
Targeting Cancer Stem Cells with Radioimmunotherapy: The Case of the Ovarian Cancer Stemness-Associated Biomarker L1CAM



Abstract
1. Introduction
2. Ovarian Cancer—Epidemiology, Histotypes, and Current Standard of Treatment of Epithelial Ovarian Cancer
2.1. Epidemiology, Histological Subtypes, and Molecular Features of HGSOC
2.2. Current Standard of Treatment of Epithelial Ovarian Cancer and Therapies under Investigation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Agent | Target | Status | Ref. |
|---|---|---|---|
| Taxane-based drugs | Tubulin (inhibition of the microtubule disassembly) | Approved | [27] |
| Platinum-based drugs | DNA cross-linking agent | Approved | |
| Bevasizumab | Anti-VEGF | Approved | [28,29,30] |
| PARP inhibitors | PARP enzymes | Approved | [16,30,31,32] |
| Mirvetuximab soravtansine | Folate receptor alpha | NCT020631876, Phase III | [33] |
| Anti-PD-1/PD-L1, anti-CTLA-4 | PD-1/PD-L1, CTLA-4 | NCT02580058, Phase III NCT02811497, Phase II NCT02657889, Phase 1/2 | [16] |
| CAR T cells | Folate receptor alpha, mesothelin, MUC16 (also known as CA125), CD70 | Multiple ongoing or completed clinical and preclinical studies | [34] |
3. Cancer Stem Cells—Definition, Concept, Study, and Identification, Ovarian Cancer Stem Cells
3.1. Definition and Concept of Cancer Stem Cells
| CSC Population | Cancer Type | Associated CSC Properties | Ref. |
|---|---|---|---|
| L1CAM+/CD133+ | Ovarian | In vitro—high clonogenicity, spherogenicity, invasiveness, radioresistance. Upregulation of stem cell and EMT genes (Oct-4, CXCR4, ABCG2, TGF-1β, β-catenin, vimentin). In vivo—high tumorigenicity, self-renewal. | [11] |
| CD44+/CD117+ | Ovarian | In vitro—anchorage-independent and self-renewing sphere formation in CSC selective conditions, chemoresistance. Expression of stem cell genes (Oct-4, Nestin, Nanog, Notch-1, Bmi-1). In vivo—tumorigenicity and serial propagation, histological recapitulation of the original tumor. | [52] |
| ALDH+/CD133+ | Ovarian | In vitro—high and long-term spherogenicity in CSC selective conditions. Upregulation of stem cell genes (Sox2, Oct4, Nanog). In vivo—generate heterogenous tumors. | [53] |
| CD44+/CD133+ | Pancreatic | In vitro—high sphere formation in CSC selective conditions, proliferation, chemoresistance, recapitulation of the other tumor populations. Upregulation of inflammation and EMT genes (Sparc, Col1a1, Ccl2, Cxcl1, Cxcl2); mRNAs * in CSC-related pathways. In vivo—high tumorigenicity. | [54] |
| CXCR4+/CD133+ | Colorectal Pancreatic | In vitro—high migratory capacity. Upregulation of EMT genes (vimentin, N-cadherin, Snail). In vivo—high tumorigenicity and metastasis formation blocked with the CXCR4 antagonist AMD3100. In vivo—high tumorigenicity, invasiveness, and metastasis formation, inhibited by AMD3100. | [55] [56] |
| CD44+/CD24+ | Gastric | In vitro—enhanced sphere formation in CSC selective conditions. Upregulation of stemness genes (Shh, Ptch1). In vivo—high tumorigenicity; regeneration of the tumor heterogeneity. | [57] |
| EpCAM+/CD166+/ CD44+ | Non-small cell lung | In vitro—higher proliferation, clonogenicity, sphere formation, migration, chemoresistance. Upregulated stem cell genes (Rex1, Ssea4). | [58] |
| CD44+/CD24− /ALDH1+ | Head and neck | In vitro—high spherogenicity in CSC selective conditions, increased invasion, radioresistance. Upregulation of the EMT and stem cell genes (Snail, Oct-4, Nanog, Sox2). In vivo—high tumorigenicity. Knockdown of Snail reduced the CSC properties. | [59] |
| CD34+/CD38- | AML | In vivo—high tumorigenicity, self-renewal, differentiation into other tumor cells populations. | [37,38] ** |
| CD44+/CD24−/low | Breast cancer | In vivo—high tumorigenicity, regeneration of the tumor heterogeneity. | [39] ** |
3.2. Study and Identification of Cancer Stem Cells
| Method | CSC Property | Method Limitations | Ref. |
|---|---|---|---|
| In vitro | |||
| Anchorage -independent cell growth (spherogenicity in non-adherent conditions) | Tumorigenicity Anchorage-independent survival Long-term self-renewal (sphere passaging) |
| [62,63,64,65,66] |
| Clonogenicity | Clonogenic survival (Unlimited proliferation capacity) |
| [67,68] |
| Radio- and chemoresistance | Therapy resistance/sensitivity |
| |
| In vivo | |||
| Limiting dilution | Tumorigenicity CSC frequency |
| [5,6,40,69,70,71] |
| Serial transplantation | Long-term self-renewal | ||
| Lineage tracing | Cancer cell of origin Tumorigenicity Clonal expansion Cellular heterogeneity CSC frequency Localization |
| |
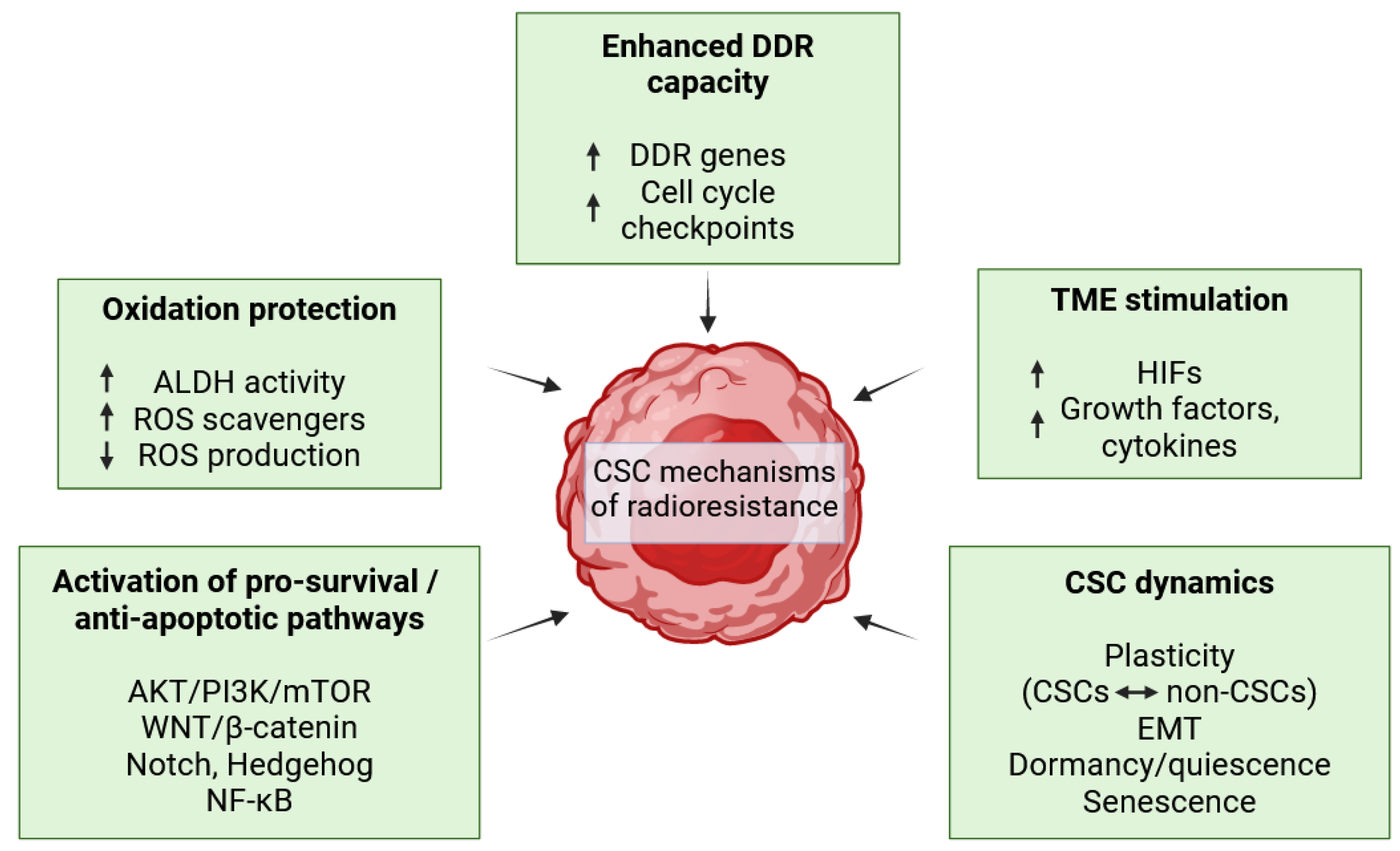
3.3. Cancer Stem Cell Determinants of Radioresistance
3.4. Ovarian Cancer Stem Cells—Origin and Biomarkers
3.5. Current Therapeutic Strategies against Ovarian Cancer Stem Cells
4. L1CAM as a Promising Target for Radioimmunotherapy against Ovarian Cancer Stem Cells
4.1. The Ovarian Cancer Stemness-Associated Biomarker L1CAM
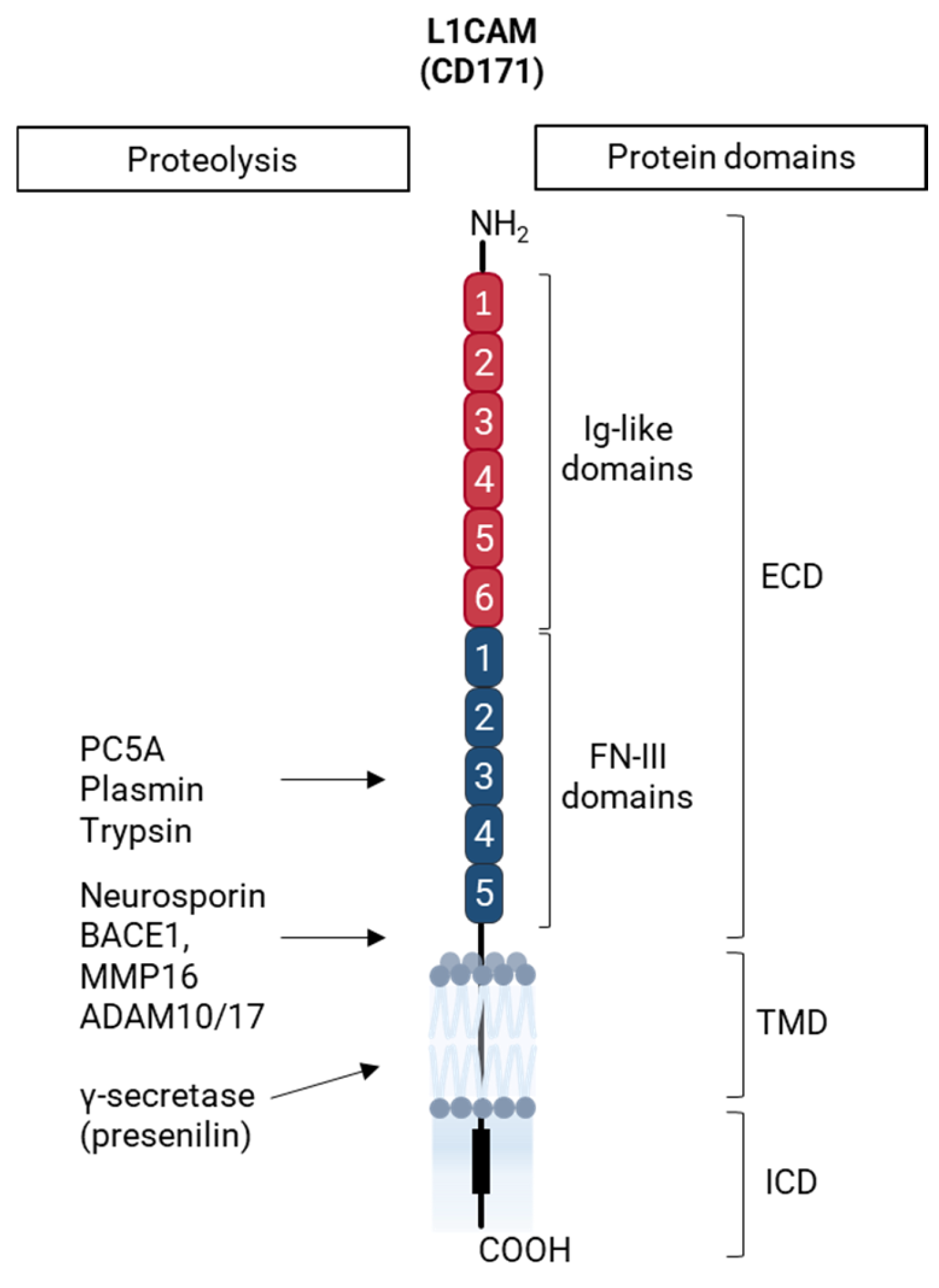
4.1.1. L1CAM in Health and Disease
4.1.2. L1CAM as a Cancer Stem Cell-Associated Biomarker in Epithelial Ovarian Cancer
4.2. Anti-L1CAM Radioimmunotherapy against Ovarian Cancer Stem Cells
4.2.1. Clinical Radioimmunotherapy against Solid Tumors
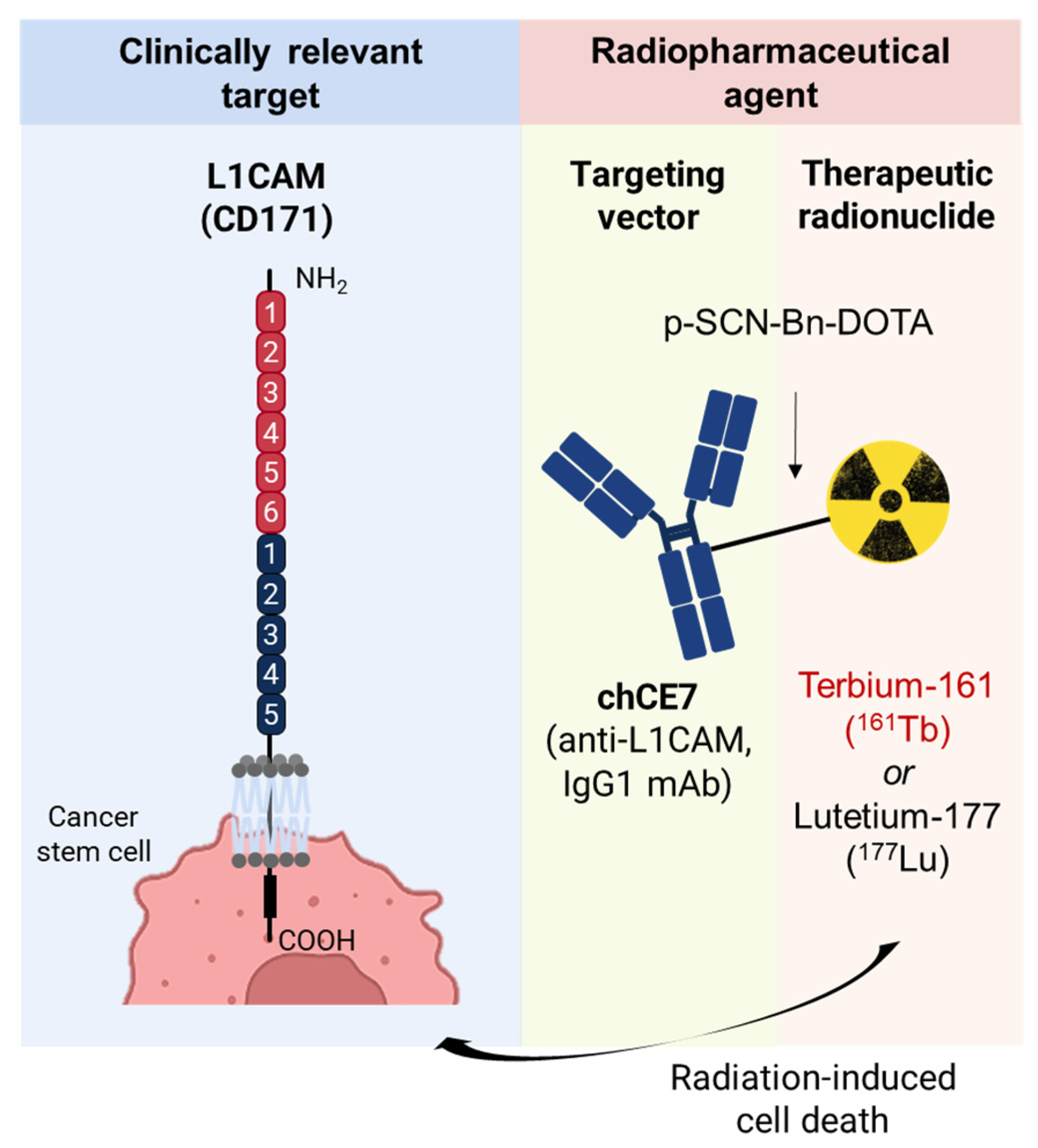
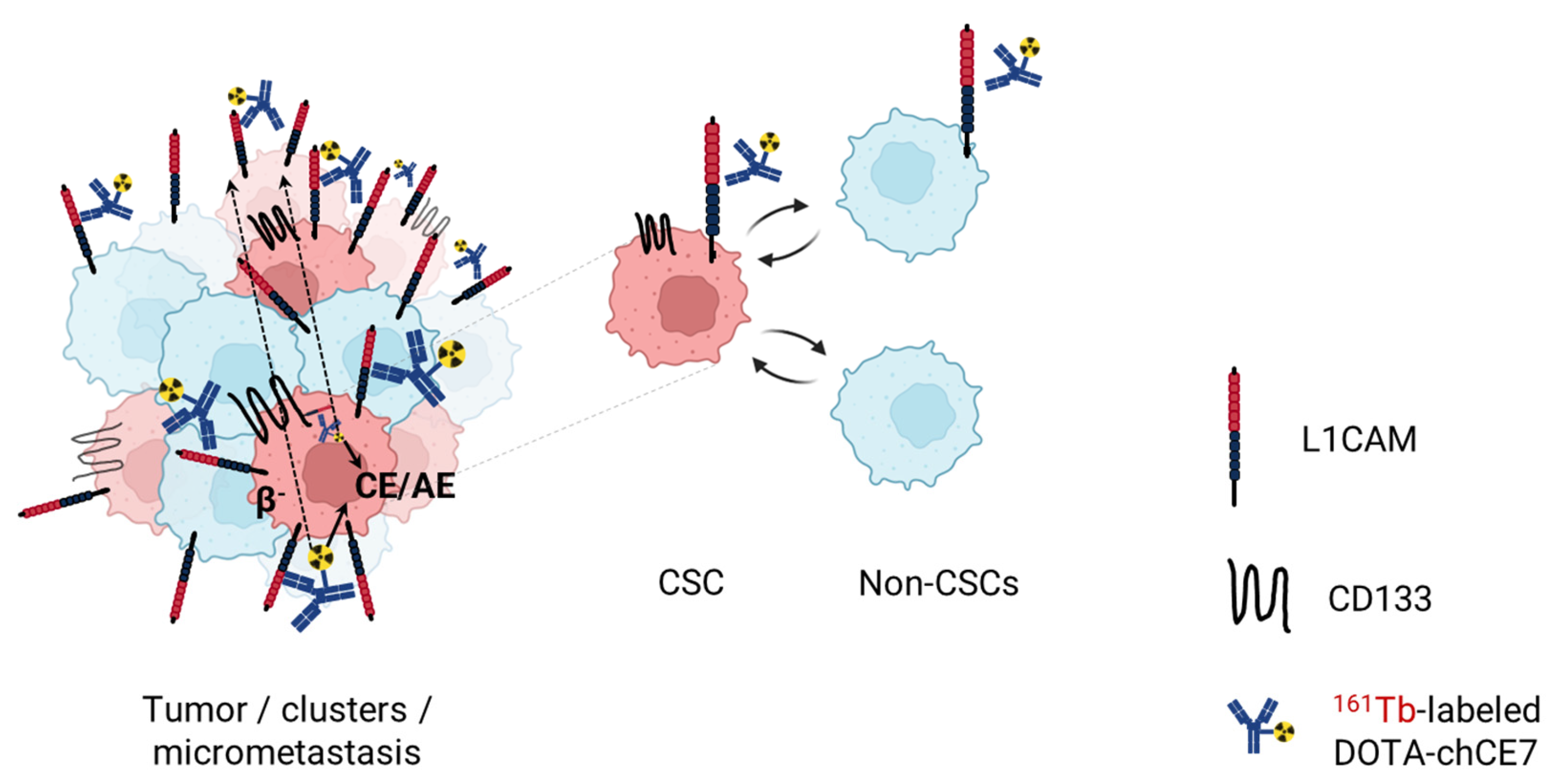
4.2.2. Anti-L1CAM Radioimmunotherapy as a Novel Therapeutic Modality against Ovarian Cancer Stem Cells
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Reid, F. World Ovarian Cancer Coalition Atlas 2023—Global Trends in Incidence, Mortality, and Survival; World Ovarian Cancer Coalition: Toronto, ON, Canada, 2023. [Google Scholar]
