
The Peptide TAT-I24 with Antiviral Activity against DNA Viruses Binds Double-Stranded DNA with High Affinity

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
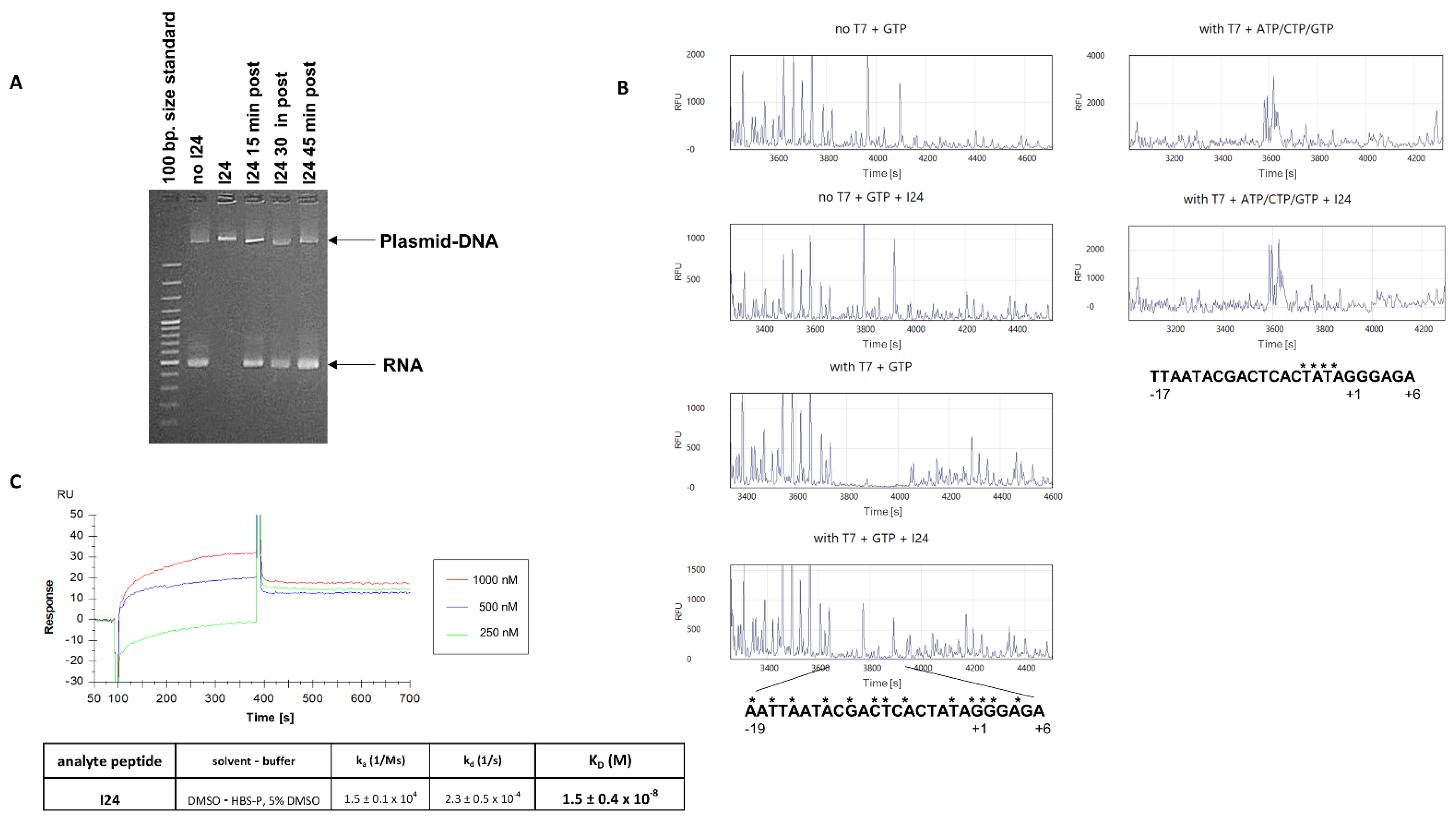
2.1. The 9-mer Peptide I24 Blocks RNA Synthesis and Initiation Complex Formation by T7 RNA Polymerase
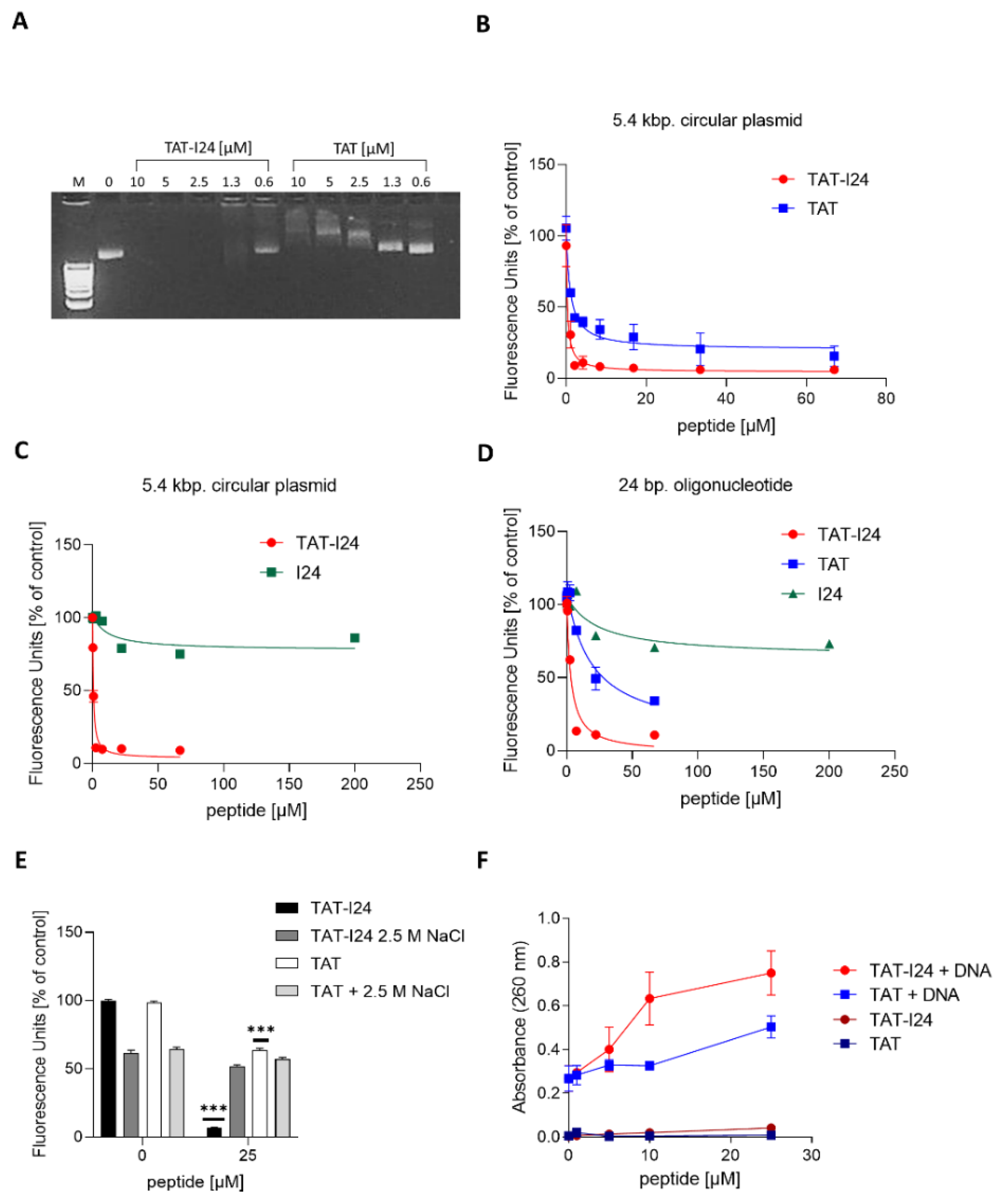
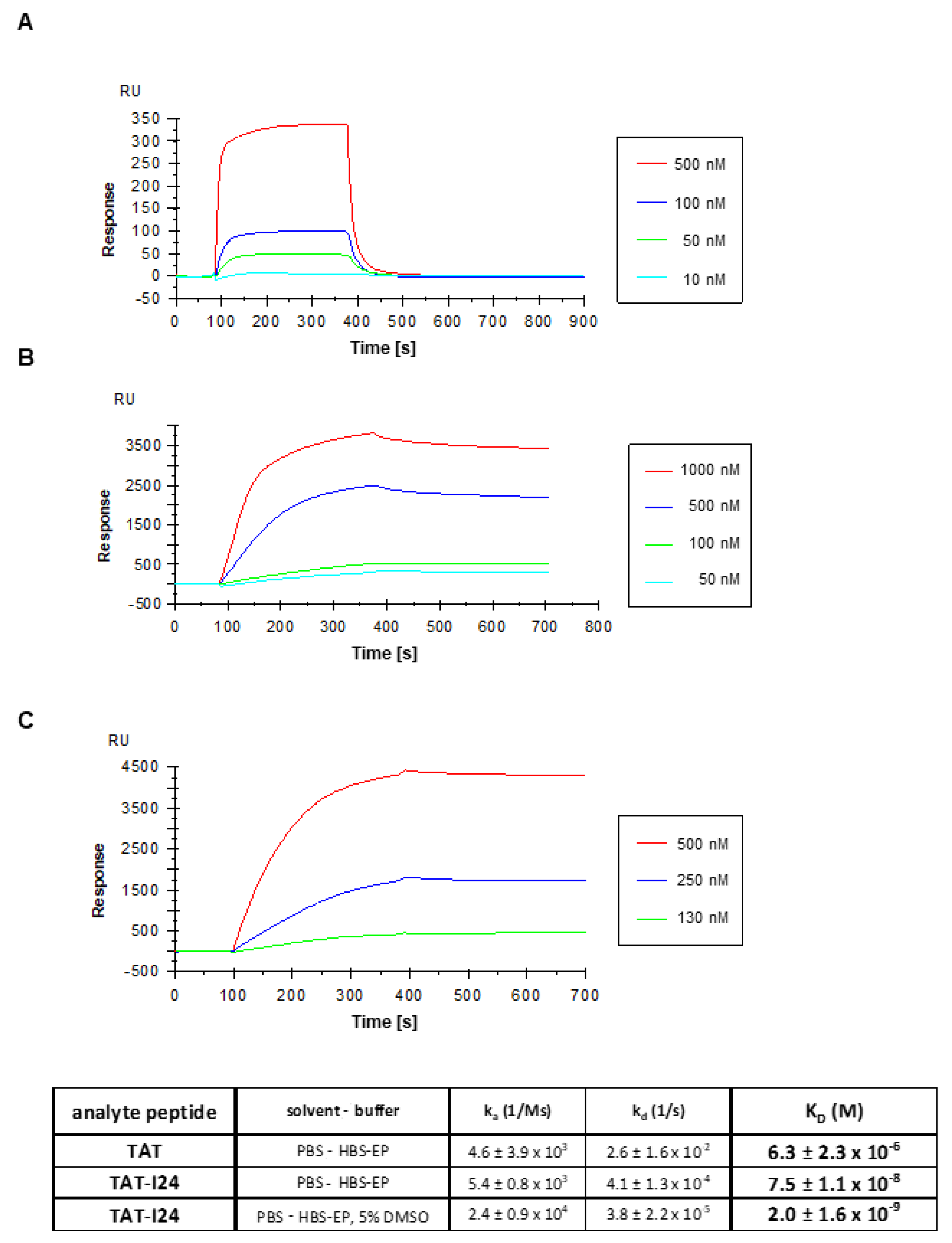
2.2. The Fusion of Peptide I24 to the TAT Peptide Binds Double-Stranded DNA with High Affinity
2.3. TAT-I24 Blocks Activity of Taq DNA Polymerase from Double-Stranded DNA
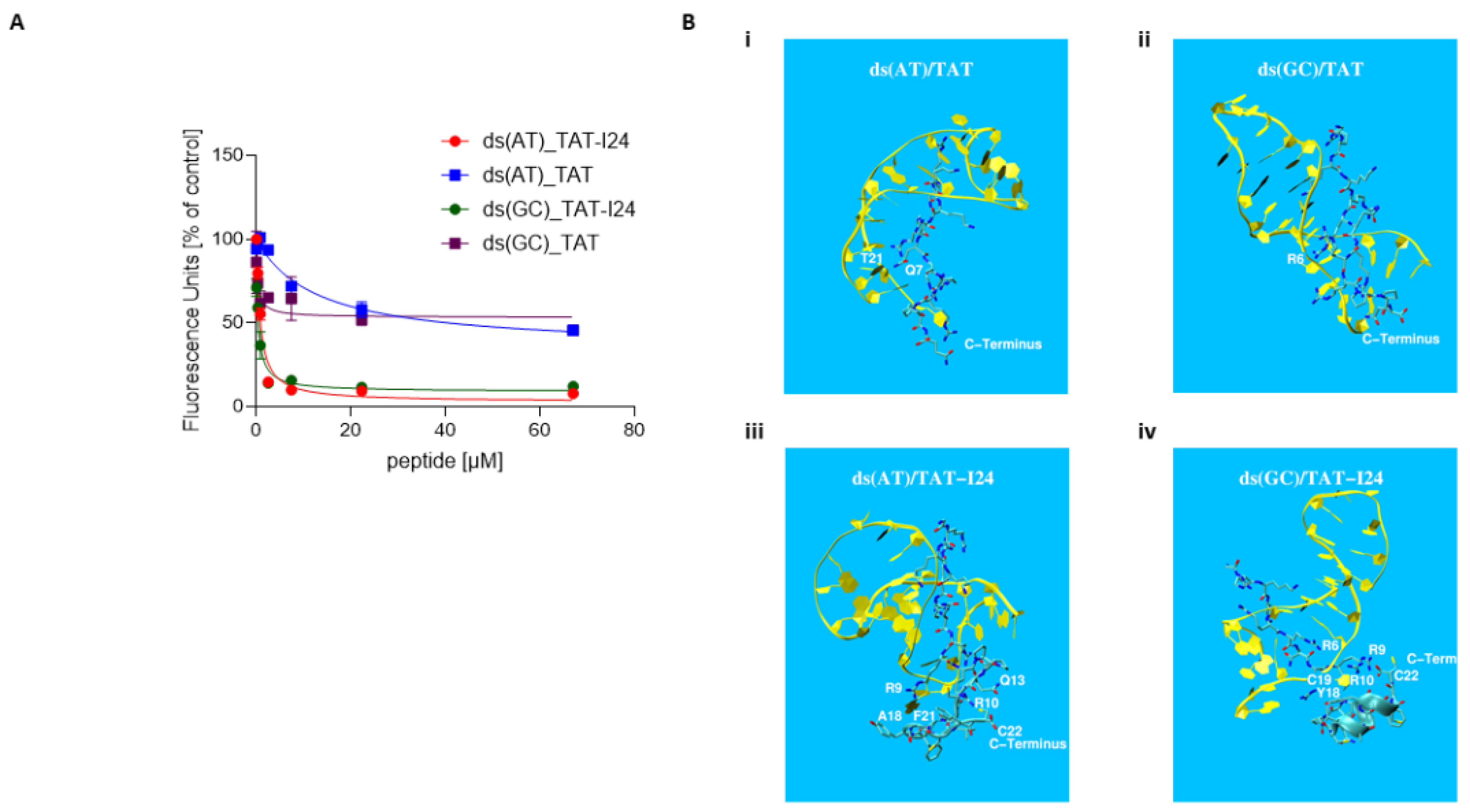
2.4. Fluorescence Measurement and Molecular Modelling of the Peptide-DNA Complexes
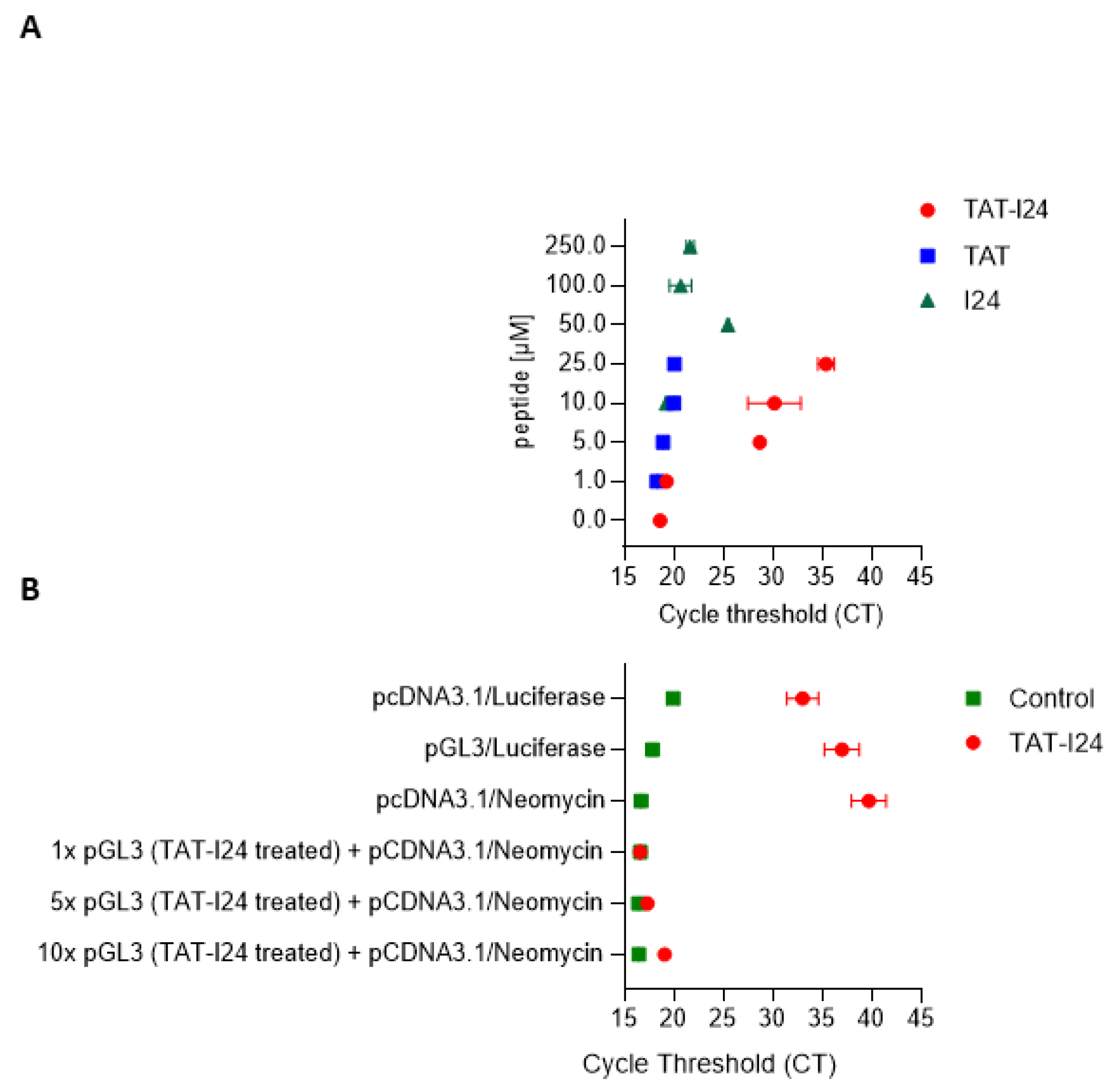
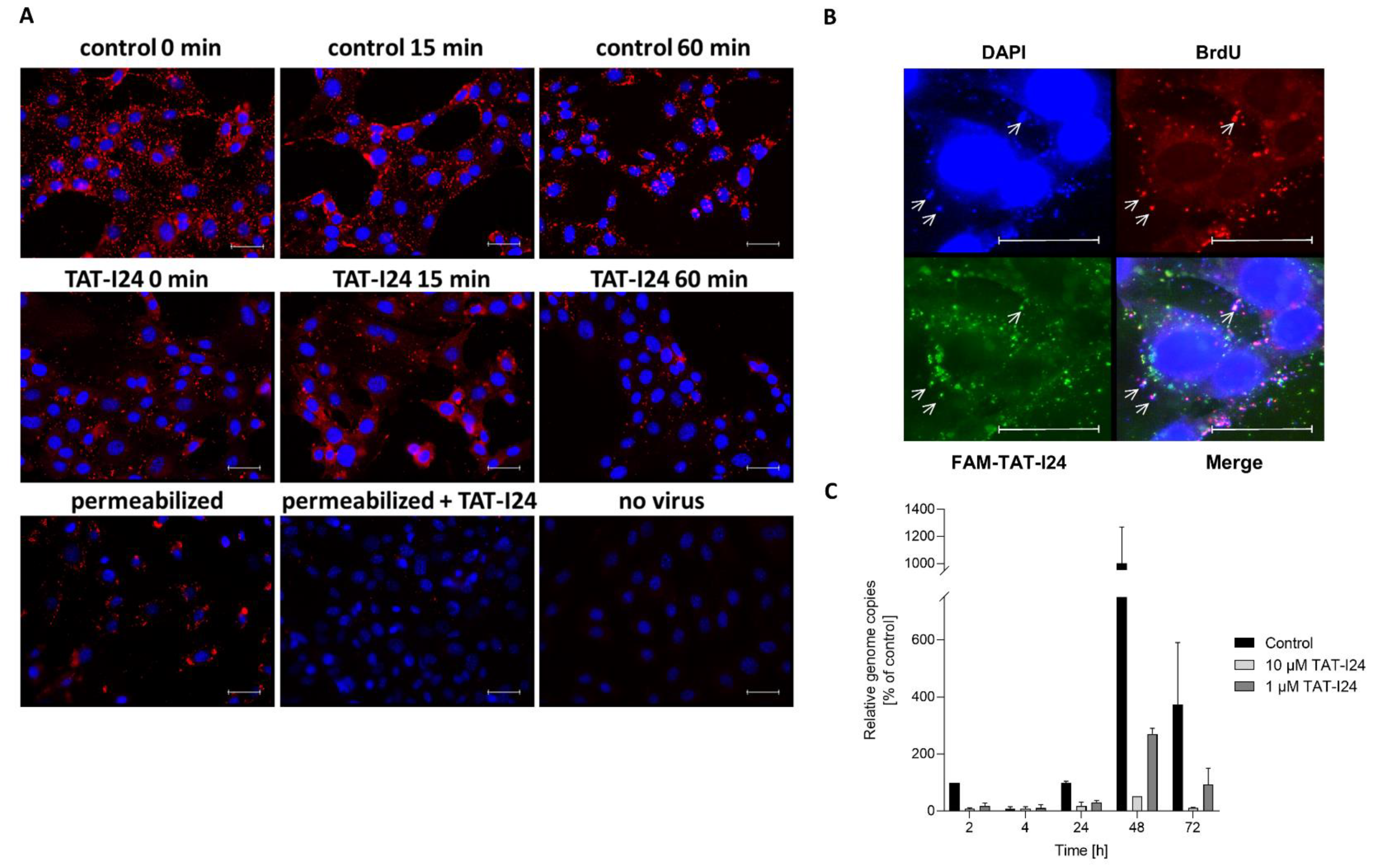
2.5. Effect of TAT-I24 on Viral DNA Upon Entry
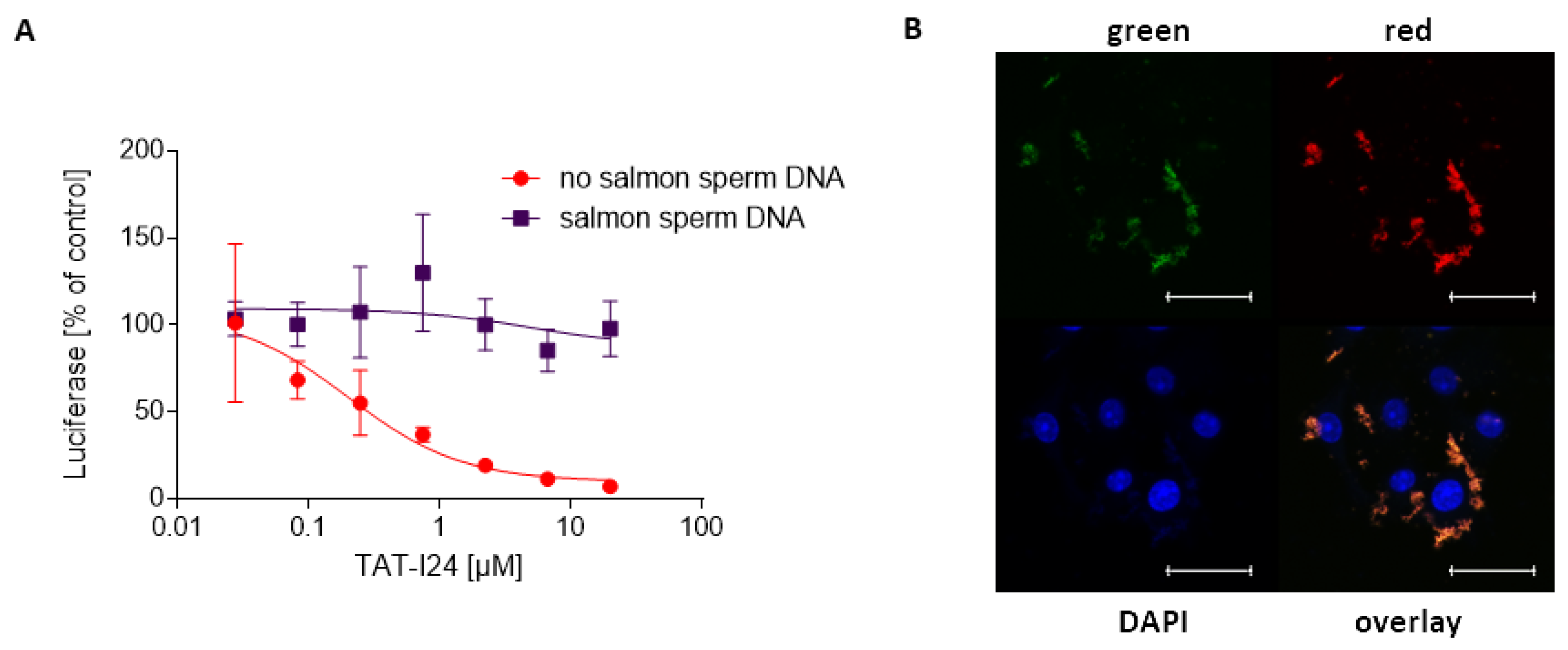
2.6. Inhibitory Effect of TAT-I24 Is Prevented by Preincubation of the Peptide with Double-Stranded DNA
3. Discussion
4. Materials and Methods
4.1. Peptides
4.2. Plasmids
4.3. RNA Synthesis and Binding of T7 RNA Polymerase
4.4. DNA Gel Retardation Assay
4.5. Fluorescence Measurements
4.6. UV Radiation
4.7. Surface Plasmon Resonance
4.8. Plasmid-DNA Binding Assay
4.9. Cell Culture
4.10. Baculovirus
4.11. Murine Cytomegalovirus
4.12. Bromodeoxyuridine-Labelled MCMV and Microscopy
4.13. Molecular Modelling
4.13.1. Setup of Structures and Geometries
4.13.2. Minimization, Equilibration, Production MD
4.13.3. MM/PBSA
4.14. Data Analysis
5. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adalja, A.; Inglesby, T. Broad-Spectrum Antiviral Agents: A Crucial Pandemic Tool. Expert Rev. Anti Infect. Ther. 2019, 17, 467–470. [Google Scholar] [CrossRef] [Green Version]
