. Immunol. 2000, 12, 1005–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Ménoret, A.; Nicholas, S.; Günther, S.; Sundberg, E.; Zhou, B.; Rodriguez, A.; Murphy, P.; Vella, A. Direct CD137 costimulation of CD8 T cells promotes retention and innate-like function within nascent atherogenic foci. Am. J. -Physiol.-Heart Circ. Physiol. 2019, 316, H1480–H1494. [Google Scholar] [CrossRef] [PubMed]
- Files, J.; Boppana, S.; Perez, M.; Sarkar, S.; Lowman, K.; Qin, K.; Sterrett, S.; Carlin, E.; Bansal, A.; Sabbaj, S.; et al. Sustained cellular immune dysregulation in individuals recovering from SARS-CoV-2 infection. J. Clin. Investig. 2021, 131, e140491. [Google Scholar] [CrossRef] [PubMed]
- Effros, R. Loss of CD28 expression on T lymphocytes: A marker of replicative senescence. Dev. Comp. Immunol. 1997, 21, 471–478. [Google Scholar] [CrossRef]
- Pontelli, M.; Castro, I.; Martins, R.; Veras, F.; La Serra, L.; Nascimento, D.; Cardoso, R.; Rosales, R.; Lima, T.; Souza, J.; et al. Infection of human lymphomononuclear cells by SARS-CoV-2. BioRxiv 2020. [Google Scholar] [CrossRef]
- Hsu, R.; Yu, W.; Peng, G.; Ye, C.; Hu, S.; Chong, P.; Yap, K.; Lee, J.; Lin, W.; Yu, S. The role of cytokines and chemokines in severe acute respiratory syndrome coronavirus 2 infections. Front. Immunol. 2022, 13, 832394. [Google Scholar] [CrossRef] [PubMed]
- Darif, D.; Hammi, I.; Kihel, A.; Saik, I.; Guessous, F.; Akarid, K. The pro-inflammatory cytokines in COVID-19 pathogenesis: What goes wrong? Microb. Pathog. 2021, 153, 104799. [Google Scholar] [CrossRef] [PubMed]
- Costela-Ruiz, V.; Illescas-Montes, R.; Puerta-Puerta, J.; Ruiz, C.; Melguizo-Rodrıiguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liu, J.; Zhang, D.; Xu, Z.; Ji, J.; Wen, C. Cytokine storm in COVID-19: The current evidence and treatment strategies. Front. Immunol. 2020, 11, 1708. [Google Scholar] [CrossRef]
- Merad, M.; Martin, J. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef]
- Feng, Z.; Diao, B.; Wang, R.; Wang, G.; Wang, C.; Tan, Y.; Liu, L.; Wang, C.; Liu, Y.; Liu, Y.; et al. The novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) directly decimates human spleens and lymph nodes. MedRxiv, 2020; in press. [Google Scholar] [CrossRef] [Green Version]
- Torres-Castro, I.; Arroyo-Camarena, U.; Martıinez-Reyes, C.; Gomez-Arauz, A.; Duenas-Andrade, Y.; Hern ez-Ruiz, J.; Bejar, Y.; Zaga-Clavellina, V.; Morales-Montor, J.; Terrazas, L.; et al. Human monocytes and macrophages undergo M1-type inflammatory polarization in response to high levels of glucose. Immunol. Lett. 2016, 176, 81–89. [Google Scholar] [CrossRef]
- Lee, J.; Boyce, S.; Powers, J.; Baer, C.; Sassetti, C.; Behar, S. CD11cHi monocyte-derived macrophages are a major cellular compartment infected by Mycobacterium tuberculosis. PLoS Pathog. 2020, 16, e1008621. [Google Scholar] [CrossRef]
- Palipane, M.; Snyder, J.; LeMessurier, K.; Schofield, A.; Woolard, S.; Samarasinghe, A. Macrophage CD14 impacts immune defenses against influenza virus in allergic hosts. Microb. Pathog. 2019, 127, 212–219. [Google Scholar] [CrossRef]