- Hollis, R.L. Molecular characteristics and clinical behaviour of epithelial ovarian cancers. Cancer Lett. 2023, 555, 216057. [Google Scholar] [CrossRef]
- Cabasag, C.J.; Fagan, P.J.; Ferlay, J.; Vignat, J.; Laversanne, M.; Liu, L.; van der Aa, M.A.; Bray, F.; Soerjomataram, I. Ovarian cancer today and tomorrow: A global assessment by world region and Human Development Index using GLOBOCAN 2020. Int. J. Cancer 2022, 151, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Duska, L.R.; Kohn, E.C. The new classifications of ovarian, fallopian tube, and primary peritoneal cancer and their clinical implications. Ann. Oncol. 2017, 28, viii8–viii12. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Bonnet, D.; De Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Rev. Cancer 2012, 12, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Loh, J.J.; Ma, S. Hallmarks of cancer stemness. Cell Stem Cell 2024, 31, 617–639. [Google Scholar] [CrossRef] [PubMed]
- Giordano, M.; Cavallaro, U. Different shades of L1CAM in the pathophysiology of cancer stem cells. J. Clin. Med. 2020, 9, 1502. [Google Scholar] [CrossRef] [PubMed]
- Altevogt, P.; Doberstein, K.; Fogel, M. L1CAM in human cancer. Int. J. Cancer 2016, 138, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Doberstein, K.; Spivak, R.; Reavis, H.D.; Hooda, J.; Feng, Y.; Kroeger, P.T., Jr.; Stuckelberger, S.; Mills, G.B.; Devins, K.M.; Schwartz, L.E.; et al. L1CAM is required for early dissemination of fallopian tube carcinoma precursors to the ovary. Commun. Biol. 2022, 5, 1362. [Google Scholar] [CrossRef] [PubMed]
- Giordano, M.; Decio, A.; Battistini, C.; Baronio, M.; Bianchi, F.; Villa, A.; Bertalot, G.; Freddi, S.; Lupia, M.; Jodice, M.G.; et al. L1CAM promotes ovarian cancer stemness and tumor initiation via FGFR1/SRC/STAT3 signaling. J. Exp. Clin. Cancer Res. 2021, 40, 319. [Google Scholar] [CrossRef]
- Terraneo, N.; Jacob, F.; Peitzsch, C.; Dubrovska, A.; Krudewig, C.; Huang, Y.L.; Heinzelmann-Schwarz, V.; Schibli, R.; Behe, M.; Grünberg, J. L1 cell adhesion molecule confers radioresistance to ovarian cancer and defines a new cancer stem cell population. Cancers 2020, 12, 217. [Google Scholar] [CrossRef]
- Grünberg, J.; Lindenblatt, D.; Dorrer, H.; Cohrs, S.; Zhernosekov, K.; Koster, U.; Turler, A.; Fischer, E.; Schibli, R. Anti-L1CAM radioimmunotherapy is more effective with the radiolanthanide terbium-161 compared to lutetium-177 in an ovarian cancer model. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 1907–1915. [Google Scholar] [CrossRef]
- Ku, A.; Facca, V.J.; Cai, Z.; Reilly, R.M. Auger electrons for cancer therapy—A review. EJNMMI Radiopharm. Chem. 2019, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Pouget, J.P.; Navarro-Teulon, I.; Bardies, M.; Chouin, N.; Cartron, G.; Pelegrin, A.; Azria, D. Clinical radioimmunotherapy—The role of radiobiology. Nat. Rev. Clin. Oncol. 2011, 8, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef] [PubMed]
- Ebell, M.H.; Culp, M.B.; Radke, T.J. A systematic review of symptoms for the diagnosis of ovarian cancer. Am. J. Prev. Med. 2016, 50, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, F.; Choudhary, B. Ovarian Cancer: Molecular Classification and Targeted Therapy; InTechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Prat, J.; FIGO Committee on Gynecologic Oncology. FIGO’s staging classification for cancer of the ovary, fallopian tube, and peritoneum: Abridged republication. J. Gynecol. Oncol. 2015, 26, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.K.; Pothuri, B. Homologous recombination deficiency (HRD) testing in ovarian cancer clinical practice: A review of the literature. Gynecol. Oncol. Res. Pract. 2017, 4, 4. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Lo Riso, P.; Villa, C.E.; Gasparoni, G.; Vingiani, A.; Luongo, R.; Manfredi, A.; Jungmann, A.; Bertolotti, A.; Borgo, F.; Garbi, A.; et al. A cell-of-origin epigenetic tracer reveals clinically distinct subtypes of high-grade serous ovarian cancer. Genome Med. 2020, 12, 94. [Google Scholar] [CrossRef]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Pignata, S.; Cecere, S.C.; Du Bois, A.; Harter, P.; Heitz, F. Treatment of recurrent ovarian cancer. Ann. Oncol. 2017, 28, viii51–viii56. [Google Scholar] [CrossRef]
- Flores-Balcazar, C.H.; Urias-Arce, D.M. Radiotherapy in women with epithelial ovarian cancer: Historical role, current advances, and indications. Chin. Clin. Oncol. 2020, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef] [PubMed]
- Sazonova, E.V.; Kopeina, G.S.; Imyanitov, E.N.; Zhivotovsky, B. Platinum drugs and taxanes: Can we overcome resistance? Cell Death Discov. 2021, 7, 155. [Google Scholar] [CrossRef] [PubMed]
- Tewari, K.S.; Burger, R.A.; Enserro, D.; Norquist, B.M.; Swisher, E.M.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Huang, H.; Homesley, H.D.; et al. Final overall survival of a randomized trial of bevacizumab for primary treatment of ovarian cancer. J. Clin. Oncol. 2019, 37, 2317–2328. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, J.; Shannon, C.M.; Baumann, K.; Rau, J.; Harter, P.; Joly, F.; Sehouli, J.; Canzler, U.; Schmalfeldt, B.; Dean, A.P.; et al. Bevacizumab and platinum-based combinations for recurrent ovarian cancer: A randomised, open-label, phase 3 trial. Lancet Oncol. 2020, 21, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Alvarez Secord, A.; O’Malley, D.M.; Sood, A.K.; Westin, S.N.; Liu, J.F. Rationale for combination PARP inhibitor and antiangiogenic treatment in advanced epithelial ovarian cancer: A review. Gynecol. Oncol. 2021, 162, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Foo, T.; George, A.; Banerjee, S. PARP inhibitors in ovarian cancer: An overview of the practice-changing trials. Genes Chromosomes Cancer 2021, 60, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Pothuri, B. Appropriate selection of PARP inhibitors in ovarian cancer. Curr. Treat. Options Oncol. 2022, 23, 887–903. [Google Scholar] [CrossRef]