- Chitalia, V.C.; Munawar, A.H. A painful lesson from the COVID-19 pandemic: The need for broad-spectrum, host-directed antivirals. J. Transl. Med. 2020, 18, 390. [Google Scholar] [CrossRef]
- Zhu, J.-D.; Meng, W.; Wang, X.-J.; Wang, H.-C.R. Broad-spectrum antiviral agents. Front. Microbiol. 2015, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, A.; Tandon, H.; Thakur, N.; Kumar, M. AVPdb: A database of experimentally validated antiviral peptides targeting medically important viruses. Nucleic Acids Res. 2013, 42, D1147–D1153. [Google Scholar] [CrossRef] [Green Version]
- Vilas Boas, L.C.P.; Campos, M.L.; Berlanda, R.L.A.; de Carvalho Neves, N.; Franco, O.L. Antiviral peptides as promising therapeutic drugs. Cell. Mol. Life Sci. 2019, 76, 3525–3542. [Google Scholar] [CrossRef]
- Ahmed, A.; Siman-Tov, G.; Hall, G.; Bhalla, N.; Narayanan, A. Human Antimicrobial Peptides as Therapeutics for Viral Infections. Viruses 2019, 11, 704. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, G.; Gabrani, R. Antiviral Peptides: Identification and Validation. Int. J. Pept. Res. Ther. 2021, 27, 149–168. [Google Scholar] [CrossRef] [PubMed]
- Skalickova, S.; Heger, Z.; Krejcova, L.; Pekarik, V.; Bastl, K.; Janda, J.; Kostolansky, F.; Vareckova, E.; Zitka, O.; Adam, V.; et al. Perspective of Use of Antiviral Peptides against Influenza Virus. Viruses 2015, 7, 5428–5442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; To, K.K.W.; Sze, K.-H.; Yung, T.T.-M.; Bian, M.; Lam, H.; Yeung, M.L.; Li, C.; Chu, H.; Yuen, K.-Y. A broad-spectrum virus- and host-targeting peptide against respiratory viruses including influenza virus and SARS-CoV-2. Nat. Commun. 2020, 11, 4252. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.C.; Turpin, E.A.; Bultmann, H.; Schultz-Cherry, S.; Brandt, C.R. Inhibition of Influenza Virus Infection by a Novel Antiviral Peptide That Targets Viral Attachment to Cells. J. Virol. 2006, 80, 11960–11967. [Google Scholar] [CrossRef] [Green Version]