- Perussia, B.; Ravetch, J. FcγRIII (CD16) on human macrophages is a functional product of the FcγRIII-2 gene. Eur. J. Immunol. 1991, 21, 425–429. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, H.; Fingerle, G.; Ströbel, M.; Schraut, W.; Stelter, F.; Schütt, C.; Passlick, B.; Pforte, A. The novel subset of CD14+/CD16+ blood monocytes exhibits features of tissue macrophages. Eur. J. Immunol. 1993, 23, 2053–2058. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Kitakawa, M.; Koshikawa, S.; Watanabe, H.; Sueki, H. CD14 and CD16 expression in noninfectious granulomatous skin diseases. J. Cutan. Immunol. Allergy 2020, 3, 10–16. [Google Scholar] [CrossRef]
- Dong, L.; Wang, S.; Chen, M.; Li, H.; Bi, W. The activation of macrophage and upregulation of CD40 costimulatory molecule in lipopolysaccharide-induced acute lung injury. J. Biomed. Biotechnol. 2008, 2008, 852571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niles, M.; Gogesch, P.; Kronhart, S.; Ortega, I.S.; Kochs, G.; Waibler, Z.; Anzaghe, M. Macrophages and dendritic cells are not the major source of pro-inflammatory cytokines upon SARS-CoV-2 infection. Front. Immunol. 2021, 12, 647824. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Park, M.; Del Valle, D.; Buckup, M.; Tabachnikova, A.; Simons, N.; Mouskas, K.; Lee, B.; Geanon, D.; D’Souza, D.; et al. Shift of lung macrophage composition is associated with COVID-19 disease severity and recovery. BioRxiv 2022. [Google Scholar] [CrossRef]
- Salina, A.; Santos, D.; Rodrigues, T.; Fortes-Rocha, M.; Freitas-Filho, E.; Alzamora-Terrel, D.; Castro, I.; Fraga, T.; Lima, M.; Nascimento, D.; et al. Efferocytosis of SARS-CoV-2-infected dying cells impairs macrophage anti-inflammatory programming and continual clearance of apoptotic cells. eLife 2022, 11, e74443. [Google Scholar] [CrossRef] [PubMed]
- Julia, A.; Bonafonte-Pardas, I.; Gomez, A.; Lopez-Lasanta, M.; Lopez-Corbeto, M.; Martinez-Mateu, S.; Llados, J.; Rodriguez-Nunez, I.; Myers, R.; Marsal, S. Targeting of the CD80/86 proinflammatory axis as a therapeutic strategy to prevent severe COVID-19. Sci. Rep. 2021, 11, 1–14. [Google Scholar] [CrossRef]
- Horenstein, A.; Faini, A.; Malavasi, F. CD38 in the age of COVID-19: A medical perspective. Physiol. Rev. 2021, 101, 1457–1486. [Google Scholar] [CrossRef]
- Amici, S.; Young, N.; Narvaez-Mir, J.; Jablonski, K.; Arcos, J.; Rosas, L.; Papenfuss, T.; Torrelles, J.; Jarjour, W.; Arellano, M. CD38 is robustly induced in human macrophages and monocytes in inflammatory conditions. Front. Immunol. 2018, 9, 1593. [Google Scholar] [CrossRef] [Green Version]
- Lam, J.; Ng, H.; Lim, C.; Sim, X.; Malavasi, F.; Li, H.; Loh, J.; Sabai, K.; Kim, J.; Ong, C.; et al. Expression of CD38 on macrophages predicts improved prognosis in hepatocellular carcinoma. Front. Immunol. 2019, 10, 2093. [Google Scholar] [CrossRef] [Green Version]
- Bonam, S.; Chauvin, C.; Mathew, M.; Bayry, J. IFN-γ Induces PD-L1 Expression in Primed Human Basophils. Cells 2022, 11, 801. [Google Scholar] [CrossRef]