- Moore, K.N.; Vergote, I.; Oaknin, A.; Colombo, N.; Banerjee, S.; Oza, A.; Pautier, P.; Malek, K.; Birrer, M.J. FORWARD I: A Phase III study of mirvetuximab soravtansine versus chemotherapy in platinum-resistant ovarian cancer. Future Oncol. 2018, 14, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Cutri-French, C.; Nasioudis, D.; George, E.; Tanyi, J.L. CAR-T cell therapy in ovarian cancer: Where are we now? Diagnostics 2024, 14, 819. [Google Scholar] [CrossRef] [PubMed]
- Terraneo, N.; Jacob, F.; Dubrovska, A.; Grunberg, J. Novel therapeutic strategies for ovarian cancer stem cells. Front. Oncol. 2020, 10, 319. [Google Scholar] [CrossRef]
- Capp, J.P. Cancer stem cells: From historical roots to a new perspective. J. Oncol. 2019, 2019, 5189232. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Mindent, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Nassar, D.; Blanpain, C. Cancer stem cells: Basic concepts and therapeutic implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Paul, R.; Dorsey, J.F.; Fan, Y. Cell plasticity, senescence, and quiescence in cancer stem cells: Biological and therapeutic implications. Pharmacol. Ther. 2022, 231, 107985. [Google Scholar] [CrossRef]
- Crea, F.; Nur Saidy, N.R.; Collins, C.C.; Wang, Y. The epigenetic/noncoding origin of tumor dormancy. Trends Mol. Med. 2015, 21, 206–211. [Google Scholar] [CrossRef]
- El Touny, L.H.; Vieira, A.; Mendoza, A.; Khanna, C.; Hoenerhoff, M.J.; Green, J.E. Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumor cells. J. Clin. Investig. 2014, 124, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauss, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer stem cells—Origins and biomarkers: Perspectives for targeted personalized therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- Zon, L.I. Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature 2008, 453, 306–313. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Was, H.; Barszcz, K.; Czarnecka, J.; Kowalczyk, A.; Bernas, T.; Uzarowska, E.; Koza, P.; Kaminska, B.; Klejman, A.; Piwocka, K.; et al. Bafilomycin A1 triggers proliferative potential of senescent cancer cells in vitro and in NOD/SCID mice. Oncotarget 2017, 8, 9303–9322. [Google Scholar] [CrossRef]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef]
- Kryczek, I.; Liu, S.; Roh, M.; Vatan, L.; Szeliga, W.; Wei, S.; Banerjee, M.; Mao, Y.; Kotarski, J.; Wicha, M.S.; et al. Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int. J. Cancer 2012, 130, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Eptaminitaki, G.C.; Zaravinos, A.; Stellas, D.; Panagopoulou, M.; Karaliota, S.; Baltsavia, I.; Iliopoulos, I.; Chatzaki, E.; Iliopoulos, D.; Baritaki, S. Genome-wide analysis of lncRNA-mRNA co-expression networks in CD133+/CD44+ stem-like PDAC cells. Cancers 2023, 15, 1053. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-s.; Han, Z.-p.; Jing, Y.-y.; Tao, S.-f.; Li, T.-j.; Wang, H.; Wang, Y.; Li, R.; Yang, Y.; Zhao, X.; et al. CD133+CXCR4+ colon cancer cells exhibit metastatic potential and predict poor prognosis of patients. BMC Med. 2012, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, C.; He, F.; Cai, Y.; Yang, H. Identification of CD44+CD24+ gastric cancer stem cells. J. Cancer Res. Clin. Oncol. 2011, 137, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Satar, N.A.; Fakiruddin, K.S.; Lim, M.N.; Mok, P.L.; Zakaria, N.; Fakharuzi, N.A.; Rahman, A.Z.A.; Zakaria, Z.; Yahaya, B.H.; Baharuddin, P. Novel triple-positive markers identified in human non-small cell lung cancer cell line with chemotherapy-resistant and putative cancer stem cell characteristics. Oncol. Rep. 2018, 40, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Chen, Y.W.; Hsu, H.S.; Tseng, L.M.; Huang, P.I.; Lu, K.H.; Chen, D.T.; Tai, L.K.; Yung, M.C.; Chang, S.C.; et al. Aldehyde dehydrogenase 1 is a putative marker for cancer stem cells in head and neck squamous cancer. Biochem. Biophys. Res. Commun. 2009, 385, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.T.; Ryu, C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef]
- Peitzsch, C.; Nathansen, J.; Schniewind, S.I.; Schwarz, F.; Dubrovska, A. Cancer stem cells in head and neck squamous cell carcinoma: Identification, characterization and clinical implications. Cancers 2019, 11, 616. [Google Scholar] [CrossRef]
- Pastrana, E.; Silva-Vargas, V.; Doetsch, F. Eyes wide open: A critical review of sphere-formation as an assay for stem cells. Cell Stem Cell 2011, 8, 486–498. [Google Scholar] [CrossRef]
- Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017, 108, 283–289. [Google Scholar] [CrossRef]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Karuppusamy Rathinam, M.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The soft agar colony formation assay. J. Vis. Exp. 2014, 92, e51998. [Google Scholar] [CrossRef]