- Altmann, S.E.; Jones, J.C.; Schultz-Cherry, S.; Brandt, C.R. Inhibition of Vaccinia virus entry by a broad spectrum antiviral peptide. Virology 2009, 388, 248–259. [Google Scholar] [CrossRef] [Green Version]
- Nicol, M.Q.; Ligertwood, Y.; Bacon, M.N.; Dutia, B.M.; Nash, A.A. A novel family of peptides with potent activity against influenza a viruses. J. Gen. Virol. 2012, 93, 980–986. [Google Scholar] [CrossRef]
- Kadam, R.U.; Juraszek, J.; Brandenburg, B.; Buyck, C.; Schepens, W.B.G.; Kesteleyn, B.; Stoops, B.; Vreeken, R.J.; Vermond, J.; Goutier, W.; et al. Potent peptidic fusion inhibitors of influenza virus. Science 2017, 358, 496–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, S.; Yan, L.; Xu, W.; Agrawal, A.S.; Algaissi, A.; Tseng, C.-T.K.; Wang, Q.; Du, L.; Tan, W.; Wilson, I.A.; et al. A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci. Adv. 2019, 5, eaav4580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Mol. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef]
- Badani, H.; Garry, R.F.; Wimley, W.C. Peptide entry inhibitors of enveloped viruses: The importance of interfacial hydrophobicity. Biochim. Biophys. Acta -Biomembr. 2014, 1838, 2180–2197. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.R.; Guha, S.; Wu, E.; Ghimire, J.; Wang, Y.; He, J.; Garry, R.F.; Wimley, W.C. Broad-Spectrum Antiviral Entry Inhibition by Interfacially Active Peptides. J. Virol. 2020, 94, 1–20. [Google Scholar] [CrossRef]