- Kammala, A.; Syed, M.; Yang, C.; Occhiuto, C.; Subramanian, H. A Critical Role for Na+/H+ Exchanger Regulatory Factor 1 in Modulating FcεRI-Mediated Mast Cell Activation. J. Immunol. 2021, 206, 471–480. [Google Scholar] [CrossRef]
- Zhang, Q.; Gefter, J.; Sneddon, W.; Mamonova, T.; Friedman, P. ACE2 interaction with cytoplasmic PDZ protein enhances SARS-CoV-2 invasion. IScience 2021, 24, 102770. [Google Scholar] [CrossRef]
- Abdel, H.R.; Cormet-Boyaka, E.; Kuebler, W.; Uddin, M.; Berdiev, B. SARS-CoV-2 may hijack GPCR signaling pathways to dysregulate lung ion and fluid transport. Am. J. -Physiol.-Lung Cell. Mol. Physiol. 2021, 320, L430–L435. [Google Scholar] [CrossRef]
- Fernández-Ayala, D.; Navas, P.; Lopez-Lluch, G. Age-related mitochondrial dysfunction as a key factor in COVID-19 disease. Exp. Gerontol. 2020, 142, 111147. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Camacho, J.; Garcia-Corzo, L.; Fernández-Ayala, D.; Navas, P.; Lopez-Lluch, G. Coenzyme Q at the Hinge of Health and Metabolic Diseases. Antioxidants 2021, 10, 1785. [Google Scholar] [CrossRef]
- Durante, W. Targeting Arginine in COVID-19-Induced Immunopathology and Vasculopathy. Metabolites 2022, 12, 240. [Google Scholar] [CrossRef] [PubMed]
- Wyler, E.; Mösbauer, K.; Franke, V.; Diag, A.; Gottula, L.; Arsiè, R.; Klironomos, F.; Koppstein, D.; Hönzke, K.; Ayoub, S.; et al. Transcriptomic profiling of SARS-CoV-2 infected human cell lines identifies HSP90 as target for COVID-19 therapy. IScience 2021, 24, 102151. [Google Scholar] [CrossRef] [PubMed]
- Russo, C.; Morello, G.; Malaguarnera, R.; Piro, S.; Furno, D.; Malaguarnera, L. Candidate genes of SARS-CoV-2 gender susceptibility. Sci. Rep. 2021, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.; Lareau, C.; Keshishian, H.; Ganskih, S.; Schneider, C.; Hennig, T.; Melanson, R.; Werner, S.; Wei, Y.; Zimmer, M.; et al. The SARS-CoV-2 RNA–protein interactome in infected human cells. Nat. Microbiol. 2021, 6, 339–353. [Google Scholar] [CrossRef]
- Park, J.; Kim, H.; Kim, S.; Kim, Y.; Lee, J.; Dan, K.; Seong, M.; Han, D. In-depth blood proteome profiling analysis revealed distinct functional characteristics of plasma proteins between severe and non-severe COVID-19 patients. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Guarnieri, J.; Dybas, J.; Fazelinia, H.; Kim, M.; Frere, J.; Zhang, Y.; Albrecht, Y.; Murdock, D.; Angelin, A.; Singh, L.; et al. Targeted down regulation of core mitochondrial genes during SARS-CoV-2 infection. BioRxiv, 2022; preprint. [Google Scholar] [CrossRef]
- Srivastava, R.; Daulatabad, S.; Srivastava, M.; Janga, S. Role of SARS-CoV-2 in altering the RNA-binding protein and miRNA-directed post-transcriptional regulatory networks in humans. Int. J. Mol. Sci. 2020, 21, 7090. [Google Scholar] [CrossRef] [PubMed]
- Ahern, D.; Ai, Z.; Ainsworth, M.; Allan, C.; Allcock, A.; Ansari, A.; Arancibia-Carcamo, C.; Aschenbrenner, D.; Attar, M.; Baillie, J.; et al. A blood atlas of COVID-19 defines hallmarks of disease severity and specificity. MedRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.; Morham, S.; Walsh, D.; Naghavi, M. Focal adhesion proteins talin-1 and vinculin negatively affect paxillin phosphorylation and limit retroviral infection. J. Mol. Biol. 2011, 410, 761–777. [Google Scholar] [CrossRef]