- Bahmad, H.F.; Cheaito, K.; Chalhoub, R.M.; Hadadeh, O.; Monzer, A.; Ballout, F.; El-Hajj, A.; Mukherji, D.; Liu, Y.N.; Daoud, G.; et al. Sphere-formation assay: Three-dimensional in vitro culturing of prostate cancer stem/progenitor sphere-forming cells. Front. Oncol. 2018, 8, 347. [Google Scholar] [CrossRef]
- Lim, Y.C.; Oh, S.Y.; Cha, Y.Y.; Kim, S.H.; Jin, X.; Kim, H. Cancer stem cell traits in squamospheres derived from primary head and neck squamous cell carcinomas. Oral Oncol. 2011, 47, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Brix, N.; Samaga, D.; Hennel, R.; Gehr, K.; Zitzelsberger, H.; Lauber, K. The clonogenic assay: Robustness of plating efficiency-based analysis is strongly compromised by cellular cooperation. Radiat. Oncol. 2020, 15, 248. [Google Scholar] [CrossRef]
- Rycaj, K.; Tang, D.G. Cell-of-origin of cancer versus cancer stem cells: Assays and interpretations. Cancer Res. 2015, 75, 4003–4011. [Google Scholar] [CrossRef]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef]
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA genes: The role in genome stability, cancer stemness and therapy resistance. J. Cancer 2019, 10, 2109–2127. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Dubrovska, A.; Linge, A.; Baumann, M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv. Drug Deliv. Rev. 2017, 109, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wei, Y.; Wang, L.; Debeb, B.G.; Yuan, Y.; Zhang, J.; Yuan, J.; Wang, M.; Chen, D.; Sun, Y.; et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell. Biol. 2014, 16, 864–875. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Wu, S.P.; Liu, J.B.; Shi, Y.S.; Huang, X.; Zhang, Q.B.; Yao, K.T. MYC regulation of CHK1 and CHK2 promotes radioresistance in a stem cell-like population of nasopharyngeal carcinoma cells. Cancer Res. 2013, 73, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Lendeckel, U.; Wolke, C. Redox-regulation in cancer stem cells. Biomedicines 2022, 10, 2413. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Chen, Y.S.; Chou, S.H.; Han, C.L.; Chen, Y.J.; Yang, C.C.; Huang, C.Y.; Lo, J.F. Distinct subpopulations of head and neck cancer cells with different levels of intracellular reactive oxygen species exhibit diverse stemness, proliferation, and chemosensitivity. Cancer Res. 2014, 74, 6291–6305. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Singh, S.; Brocker, C.; Koppaka, V.; Chen, Y.; Jackson, B.C.; Matsumoto, A.; Thompson, D.C.; Vasiliou, V. Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free Radic. Biol. Med. 2013, 56, 89–101. [Google Scholar] [CrossRef]
- Schwarz, F.M.; Schniewind, I.; Besso, M.J.; Lange, S.; Linge, A.; Patil, S.G.; Lock, S.; Klusa, D.; Dietrich, A.; Voss-Bohme, A.; et al. Plasticity within aldehyde dehydrogenase-positive cells determines prostate cancer radiosensitivity. Mol. Cancer Res. 2022, 20, 794–809. [Google Scholar] [CrossRef] [PubMed]
- Matsui, W.H. Cancer stem cell signaling pathways. Medicine 2016, 95, S8–S19. [Google Scholar] [CrossRef] [PubMed]
- Jarosz-Biej, M.; Smolarczyk, R.; Cichon, T.; Kulach, N. Tumor microenvironment as a “Game Changer” in cancer radiotherapy. Int. J. Mol. Sci. 2019, 20, 3212. [Google Scholar] [CrossRef] [PubMed]
- Muller, L.; Tunger, A.; Plesca, I.; Wehner, R.; Temme, A.; Westphal, D.; Meier, F.; Bachmann, M.; Schmitz, M. Bidirectional crosstalk between cancer stem cells and immune cell subsets. Front. Immunol. 2020, 11, 140. [Google Scholar] [CrossRef] [PubMed]
- Schoning, J.P.; Monteiro, M.; Gu, W. Drug resistance and cancer stem cells: The shared but distinct roles of hypoxia-inducible factors HIF1alpha and HIF2alpha. Clin. Exp. Pharmacol. Physiol. 2017, 44, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-induced reprogramming of breast cancer cells. Stem Cells 2012, 30, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Sishc, B.J.; Nelson, C.B.; Hahnfeldt, P.; Bailey, S.M.; Hlatky, L. Radiation-induced reprogramming of pre-senescent mammary epithelial cells enriches putative CD44+/CD24−/low stem cell phenotype. Front. Oncol. 2016, 6, 138. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Chargari, C.; Galluzzi, L.; Kroemer, G. Optimising efficacy and reducing toxicity of anticancer radioimmunotherapy. Lancet Oncol. 2019, 20, e452–e463. [Google Scholar] [CrossRef] [PubMed]
- Lupia, M.; Cavallaro, U. Ovarian cancer stem cells: Still an elusive entity? Mol. Cancer 2017, 16, 64. [Google Scholar] [CrossRef]
- Varier, L.; Sundaram, S.M.; Gamit, N.; Warrier, S. An overview of ovarian cancer: The role of cancer stem cells in chemoresistance and a precision medicine approach targeting the Wnt pathway with the antagonist sFRP4. Cancers 2023, 15, 1275. [Google Scholar] [CrossRef]
- Brown, J.R.; Chan, D.K.; Shank, J.J.; Griffith, K.A.; Fan, H.; Szulawski, R.; Yang, K.; Reynolds, R.K.; Johnston, C.; McLean, K.; et al. Phase II clinical trial of metformin as a cancer stem cell-targeting agent in ovarian cancer. JCI Insight 2020, 5, e133247. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, P.; Wang, H.; Hou, D.; Li, W.; Xiao, G.; Li, C. Inhibitory effects of metformin at low concentration on epithelial-mesenchymal transition of CD44+CD117+ ovarian cancer stem cells. Stem Cell Res. Ther. 2015, 6, 262. [Google Scholar] [CrossRef]