- Thakur, N.; Qureshi, A.; Kumar, M. AVPpred: Collection and prediction of highly effective antiviral peptides. Nucleic Acids Res. 2012, 40, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, A.S.; Reehl, S.M.; Kehn-Hall, K.; Bishop, B.; Webb-Robertson, B.J.M. Better understanding and prediction of antiviral peptides through primary and secondary structure feature importance. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Matthews, T.; Salgo, M.; Greenberg, M.; Chung, J.; DeMasi, R.; Bolognesi, D. Enfuvirtide: The first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat. Rev. Drug Discov. 2004, 3, 215–225. [Google Scholar] [CrossRef]
- Blank, A.; Markert, C.; Hohmann, N.; Carls, A.; Mikus, G.; Lehr, T.; Alexandrov, A.; Haag, M.; Schwab, M.; Urban, S.; et al. First-in-human application of the novel hepatitis B and hepatitis D virus entry inhibitor myrcludex B. J. Hepatol. 2016, 65, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Ruzsics, Z.; Hoffmann, K.; Riedl, A.; Krawczyk, A.; Widera, M.; Sertznig, H.; Schipper, L.; Kapper-Falcone, V.; Debreczeny, M.; Ernst, W.; et al. A Novel, Broad-Acting Peptide Inhibitor of Double-Stranded DNA Virus Gene Expression and Replication. Front. Microbiol. 2020, 11, 2934. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.B.; Gamper, H.; Hearst, J.E. Interaction of T7 RNA polymerase with DNA in an elongation complex arrested at a specific psoralen adduct site. J. Biol. Chem. 1988, 263, 527–534. [Google Scholar] [CrossRef]
- Place, C.; Oddos, J.; Buc, H.; McAllister, W.T.; Buckle, M. Studies of contacts between T7 RNA polymerase and its promoter reveal features in common with multisubunit RNA polymerases. Biochemistry 1999, 38, 4948–4957. [Google Scholar] [CrossRef] [PubMed]
- Yindeeyoungyeon, W.; Schell, M.A. Footprinting with an Automated Capillary DNA Sequencer. Biotechniques 2000, 29, 1034–1041. [Google Scholar] [CrossRef]