- Nathan, K.; Lal, S. The multifarious role of 14–3-3 family of proteins in viral replication. Viruses 2020, 12, 436. [Google Scholar] [CrossRef] [Green Version]
- Souza, D.; Guest, P.; Mann, D.; Roeber, S.; Rahmoune, H.; Bauder, C.; Kretzschmar, H.; Volk, B.; Baborie, A.; Bahn, S. Proteomic analysis identifies dysfunction in cellular transport, energy, and protein metabolism in different brain regions of atypical frontotemporal lobar degeneration. J. Proteome Res. 2012, 11, 2533–2543. [Google Scholar] [CrossRef]
- Chan, C.; Wong, S.; Lam, M.; Hui, E.; Chan, J.; Lo, E.; Cheuk, W.; Wong, M.; Tsao, S.; Chan, A. Proteomic comparison of nasopharyngeal cancer cell lines C666-1 and NP69 identifies down-regulation of annexin II and β2-tubulin for nasopharyngeal carcinoma. Arch. Pathol. Lab. Med. 2008, 132, 675–683. [Google Scholar] [CrossRef]
- Danielsson, F.; Peterson, M.; Caldeira, A.H.; Lautenschläger, F.; Gad, A. Vimentin diversity in health and disease. Cells 2018, 7, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Paulin, D.; Lacolley, P.; Coletti, D.; Agbulut, O. Vimentin as a target for the treatment of COVID-19. BMJ Open Respir. Res. 2020, 7, e000623. [Google Scholar] [CrossRef] [PubMed]
- Amraei, R.; Xia, C.; Olejnik, J.; White, M.; Napoleon, M.; Lotfollahzadeh, S.; Hauser, B.; Schmidt, A.; Chitalia, V.; Mühlberger, E.; et al. Extracellular vimentin is an attachment factor that facilitates SARS-CoV-2 entry into human endothelial cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2113874119. [Google Scholar] [CrossRef]
- Saadat, M. The morbidity and mortality of COVID-19 are correlated with the Ile105Val glutathione S-transferase P1 polymorphism. Egypt. J. Med Hum. Genet. 2020, 21, 1–5. [Google Scholar] [CrossRef]
- Djukic, T.; Stevanovic, G.; Coric, V.; Bukumiric, Z.; Pljesa-Ercegovac, M.; Matic, M.; Jerotic, D.; Todorovic, N.; Asanin, M.; Ercegovac, M.; et al. GSTO1, GSTO2 and ACE2 Polymorphisms Modify Susceptibility to Developing COVID-19. J. Pers. Med. 2022, 12, 458. [Google Scholar] [CrossRef]
- Coric, V.; Milosevic, I.; Djukic, T.; Bukumiric, Z.; Savic-Radojevic, A.; Matic, M.; Jerotic, D.; Todorovic, N.; Asanin, M.; Ercegovac, M.; et al. GSTP1 and GSTM3 variant alleles affect susceptibility and severity of COVID-19. Front. Mol. Biosci. 2021, 8, 747493. [Google Scholar] [CrossRef]
- Simons, P.; Rinaldi, D.; Bondu, V.; Kell, A.; Bradfute, S.; Lidke, D.; Buranda, T. Integrin activation is an essential component of SARS-CoV-2 infection. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Atik, N.; Wirawan, F.; Amalia, R.; Khairani, A.; Pradini, G. Differences in endosomal Rab gene expression between positive and negative COVID-19 patients. BMC Res. Notes 2022, 15, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Cheng, L.; Wang, Z.; Wu, Y. Desmoplakin and clinical manifestations of desmoplakin cardiomyopathy. Chin. Med. J. 2021, 134, 1771–1779. [Google Scholar] [CrossRef] [PubMed]