- Ma, H.; Tian, T.; Cui, Z. Targeting ovarian cancer stem cells: A new way out. Stem Cell Res. Ther. 2023, 14, 28. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Galvan, S.; Carnero, A. Targeting cancer stem cells to overcome therapy resistance in ovarian cancer. Cells 2020, 9, 1402. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Padilla, I.; Wilson, M.K.; Clarke, B.A.; Hirte, H.W.; Welch, S.A.; Mackay, H.J.; Biagi, J.J.; Reedijk, M.; Weberpals, J.I.; Fleming, G.F.; et al. A phase II study of single-agent RO4929097, a gamma-secretase inhibitor of Notch signaling, in patients with recurrent platinum-resistant epithelial ovarian cancer: A study of the Princess Margaret, Chicago and California phase II consortia. Gynecol. Oncol. 2015, 137, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Rizvi, A.; Banerjee, A.; Naseem, I.; Cui, T.; Zheng, Y.; Wani, A.A.; Han, C.; Wang, Q.-E. Depleting ovarian cancer stem cells with calcitriol. Oncotarget 2018, 9, 14481–14491. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.N.; Gunderson, C.C.; Sabbatini, P.; McMeekin, D.S.; Mantia-Smaldone, G.; Burger, R.A.; Morgan, M.A.; Kapoun, A.M.; Brachmann, R.K.; Stagg, R.; et al. A phase 1b dose escalation study of ipafricept (OMP54F28) in combination with paclitaxel and carboplatin in patients with recurrent platinum-sensitive ovarian cancer. Gynecol. Oncol. 2019, 154, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Hess, D.; von Moos, R.; Homicsko, K.; Griguolo, G.; Joerger, M.; Mark, M.; Ackermann, C.J.; Allegrini, S.; Catapano, C.V.; et al. Phase I trial of the oral smoothened inhibitor sonidegib in combination with paclitaxel in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.B.; Fehrenbacher, L.; Holloway, R.; Amit, A.; Karlan, B.; Slomovitz, B.; Sabbatini, P.; Fu, L.; Yauch, R.L.; Chang, I.; et al. A phase II, randomized, placebo-controlled study of vismodegib as maintenance therapy in patients with ovarian cancer in second or third complete remission. Clin. Cancer Res. 2012, 18, 6509–6518. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.X.; Siu, M.K.Y.; Wang, J.J.; Leung, T.H.Y.; Chan, D.W.; Cheung, A.N.Y.; Ngan, H.Y.S.; Chan, K.K.L. PFKFB3 regulates chemoresistance, metastasis and stemness via IAP proteins and the NF-kappaB signaling pathway in ovarian cancer. Front. Oncol. 2022, 12, 748403. [Google Scholar] [CrossRef]
- Kim, D.; Choi, B.H.; Ryoo, I.G.; Kwak, M.K. High NRF2 level mediates cancer stem cell-like properties of aldehyde dehydrogenase (ALDH)-high ovarian cancer cells: Inhibitory role of all-trans retinoic acid in ALDH/NRF2 signaling. Cell Death Dis. 2018, 9, 896. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.; Srivastava, A.K.; Han, C.; Wu, D.; Wani, N.; Liu, L.; Gao, Z.; Qu, M.; Zou, N.; Zhang, X.; et al. DDB2 represses ovarian cancer cell dedifferentiation by suppressing ALDH1A1. Cell Death Dis. 2018, 9, 561. [Google Scholar] [CrossRef]
- Matei, D.; Ghamande, S.; Roman, L.; Alvarez Secord, A.; Nemunaitis, J.; Markham, M.J.; Nephew, K.P.; Jueliger, S.; Oganesian, A.; Naim, S.; et al. A Phase I clinical trial of guadecitabine and carboplatin in platinum-resistant, recurrent ovarian cancer: Clinical, pharmacokinetic, and pharmacodynamic analyses. Clin. Cancer Res. 2018, 24, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cardenas, H.; Fang, F.; Condello, S.; Taverna, P.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. Epigenetic targeting of ovarian cancer stem cells. Cancer Res. 2014, 74, 4922–4936. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: Achieving the full therapeutic potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Zhu, H.; Lee, J.H.; Kossenkov, A.V.; Wu, S.Y.; Wickramasinghe, J.M.; Yin, X.; Palozola, K.C.; Gardini, A.; Showe, L.C.; et al. BET inhibitors suppress ALDH activity by targeting ALDH1A1 super-enhancer in ovarian cancer. Cancer Res. 2016, 76, 6320–6330. [Google Scholar] [CrossRef]
- Klapdor, R.; Wang, S.; Morgan, M.; Dork, T.; Hacker, U.; Hillemanns, P.; Buning, H.; Schambach, A. Characterization of a novel third-generation anti-CD24-CAR against ovarian cancer. Int. J. Mol. Sci. 2019, 20, 660. [Google Scholar] [CrossRef]
- Muinao, T.; Deka Boruah, H.P.; Pal, M. Diagnostic and prognostic biomarkers in ovarian cancer and the potential roles of cancer stem cells—An updated review. Exp. Cell Res. 2018, 362, 1–10. [Google Scholar] [CrossRef]
- Coelho, R.; Ricardo, S.; Amaral, A.L.; Huang, Y.L.; Nunes, M.; Neves, J.P.; Mendes, N.; Lopez, M.N.; Bartosch, C.; Ferreira, V.; et al. Regulation of invasion and peritoneal dissemination of ovarian cancer by mesothelin manipulation. Oncogenesis 2020, 9, 61. [Google Scholar] [CrossRef]
- Chen, J.; Hu, J.; Gu, L.; Ji, F.; Zhang, F.; Zhang, M.; Li, J.; Chen, Z.; Jiang, L.; Zhang, Y.; et al. Anti-mesothelin CAR-T immunotherapy in patients with ovarian cancer. Cancer Immunol. Immunother. 2023, 72, 409–425. [Google Scholar] [CrossRef]
- Sacks, J.D.; Barbolina, M.V. Expression and function of CD44 in epithelial ovarian carcinoma. Biomolecules 2015, 5, 3051–3066. [Google Scholar] [CrossRef]
- Shah, V.; Taratula, O.; Garbuzenko, O.B.; Taratula, O.R.; Rodriguez-Rodriguez, L.; Minko, T. Targeted nanomedicine for suppression of CD44 and simultaneous cell death induction in ovarian cancer: An optimal delivery of siRNA and anticancer drug. Clin. Cancer Res. 2013, 19, 6193–6204. [Google Scholar] [CrossRef]
- Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Neural cell adhesion molecules of the immunoglobulin superfamily regulate synapse formation, maintenance, and function. Trends Neurosci. 2017, 40, 295–308. [Google Scholar] [CrossRef]
- Maten, M.V.; Reijnen, C.; Pijnenborg, J.M.A.; Zegers, M.M. L1 cell adhesion molecule in cancer, a systematic review on domain-specific functions. Int. J. Mol. Sci. 2019, 20, 4180. [Google Scholar] [CrossRef]