- Severinov, K.; Darst, S.A. A mutant RNA polymerase that forms unusual open promoter complexes. Proc. Natl. Acad. Sci. USA 1997, 94, 13481–13486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Vivès, E.; Brodin, P.; Lebleu, B. A Truncated HIV-1 Tat Protein Basic Domain Rapidly Translocates through the Plasma Membrane and Accumulates in the Cell Nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [Green Version]
- Ignatovich, I.A.; Dizhe, E.B.; Pavlotskaya, A.V.; Akifiev, B.N.; Burov, S.V.; Orlov, S.V.; Perevozchikov, A.P. Complexes of Plasmid DNA with Basic Domain 47-57 of the HIV-1 Tat Protein Are Transferred to Mammalian Cells by Endocytosis-mediated Pathways. J. Biol. Chem. 2003, 278, 42625–42636. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, A.; Seelig, J. High affinity of the cell-penetrating peptide HIV-1 Tat-PTD for DNA. Biochemistry 2007, 46, 8138–8145. [Google Scholar] [CrossRef]
- Lv, M.X.; Duan, B.C.; Lu, K.; Wu, Y.J.; Zhao, Y.F. Synthesis, DNA-Binding and Antibacterial Activity of the Cell-Penetrating Peptide HIV-1 Tat (49–57). Indian J. Pharm. Sci. 2017, 79, 893–899. [Google Scholar] [CrossRef]
- Hsu, C.-H. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: Evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 2005, 33, 4053–4064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze-Gahmen, U.; Hurley, J.H. Structural mechanism for HIV-1 TAR loop recognition by Tat and the super elongation complex. Proc. Natl. Acad. Sci. USA 2018, 115, 12973–12978. [Google Scholar] [CrossRef] [Green Version]
- Pham, V.V.; Salguero, C.; Khan, S.N.; Meagher, J.L.; Brown, W.C.; Humbert, N.; de Rocquigny, H.; Smith, J.L.; D’Souza, V.M. HIV-1 Tat interactions with cellular 7SK and viral TAR RNAs identifies dual structural mimicry. Nat. Commun. 2018, 9, 4266. [Google Scholar] [CrossRef] [Green Version]