- Hok, L.; Rimac, H.; Mavri, J.; Vianello, R. COVID-19 infection and neurodegeneration: Computational evidence for interactions between the SARS-CoV-2 spike protein and monoamine oxidase enzymes. Comput. Struct. Biotechnol. J. 2022, 20, 1254–1263. [Google Scholar] [CrossRef]
- Wyles, J.; McMaster, C.; Ridgway, N. Vesicle-associated membrane protein-associated protein-A (VAP-A) interacts with the oxysterol-binding protein to modify export from the endoplasmic reticulum. J. Biol. Chem. 2002, 277, 29908–29918. [Google Scholar] [CrossRef] [Green Version]
- Galber, C.; Carissimi, S.; Baracca, A.; Giorgio, V. The ATP synthase deficiency in human diseases. Life 2021, 11, 325. [Google Scholar] [CrossRef]
- Panfoli, I.; Esposito, A. Beneficial effect of polyphenols in COVID-19 and the ectopic F1FO-ATP synthase: Is there a link? J. Cell. Biochem. 2022, 123, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, S.; Nordqvist, H.; Ambikan, A.; Gupta, S.; Sperk, M.; Svensson-Akusjärvi, S.; Mikaeloff, F.; Benfeitas, R.; Saccon, E.; Ponnan, S.; et al. Metabolic perturbation associated with COVID-19 disease severity and SARS-CoV-2 replication. Mol. Cell. Proteom. 2021, 20, 100159. [Google Scholar] [CrossRef] [PubMed]
- Ceperuelo-Mallafré, V.; Reverté, L.; Peraire, J.; Madeira, A.; Maymo-Masip, E.; Lopez-Dupla, M.; Gutierrez-Valencia, A.; Ruiz-Mateos, E.; Buzon, M.; Jorba, R.; et al. Circulating pyruvate is a potent prognostic marker for critical COVID-19 outcomes. Front Immunol. 2022, 13, 912579. [Google Scholar] [CrossRef]
- Yoo, J.; Sasaki, M.; Cho, S.; Kasuga, Y.; Zhu, B.; Ouda, R.; Orba, Y.; Figueiredo, P.; Sawa, H.; Kobayashi, K. SARS-CoV-2 inhibits induction of the MHC class I pathway by targeting the STAT1-IRF1-NLRC5 axis. Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Choudhury, I.; Han, H.; Manthani, K.; Gandhi, S.; Dabhi, R. COVID-19 as a possible cause of functional exhaustion of CD4 and CD8 T-cells and persistent cause of methicillin-sensitive Staphylococcus aureus bacteremia. Cureus 2020, 12, e9000. [Google Scholar] [CrossRef]
- Abramenko, N.; Vellieux, F.; Tesařová, P.; Kejik, Z.; Kaplánek, R.; Lacina, L.; Dvořánková, B.; Rösel, D.; Brábek, J.; Tesař, A.; et al. Estrogen Receptor Modulators in Viral Infections Such as SARS-CoV-2: Therapeutic Consequences. Int. J. Mol. Sci. 2021, 22, 6551. [Google Scholar] [CrossRef]
- Ansone, L.; Briviba, M.; Silamikelis, I.; Terentjeva, A.; Perkons, I.; Birzniece, L.; Rovite, V.; Rozentale, B.; Viksna, L.; Kolesova, O.; et al. Amino Acid Metabolism is Significantly Altered at the Time of Admission in Hospital for Severe COVID-19 Patients: Findings from Longitudinal Targeted Metabolomics Analysis. Microbiol. Spectr. 2021, 9, e00338-21. [Google Scholar] [CrossRef]
- Deb, S.; Arrighi, S. Potential effects of COVID-19 on cytochrome P450-mediated drug metabolism and disposition in infected patients. Eur. J. Drug Metab. Pharmacokinet. 2021, 46, 185–203. [Google Scholar] [CrossRef]
- Aluganti Narasimhulu, C.; Singla, D. Mechanisms of COVID-19 pathogenesis in diabetes. Am. J. -Physiol.-Heart Circ. Physiol. 2022, 323, H403–H420. [Google Scholar] [CrossRef]