- Meli, M.L.; Carrel, F.O.; Waibel, R.; Amstutz, H.; Crompton, N.; Jaussi, R.; Moch, H.; Schubiger, P.A.; Novak-Hofer, I. Anti-neuroblastoma antibody chCE7 binds to an isoform of L1-CAM present in renal carcinoma cells. Int. J. Cancer 1999, 83, 401–408. [Google Scholar] [CrossRef]
- Grünberg, J.; Knogler, K.; Waibel, R.; Hofer-Novak, I. High-yield production of recombinant antibody fragments in HEK-293 cells using sodium butyrate. BioTechniques 2003, 35, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Fogel, M.; Harari, A.; Muller-Holzner, E.; Zeimet, A.G.; Moldenhauer, G.; Altevogt, P. A standardized staining protocol for L1CAM on formalin-fixed, paraffin-embedded tissues using automated platforms. Int. J. Biol. Markers 2014, 29, e180–e183. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Eggle, D.; Klostermann, S. L1-CAM as a target for treatment of cancer with monoclonal antibodies. Anticancer Res. 2009, 29, 4919–4932. [Google Scholar]
- Rathjen, F.G.; Schachner, M. Immunocytological and biochemical characterization of a new neuronal cell surface component (L1 antigen) which is involved in cell adhesion. EMBO J. 1984, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Moos, M.; Tacke, R.; Scherer, H.; Teplowt, D.; Friih, K.; Schachner, M. Neural adhesion molecule L1 as a member of the immunoglobulin superfamily with binding domains similar to fibronectin. Nature 1988, 334, 701–703. [Google Scholar] [CrossRef]
- Patzke, C.; Acuna, C.; Giam, L.R.; Wernig, M.; Sudhof, T.C. Conditional deletion of L1CAM in human neurons impairs both axonal and dendritic arborization and action potential generation. J. Exp. Med. 2016, 213, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Schmid, R.S.; Maness, P.F. L1 and NCAM adhesion molecules as signaling coreceptors in neuronal migration and process outgrowth. Curr. Opin. Neurobiol. 2008, 18, 245–250. [Google Scholar] [CrossRef]
- Dahme, M.; Bartsch, U.; Martini, R.; Anliker, B.; Schachner, M.; Mantei, N. Disruption of the mouse L1 gene leads to malformations of the nervous system. Nat. Genet. 1997, 17, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Brümmendorf, T.; Kenwrickt, S.; Rathjen, F.G. Neural cell recognition molecule L1: From cell biology to human hereditary brain malformations. Development 1998, 8, 87–97. [Google Scholar] [CrossRef]
- Valente, P.; Lignani, G.; Medrihan, L.; Bosco, F.; Contestabile, A.; Lippiello, P.; Ferrea, E.; Schachner, M.; Benfenati, F.; Giovedi, S.; et al. Cell adhesion molecule L1 contributes to neuronal excitability regulating the function of voltage-gated Na+ channels. J. Cell Sci. 2016, 129, 1878–1891. [Google Scholar] [CrossRef]
- Isik, E.; Onay, H.; Atik, T.; Akgun, B.; Cogulu, O.; Ozkinay, F. Clinical and genetic features of L1 syndrome patients: Definition of two novel mutations. Clin. Neurol. Neurosurg. 2018, 172, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Pechriggl, E.J.; Concin, N.; Blumer, M.J.; Bitsche, M.; Zwierzina, M.; Dudas, J.; Koziel, K.; Altevogt, P.; Zeimet, A.G.; Fritsch, H. L1CAM in the early enteric and urogenital system. J. Histochem. Cytochem. 2017, 65, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Debiec, H.; Christensen, E.I.; Ronco, P.M. The cell adhesion molecule L1 is developmentally regulated in the renal epithelium and is involved in kidney branching morphogenesis. J. Cell Biol. 1998, 143, 2067–2079. [Google Scholar] [CrossRef]
- Flaviana, C.; Monica, P.; Terenzio, C.; Raffaele, M.; Valentina, A.; Giulia, C.; Peter, V.E.; Giorgio, N.; Massimo, C.; Ferdinando, C.; et al. L1CAM expression in human gastrointestinal tract development: From tongue to colon-rectum. J. Public Health Res. 2023, 12, 22799036231165624. [Google Scholar] [CrossRef]
- Huszar, M.; Moldenhauer, G.; Gschwend, V.; Ben-Arie, A.; Altevogt, P.; Fogel, M. Expression profile analysis in multiple human tumors identifies L1 (CD171) as a molecular marker for differential diagnosis and targeted therapy. Hum. Pathol. 2006, 37, 1000–1008. [Google Scholar] [CrossRef]
- Maddaluno, L.; Verbrugge, S.E.; Martinoli, C.; Matteoli, G.; Chiavelli, A.; Zeng, Y.; Williams, E.D.; Rescigno, M.; Cavallaro, U. The adhesion molecule L1 regulates transendothelial migration and trafficking of dendritic cells. J. Exp. Med. 2009, 206, 623–635. [Google Scholar] [CrossRef]
- Ganesh, K.; Basnet, H.; Kaygusuz, Y.; Laughney, A.M.; He, L.; Sharma, R.; O’Rourke, K.P.; Reuter, V.P.; Huang, Y.H.; Turkekul, M.; et al. L1CAM defines the regenerative origin of metastasis-initiating cells in colorectal cancer. Nat. Cancer 2020, 1, 28–45. [Google Scholar] [CrossRef]
- Er, E.E.; Valiente, M.; Ganesh, K.; Zou, Y.; Agrawal, S.; Hu, J.; Griscom, B.; Rosenblum, M.; Boire, A.; Brogi, E.; et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat. Cell Biol. 2018, 20, 966–978. [Google Scholar] [CrossRef]
- Kiefel, H.; Pfeifer, M.; Bondong, S.; Hazin, J.; Altevogt, P. Linking L1CAM-mediated signaling to NF-kappaB activation. Trends Mol. Med. 2011, 17, 178–187. [Google Scholar] [CrossRef]
- Islam, R.; Kristiansen, L.V.; Romani, S.; Garcia-Alonso, L.; Hortsch, M. Activation of EGF receptor kinase by L1-mediated homophilic cell interactions. Mol. Biol. Cell 2004, 15, 2003–2012. [Google Scholar] [CrossRef]
- Kulahin, N.; Li, S.; Hinsby, A.; Kiselyov, V.; Berezin, V.; Bock, E. Fibronectin type III (FN3) modules of the neuronal cell adhesion molecule L1 interact directly with the fibroblast growth factor (FGF) receptor. Mol. Cell Neurosci. 2008, 37, 528–536. [Google Scholar] [CrossRef]