- Sim, S.; Wang, P.; Beyer, B.N.; Cutrona, K.J.; Radhakrishnan, M.L.; Elmore, D.E. Investigating the nucleic acid interactions of histone-derived antimicrobial peptides. FEBS Lett. 2017, 591, 706–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.; Seel, M.; Weidemann, T.; Höfinger, S. Theoretical mimicry of biomembranes. FEBS Lett. 2009, 583, 1909–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, P.; Seel, M.; Hansmann, U.H.E.; Höfinger, S. Dispersion Terms and Analysis of Size- and Charge Dependence in an Enhanced Poisson−Boltzmann Approach. J. Phys. Chem. B 2007, 111, 8910–8918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, R.; Kranzlmüller, D.; Volkert, J.; Hansmann, U.H.E.; Höfinger, S. Computational assessment of the entropy of solvation of small-sized hydrophobic entities. Phys. Chem. Chem. Phys. 2006, 8, 5515–5521. [Google Scholar] [CrossRef] [Green Version]
- Trilling, M.; Le, V.T.K.; Fiedler, M.; Zimmermann, A.; Bleifuß, E.; Hengel, H. Identification of DNA-Damage DNA-Binding Protein 1 as a Conditional Essential Factor for Cytomegalovirus Replication in Interferon-γ-Stimulated Cells. PLoS Pathog. 2011, 7, e1002069. [Google Scholar] [CrossRef] [Green Version]
- Loughran, S.P.; McCrudden, C.M.; McCarthy, H.O. Designer peptide delivery systems for gene therapy. Eur. J. Nanomed. 2015, 7, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Bultmann, H.; Brandt, C.R. Peptides Containing Membrane-transiting Motifs Inhibit Virus Entry. J. Biol. Chem. 2002, 277, 36018–36023. [Google Scholar] [CrossRef] [Green Version]
- Bultmann, H.; Teuton, J.; Brandt, C.R. Addition of a C-terminal cysteine improves the anti-herpes simplex virus activity of a peptide containing the human immunodeficiency virus type 1 TAT protein transduction domain. Antimicrob. Agents Chemother. 2007, 51, 1596–1607. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, M.; Wikström, S.; Johansson, M. Cell surface adherence and endocytosis of protein transduction domains. Mol. Ther. 2003, 8, 143–150. [Google Scholar] [CrossRef]