- Dorantes, J.; Lopez-Becerril, J.; Zavala-Cerna, M. Fatal attraction: Intestinal amebiasis and COVID-19 as risk factors for colonic perforation. J. Surg. Case Rep. 2021, 2021, rjab301. [Google Scholar] [CrossRef]
- Consortium, G. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef] [Green Version]
- Mukund, K.; Nayak, P.; Ashokkumar, C.; Rao, S.; Almeda, J.; Betancourt-Garcia, M.; Sindhi, R.; Subramaniam, S. Immune Response in Severe and Non-Severe Coronavirus Disease 2019 (COVID-19) Infection: A Mechanistic Landscape. Front. Immunol. 2021, 12, 738073. [Google Scholar] [CrossRef] [PubMed]
- Daamen, A.; Bachali, P.; Owen, K.; Kingsmore, K.; Hubbard, E.; Labonte, A.; Robl, R.; Shrotri, S.; Grammer, A.; Lipsky, P. Comprehensive transcriptomic analysis of COVID-19 blood, lung, and airway. Sci. Rep. 2021, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.; Baraniuk, J. Understanding COVID-19 Pathogenesis: A Drug-Repurposing Effort to Disrupt Nsp-1 Binding to Export Machinery Receptor Complex. Pathogens 2021, 10, 1634. [Google Scholar] [CrossRef]
- Bojkova, D.; Costa, R.; Reus, P.; Bechtel, M.; Jaboreck, M.; Olmer, R.; Martin, U.; Ciesek, S.; Michaelis, M.; Cinatl, J., Jr. Targeting the pentose phosphate pathway for SARS-CoV-2 therapy. Metabolites 2021, 11, 699. [Google Scholar] [CrossRef]
- Beltrami, A.; De Martino, M.; Dalla, E.; Malfatti, M.; Caponnetto, F.; Codrich, M.; Stefanizzi, D.; Fabris, M.; Sozio, E.; D’Aurizio, F.; et al. Combining Deep Phenotyping of Serum Proteomics and Clinical Data via Machine Learning for COVID-19 Biomarker Discovery. Int. J. Mol. Sci. 2022, 23, 9161. [Google Scholar] [CrossRef]
- Jiang, H.; Sun, H.; Gao, S.; Li, L.; Huang, S.; Hu, X.; Liu, S.; Wu, J.; Shao, Z.; Jin, W. SSBP1 Suppresses TGFβ-Driven Epithelial-to-Mesenchymal Transition and Metastasis in Triple-Negative Breast Cancer by Regulating Mitochondrial Retrograde SignalingSSBP1 Suppresses Triple-Negative Breast Cancer Metastasis. Cancer Res. 2016, 76, 952–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broos, C.; Koth, L.; Nimwegen, M.; Paulissen, S.; Hamburg, J.; Annema, J.; Heller-Baan, R.; Kleinjan, A.; Hoogsteden, H.; Wijsenbeek, M.; et al. Increased T-helper 17.1 cells in sarcoidosis mediastinal lymph nodes. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef] [Green Version]
- Ramstein, J.; Broos, C.; Simpson, L.; Ansel, K.; Sun, S.; Ho, M.; Woodruff, P.; Bhakta, N.; Christian, L.; Nguyen, C.; et al. IFN-γ–producing T-helper 17.1 cells are increased in sarcoidosis and are more prevalent than T-helper type 1 cells. Am. J. Respir. Crit. Care Med. 2016, 193, 1281–1291. [Google Scholar] [CrossRef] [Green Version]
- Richard, M.; Br, J.; Bestebroer, T.; Lexmond, P.; Meulder, D.; Fouchier, R.; Lowen, A.; Herfst, S. Influenza A viruses are transmitted via the air from the nasal respiratory epithelium of ferrets. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lehtinen, M.; Hibberd, A.; Männikkö, S.; Yeung, N.; Kauko, T.; Forssten, S.; Lehtoranta, L.; Lahtinen, S.; Stahl, B.; Lyra, A.; et al. Nasal microbiota clusters associate with inflammatory response, viral load, and symptom severity in experimental rhinovirus challenge. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nochi, T.; Kiyono, H. Innate immunity in the mucosal immune system. Curr. Pharm. Des. 2006, 12, 4203–4213. [Google Scholar] [CrossRef] [PubMed]