- Gutwein, P.; Mechtersheimer, S.; Riedle, S.; Stoeck, A.; Gast, D.; Joumaa, S.; Zentgraf, H.; Fogel, M.; Altevogt, D.P. ADAM10-mediated cleavage of L1 adhesion molecule at the cell surface and in released membrane vesicles. FASEB J. 2003, 17, 292–294. [Google Scholar] [CrossRef]
- Riedle, S.; Kiefel, H.; Gast, D.; Bondong, S.; Wolterink, S.; Gutwein, P.; Altevogt, P. Nuclear translocation and signalling of L1-CAM in human carcinoma cells requires ADAM10 and presenilin/gamma-secretase activity. Biochem. J. 2009, 420, 391–402. [Google Scholar] [CrossRef]
- Linneberg, C.; Toft, C.L.F.; Kjaer-Sorensen, K.; Laursen, L.S. L1cam-mediated developmental processes of the nervous system are differentially regulated by proteolytic processing. Sci. Rep. 2019, 9, 3716. [Google Scholar] [CrossRef]
- Stoeck, A.; Gast, D.; Sanderson, M.P.; Issa, Y.; Gutwein, P.; Altevogt, P. L1-CAM in a membrane-bound or soluble form augments protection from apoptosis in ovarian carcinoma cells. Gynecol. Oncol. 2007, 104, 461–469. [Google Scholar] [CrossRef]
- Friedli, A.; Fischer, E.; Novak-Hofer, I.; Cohrs, S.; Ballmer-Hofer, K.; Schubiger, P.A.; Schibli, R.; Grunberg, J. The soluble form of the cancer-associated L1 cell adhesion molecule is a pro-angiogenic factor. Int. J. Biochem. Cell Biol. 2009, 41, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Gast, D.; Riedle, S.; Issa, Y.; Pfeifer, M.; Beckhove, P.; Sanderson, M.P.; Arlt, M.; Moldenhauer, G.; Fogel, M.; Kruger, A.; et al. The cytoplasmic part of L1-CAM controls growth and gene expression in human tumors that is reversed by therapeutic antibodies. Oncogene 2008, 27, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Gavert, N.; Ben-Shmuel, A.; Lemmon, V.; Brabletz, T.; Ben-Ze’ev, A. Nuclear factor-kappaB signaling and ezrin are essential for L1-mediated metastasis of colon cancer cells. J. Cell Sci. 2010, 123, 2135–2143. [Google Scholar] [CrossRef] [PubMed]
- Kiefel, H.; Bondong, S.; Erbe-Hoffmann, N.; Hazin, J.; Riedle, S.; Wolf, J.; Pfeifer, M.; Arlt, A.; Schafer, H.; Muerkoster, S.S.; et al. L1CAM-integrin interaction induces constitutive NF-kappaB activation in pancreatic adenocarcinoma cells by enhancing IL-1beta expression. Oncogene 2010, 29, 4766–4778. [Google Scholar] [CrossRef]
- Kiefel, H.; Bondong, S.; Pfeifer, M.; Schirmer, U.; Erbe-Hoffmann, N.; Schafer, H.; Sebens, S.; Altevogt, P. EMT-associated up-regulation of L1CAM provides insights into L1CAM-mediated integrin signalling and NF-kappaB activation. Carcinogenesis 2012, 33, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Zuo, C.; Hong, Y.; Qiu, X.; Yang, D.; Liu, N.; Sheng, X.; Zhou, K.; Tang, B.; Xiong, S.; Ma, M.; et al. Celecoxib suppresses proliferation and metastasis of pancreatic cancer cells by down-regulating STAT3/NF-kB and L1CAM activities. Pancreatology 2018, 18, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Tatti, O.; Gucciardo, E.; Pekkonen, P.; Holopainen, T.; Louhimo, R.; Repo, P.; Maliniemi, P.; Lohi, J.; Rantanen, V.; Hautaniemi, S.; et al. MMP16 mediates a proteolytic switch to promote cell-cell adhesion, collagen alignment, and lymphatic invasion in melanoma. Cancer Res. 2015, 75, 2083–2094. [Google Scholar] [CrossRef]
- Matsumoto-Miyai, K.; Ninomiya, A.; Yamasaki, H.; Tamura, H.; Nakamura, Y.; Shiosaka, S. NMDA-dependent proteolysis of presynaptic adhesion molecule L1 in the hippocampus by neuropsin. J. Neurosci. 2003, 23, 7727–7736. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.; Haspel, J.; Kane-Goldsmith, N.; Grumet, M. L1 mediated homophilic binding and neurite outgrowth are modulated by alternative splicing of exon 2. J. Neurobiol. 2002, 51, 177–189. [Google Scholar] [CrossRef]
- Angiolini, F.; Belloni, E.; Giordano, M.; Campioni, M.; Forneris, F.; Paronetto, M.P.; Lupia, M.; Brandas, C.; Pradella, D.; Di Matteo, A.; et al. A novel L1CAM isoform with angiogenic activity generated by NOVA2-mediated alternative splicing. eLife 2019, 8, e44305. [Google Scholar] [CrossRef]
- Altevogt, P.; Ben-Ze’ev, A.; Gavert, N.; Schumacher, U.; Schafer, H.; Sebens, S. Recent insights into the role of L1CAM in cancer initiation and progression. Int. J. Cancer 2020, 147, 3292–3296. [Google Scholar] [CrossRef] [PubMed]
- Angiolini, F.; Cavallaro, U. The pleiotropic role of L1CAM in tumor vasculature. Int. J. Mol. Sci. 2017, 18, 254. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, K.W.; Ahn, D.G.; Oh, K.Y.; Yoon, H.J. Clinical significance of L1CAM expression and its biological role in the progression of oral squamous cell carcinoma. Oncol. Rep. 2023, 49, 67. [Google Scholar] [CrossRef]
- Bosse, T.; Nout, R.A.; Stelloo, E.; Dreef, E.; Nijman, H.W.; Jurgenliemk-Schulz, I.M.; Jobsen, J.J.; Creutzberg, C.L.; Smit, V.T. L1 cell adhesion molecule is a strong predictor for distant recurrence and overall survival in early stage endometrial cancer: Pooled PORTEC trial results. Eur. J. Cancer 2014, 50, 2602–2610. [Google Scholar] [CrossRef]
- Li, Y.; Galileo, D.S. Soluble L1CAM promotes breast cancer cell adhesion and migration in vitro, but not invasion. Cancer Cell Int. 2010, 10, 34. [Google Scholar] [CrossRef]
- Sung, S.-Y.; Wu, I.-H.; Chuang, P.-H.; Petros, J.A.; Wu, H.-C.; Zeng, H.-J.; Huang, W.-C.; Chung, L.W.K.; Hsieh, C.-L. Targeting L1 cell adhesion molecule expression using liposomeencapsulated siRNA suppresses prostate cancer bone metastasis and growth. Oncotarget 2014, 5, 9911–9929. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Massague, J. Targeting metastatic cancer. Nat. Med. 2021, 27, 34–44. [Google Scholar] [CrossRef]