- Richard, J.P.; Melikov, K.; Brooks, H.; Prevot, P.; Lebleu, B.; Chernomordik, L.V. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J. Biol. Chem. 2005, 280, 15300–15306. [Google Scholar] [CrossRef] [Green Version]
- Lo, S.L.; Wang, S. An endosomolytic Tat peptide produced by incorporation of histidine and cysteine residues as a nonviral vector for DNA transfection. Biomaterials 2008, 29, 2408–2414. [Google Scholar] [CrossRef]
- Jeong, C.; Yoo, J.; Lee, D.; Kim, Y.-C. A branched TAT cell-penetrating peptide as a novel delivery carrier for the efficient gene transfection. Biomater. Res. 2016, 20, 28. [Google Scholar] [CrossRef] [Green Version]
- Wilson, S.S.; Wiens, M.E.; Smith, J.G. Antiviral Mechanisms of Human Defensins. J. Mol. Biol. 2013, 425, 4965–4980. [Google Scholar] [CrossRef]
- Hazrati, E.; Galen, B.; Lu, W.; Wang, W.; Ouyang, Y.; Keller, M.J.; Lehrer, R.I.; Herold, B.C. Human α- and β-Defensins Block Multiple Steps in Herpes Simplex Virus Infection. J. Immunol. 2006, 177, 8658–8666. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Zhou, J.; Zhang, K.; Chu, H.; Liu, D.; Poon, V.K.-M.; Chan, C.C.-S.; Leung, H.-C.; Fai, N.; Lin, Y.-P.; et al. A novel peptide with potent and broad-spectrum antiviral activities against multiple respiratory viruses. Sci. Rep. 2016, 6, 22008. [Google Scholar] [CrossRef]
- Pul, Ü.; Wurm, R.; Wagner, R. KMnO4 Footprinting. Bio-Protocol 2012, 2, 1–5. [Google Scholar] [CrossRef]
- Hausner, W.; Thomm, M. Events during initiation of archaeal transcription: Open complex formation and DNA-protein interactions. J. Bacteriol. 2001, 183, 3025–3031. [Google Scholar] [CrossRef] [Green Version]