- Giavina-Bianchi, P.; Aun, M.; Takejima, P.; Kalil, J.; Agondi, R. United airway disease: Current perspectives. J. Asthma Allergy 2016, 9, 93. [Google Scholar] [CrossRef] [Green Version]
- Hua, X.; Vijay, R.; Channappanavar, R.; Athmer, J.; Meyerholz, D.; Pagedar, N.; Tilley, S.; Perlman, S. Nasal priming by a murine coronavirus provides protective immunity against lethal heterologous virus pneumonia. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Caldeira-Dantas, S.; Smith, C.; Snyder, C. Persistent viral replication and the development of T-cell responses after intranasal infection by MCMV. Med Microbiol. Immunol. 2019, 208, 457–468. [Google Scholar] [CrossRef]
- Smith, N.; Goncalves, P.; Charbit, B.; Grzelak, L.; Beretta, M.; Planchais, C.; Bruel, T.; Rouilly, V.; Bondet, V.; Hadjadj, J.; et al. Distinct systemic and mucosal immune responses during acute SARS-CoV-2 infection. Nat. Immunol. 2021, 22, 1428–1439. [Google Scholar] [CrossRef]
- Yu, Y.; Kong, W.; Yin, Y.; Dong, F.; Huang, Z.; Yin, G.; Dong, S.; Salinas, I.; Zhang, Y.; Xu, Z. Mucosal immunoglobulins protect the olfactory organ of teleost fish against parasitic infection. PLoS Pathog. 2018, 14, e1007251. [Google Scholar] [CrossRef] [Green Version]
- Ter Horst, R.; Jaeger, M.; Smeekens, S.; Oosting, M.; Swertz, M.; Li, Y.; Kumar, V.; Diavatopoulos, D.; Jansen, A.; Lemmers, H.; et al. Host and environmental factors influencing individual human cytokine responses. Cell 2016, 167, 1111–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, M. Cytokines, neurophysiology, neuropsychology, and psychiatric symptoms. Dialogues Clin. Neurosci. 2022, 5, 139–153. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayass, M.A.; Tripathi, T.; Griko, N.; Ramankutty Nair, R.; Zhang, J.; Zhu, K.; Cao, W.; Pashkov, V.; Okyay, T.; Singh, S.K.; et al. Macrophage-Induced Exacerbation of Nasopharyngeal Inflammatory Lymphocytes in COVID-19 Disease. COVID 2023, 3, 567-591. https://doi.org/10.3390/covid3040041
Ayass MA, Tripathi T, Griko N, Ramankutty Nair R, Zhang J, Zhu K, Cao W, Pashkov V, Okyay T, Singh SK, et al. Macrophage-Induced Exacerbation of Nasopharyngeal Inflammatory Lymphocytes in COVID-19 Disease. COVID. 2023; 3(4):567-591. https://doi.org/10.3390/covid3040041
Chicago/Turabian StyleAyass, Mohamad Ammar, Trivendra Tripathi, Natalya Griko, Ramya Ramankutty Nair, Jin Zhang, Kevin Zhu, Wanying Cao, Victor Pashkov, Tutku Okyay, Sharda Kalla Singh, and et al. 2023. "Macrophage-Induced Exacerbation of Nasopharyngeal Inflammatory Lymphocytes in COVID-19 Disease" COVID 3, no. 4: 567-591. https://doi.org/10.3390/covid3040041
APA StyleAyass, M. A., Tripathi, T., Griko, N., Ramankutty Nair, R., Zhang, J., Zhu, K., Cao, W., Pashkov, V., Okyay, T., Singh, S. K., & Abi-Mosleh, L. (2023). Macrophage-Induced Exacerbation of Nasopharyngeal Inflammatory Lymphocytes in COVID-19 Disease. COVID, 3(4), 567-591. https://doi.org/10.3390/covid3040041