- Wolterink, S.; Moldenhauer, G.; Fogel, M.; Kiefel, H.; Pfeifer, M.; Luttgau, S.; Gouveia, R.; Costa, J.; Endell, J.; Moebius, U.; et al. Therapeutic antibodies to human L1CAM: Functional characterization and application in a mouse model for ovarian carcinoma. Cancer Res. 2010, 70, 2504–2515. [Google Scholar] [CrossRef]
- Fischer, E.; Grünberg, J.; Cohrs, S.; Hohn, A.; Waldner-Knogler, K.; Jeger, S.; Zimmermann, K.; Novak-Hofer, I.; Schibli, R. L1-CAM-targeted antibody therapy and 177Lu-radioimmunotherapy of disseminated ovarian cancer. Int. J. Cancer 2012, 130, 2715–2721. [Google Scholar] [CrossRef]
- Zecchini, S.; Bianchi, M.; Colombo, N.; Fasani, R.; Goisis, G.; Casadio, C.; Viale, G.; Liu, J.; Herlyn, M.; Godwin, A.K.; et al. The differential role of L1 in ovarian carcinoma and normal ovarian surface epithelium. Cancer Res. 2008, 68, 1110–1118. [Google Scholar] [CrossRef]
- Gast, D.; Riedle, S.; Riedle, S.; Schabath, H.; Schlich, S.; Schneider, A.; Issa, Y.; Stoeck, A.; Fogel, M.; Joumaa, S.; et al. L1 augments cell migration and tumor growth but not beta3 integrin expression in ovarian carcinomas. Int. J. Cancer 2005, 115, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Larson, S.M.; Carrasquillo, J.A.; Cheung, N.K.; Press, O.W. Radioimmunotherapy of human tumours. Nat. Rev. Cancer 2015, 15, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Funeh, C.N.; Bridoux, J.; Ertveldt, T.; De Groof, T.W.M.; Chigoho, D.M.; Asiabi, P.; Covens, P.; D’Huyvetter, M.; Devoogdt, N. Optimizing the Safety and Efficacy of Bio-Radiopharmaceuticals for Cancer Therapy. Pharmaceutics 2023, 15, 1378. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, J.; Kim, H.; Lee, Y.J.; Kim, J.S.; Lim, S.M. Combination radioimmunotherapy strategies for solid tumors. Int. J. Mol. Sci. 2019, 20, 5579. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.K.; Dalal, V.; Jain, M.; Batra, S.K. Delivery of radioimmunotherapy for solid tumors. In Systemic Drug Delivery Strategies; Academic Press: Cambridge, MA, USA, 2022; pp. 437–461. [Google Scholar]
- Cheal, S.M.; Chung, S.K.; Vaughn, B.A.; Cheung, N.V.; Larson, S.M. Pretargeting: A path forward for radioimmunotherapy. J. Nucl. Med. 2022, 63, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Rondon, A.; Rouanet, J.; Degoul, F. Radioimmunotherapy in oncology: Overview of the last decade clinical trials. Cancers 2021, 13, 5570. [Google Scholar] [CrossRef] [PubMed]
- Hallqvist, A.; Bergmark, K.; Back, T.; Andersson, H.; Dahm-Kahler, P.; Johansson, M.; Lindegren, S.; Jensen, H.; Jacobsson, L.; Hultborn, R.; et al. Intraperitoneal alpha-emitting radioimmunotherapy with 211At in relapsed ovarian cancer: Long-term follow-up with individual absorbed dose estimations. J. Nucl. Med. 2019, 60, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Nimmagadda, S.; Penet, M.F. Ovarian cancer targeted theranostics. Front. Oncol. 2019, 9, 1537. [Google Scholar] [CrossRef]
- Chung, S.K.; Vargas, D.B.; Chandler, C.S.; Katugampola, S.; Veach, D.R.; McDevitt, M.R.; Seo, S.H.; Vaughn, B.A.; Rinne, S.S.; Punzalan, B.; et al. Efficacy of HER2-targeted Intraperitoneal 225Ac alpha-pretargeted radioimmunotherapy for small-volume ovarian peritoneal carcinomatosis. J. Nucl. Med. 2023, 64, 1439–1445. [Google Scholar] [CrossRef]
- Hoefnagel, C.A.; Rutgers, M.; Buitenhuis, C.K.; Smets, L.A.; Kraker, J.; Meli, M.; Carrel, F.; Amstutz, H.; Schubiger, P.A.; Novak-Hofer, I. A comparison of targetting of neuroblastoma with mIBG and anti L1-CAM antibody mAb chCE7: Therapeutic efficacy in a neuroblastoma xenograft model and imaging of neuroblastoma patients. Eur. J. Nucl. Med. 2001, 28, 359–368. [Google Scholar] [CrossRef]
- Lindenblatt, D.; Terraneo, N.; Pellegrini, G.; Cohrs, S.; Spycher, P.R.; Vukovic, D.; Behe, M.; Schibli, R.; Grünberg, J. Combination of lutetium-177 labeled anti-L1CAM antibody chCE7 with the clinically relevant protein kinase inhibitor MK1775: A novel combination against human ovarian carcinoma. BMC Cancer 2018, 18, 922. [Google Scholar] [CrossRef] [PubMed]
- Lindenblatt, D.; Fischer, E.; Cohrs, S.; Schibli, R.; Grünberg, J. Paclitaxel improved anti-L1CAM lutetium-177 radioimmunotherapy in an ovarian cancer xenograft model. EJNMMI Res. 2014, 4, 54. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todorov, T.Z.; Schibli, R.; Béhé, M.; Grünberg, J. Targeting Cancer Stem Cells with Radioimmunotherapy: The Case of the Ovarian Cancer Stemness-Associated Biomarker L1CAM. Int. J. Transl. Med. 2024, 4, 463-485. https://doi.org/10.3390/ijtm4030031
Todorov TZ, Schibli R, Béhé M, Grünberg J. Targeting Cancer Stem Cells with Radioimmunotherapy: The Case of the Ovarian Cancer Stemness-Associated Biomarker L1CAM. International Journal of Translational Medicine. 2024; 4(3):463-485. https://doi.org/10.3390/ijtm4030031
Chicago/Turabian StyleTodorov, Tihomir Zh., Roger Schibli, Martin Béhé, and Jürgen Grünberg. 2024. "Targeting Cancer Stem Cells with Radioimmunotherapy: The Case of the Ovarian Cancer Stemness-Associated Biomarker L1CAM" International Journal of Translational Medicine 4, no. 3: 463-485. https://doi.org/10.3390/ijtm4030031
APA StyleTodorov, T. Z., Schibli, R., Béhé, M., & Grünberg, J. (2024). Targeting Cancer Stem Cells with Radioimmunotherapy: The Case of the Ovarian Cancer Stemness-Associated Biomarker L1CAM. International Journal of Translational Medicine, 4(3), 463-485. https://doi.org/10.3390/ijtm4030031