- Rosenke, K.; Fortunato, E.A. Bromodeoxyuridine-Labeled Viral Particles as a Tool for Visualization of the Immediate-Early Events of Human Cytomegalovirus Infection. J. Virol. 2004, 78, 7818–7822. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.I.; Cruzeiro, V.W.D.; Darden, T.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. Amber 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- van Gunsteren, W.F.; Berendsen, H.J.C. Algorithms for macromolecular dynamics and constraint dynamics. Mol. Phys. 1977, 34, 1311–1327. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harant, H.; Höfinger, S.; Kricek, F.; Ruf, C.; Ruzsics, Z.; Hengel, H.; Lindley, I.J.D. The Peptide TAT-I24 with Antiviral Activity against DNA Viruses Binds Double-Stranded DNA with High Affinity. Biologics 2021, 1, 41-60. https://doi.org/10.3390/biologics1010003
Harant H, Höfinger S, Kricek F, Ruf C, Ruzsics Z, Hengel H, Lindley IJD. The Peptide TAT-I24 with Antiviral Activity against DNA Viruses Binds Double-Stranded DNA with High Affinity. Biologics. 2021; 1(1):41-60. https://doi.org/10.3390/biologics1010003
Chicago/Turabian StyleHarant, Hanna, Siegfried Höfinger, Franz Kricek, Christine Ruf, Zsolt Ruzsics, Hartmut Hengel, and Ivan J. D. Lindley. 2021. "The Peptide TAT-I24 with Antiviral Activity against DNA Viruses Binds Double-Stranded DNA with High Affinity" Biologics 1, no. 1: 41-60. https://doi.org/10.3390/biologics1010003
APA StyleHarant, H., Höfinger, S., Kricek, F., Ruf, C., Ruzsics, Z., Hengel, H., & Lindley, I. J. D. (2021). The Peptide TAT-I24 with Antiviral Activity against DNA Viruses Binds Double-Stranded DNA with High Affinity. Biologics, 1(1), 41-60. https://doi.org/10.3390/biologics1010003