Abstract
Microalgae biomass is considered a promising alternative feedstock for biodiesel production due to its high productivity of neutral lipids, especially under abiotic stress conditions. Among the unicellular microalgae that show this characteristic, Chlamydomonas reinhardtii appears as one of the most important model species with increased lipid production under abiotic stress conditions. In this study, we show that C. reinhardtii cells cultivated under mixotrophic condition supplemented with 0.1 M of NaCl rapidly raise their intracellular amount of neutral lipids without a reduction in their cellular growth rate, representing a promising condition for biomass production toward bioenergy applications. The nuclear proteome of these cells was investigated, and we identified 323 proteins with an enrichment of almost 60% of nuclear proteins in the total dataset. We found 61 proteins differentially regulated upon salt treatment, including proteins annotated in functional categories related to translation and nucleosome assembly functions. Additionally, we identified transcription factor proteins (TFs) and analyzed their likely transcription factor-binding regulatory elements, identifying target genes related to lipid metabolism and kinase functions, indicating possible regulatory pathways of lipid biosynthesis. Together, these data can help understand regulatory nuclear mechanisms, leading to an increase in lipids in the first 24 h of salt stress.
1. Introduction
In recent decades, research on renewable energy has received increased interest and funding. This has been driven mainly by climate change-related concerns which are demanding a transition from fossil fuel dependency to alternative and more sustainable energy sources, as indiscriminate fossil fuel usage has led to a worsening of the greenhouse effect and atmospheric pollution [,]. In this context, biomass production from microalgal cultures poses a valuable solution for sustainable consumption of atmospheric carbon dioxide coupled to bioenergy generation [,]. Through photosynthesis, these fast-growing microorganisms are capable of assimilating carbon dioxide into their biomass while generating, for instance, starch and lipids which serve as potential feedstock for biofuel production. Furthermore, microalgal biomass might also be converted to a plethora of industrialized products such as bioplastics (biopolymer industry), pigments (food and cosmetic industry), and nutraceuticals (food industry) [].
When facing salt stress and nutrient deprivation, some microalgae species can increase lipid biosynthesis and storage [,,,]. However, this stress can also limit cell growth, consequently hindering the biomass production []. Chlamydomonas reinhardtii is a green unicellular flagellate microalga that has been widely employed as a model organism for chloroplast and, more recently, systems biology studies of cellular stress responses [,]. The genome of C. reinhardtii was sequenced in 2007 [], which has since allowed expanding systems and genetic analysis, as well as enabled genome annotation and functional genomics analysis through omics studies []. Aiming to increase lipid yields, such studies have been pivotal to unravel the pathways and molecular mechanisms associated with stress-related responses to and consequences for biomass production in microalgae.
The cellular response to salt stress in C. reinhardtii has been widely studied [,,]. The exposure of cells to 0.2 M NaCl is highly toxic, as it raises the levels of intracellular peroxides with subsequent inhibition of photosynthesis and cell growth []. In contrast, the exposure of cells to lower concentrations of NaCl, specifically 0.1 M, drives increased production of lipids []. However, the regulatory proteins involved in the adaptation to salt stress and increased lipid yields are not yet fully characterized. Although some studies have already reported a nuclear proteome analysis during physiological conditions and under CO2 deprivation [,], the identity of the transcriptional regulator proteins (TRs) and transcription factor proteins (TFs) involved in the cell response to salt stress (0.1 M NaCl) are still mostly unknown. In this study, we analyzed the nuclear proteome of C. reinhardtii during acclimation to salt stress, identifying differentially expressed proteins and stress response-associated target genes that may be regulated by the TFs identified in the nuclear fraction analyzed.
2. Material and Methods
2.1. Cell Culture and Growth Conditions
Chlamydomonas reinhardtii (strain cc-503 cw92 mt+) was obtained from the Chlamydomonas Resource Center (University of Minnesota, Minneapolis, MN, USA). Cells were cultivated under mixotrophic and temperature-controlled conditions in Tris acetate phosphate (TAP) [] medium with continuous shaking (100 rpm) under continuous light (~100 µE·m−2·s−1) conditions.
Cells were cultivated in 2 L flasks containing 0.5 L of medium with an initial cell density of 1 × 105 cells/mL and were cultivated up to the exponential growth phase with either TAP medium (Control) or TAP supplemented with 0.1 M NaCl (Treatment). Cells were harvested every 24 h for cell growth monitoring and counting using a Countess II FL Automated Cell Counter (Thermo Fisher Scientific, Waltham, MA, USA). Nuclear proteomic studies were performed for the cells under control and treatment (0.1 M NaCl) conditions sampled 24 h after the initial cell inoculation. All experimental data are expressed as the mean value of three independently performed experiments with their standard deviation (SD).
2.2. Neutral Lipid Quantification
Neutral lipids were quantified by fluorescence measurement adapted from a method previously described by Gorman and colleagues []. Briefly, 1 × 106 cells were centrifuged at 5000× g for 10 min at 4 °C and resuspended in TAP medium. Then, DMSO was added to the samples to the final concentration of 1% and incubated at room temperature for 5 min. After this time, Nile Red solution (1 µg/mL) was added to the cell suspension, and the samples were incubated for 10 min in an amber flask at room temperature. Fluorescence was measured in 96-well plates with wavelengths at 530 nm excitation and 580 nm emission in a Synergy H1 Hybrid Reader (BioTek Technologies, Winooski, VT, USA). Each measurement was repeated three times.
2.3. Nucleus Isolation and Extraction of Nuclear Proteins
The nuclear protein extraction was performed as previously described by Winck and colleagues [] with a CelLytic PN extraction kit (Sigma-Aldrich, St. Louis, MO, USA). To avoid proteolytic degradation, plant protease inhibitor (Sigma, USA) was added, and all steps were performed at 4° C. A pellet of 250 mL of cell culture was resuspended in 600 µL of NIB solution and macerated in liquid nitrogen using a mortar and pestle. The powdered material was suspended in 10 mL of NIB solution, filtered through a miracloth membrane (22–25 μm; Calbiochem, Bad Soden, Germany), and centrifuged at 1260× g for 10 min. To the cell pellet, NIBA buffer containing 1% Triton X-100 was added. After homogenization, the sample was centrifuged at 1000× g for 30 min. The cell pellet was washed twice by homogenization in 1 mL of NIB solution and centrifuged at 600× g for 10 min. The pellet enriched with nuclei was resuspended in 1 mL of NIBA and extraction buffer (1.2 M potassium acetate, 25 mM Hepes/KOH, 10% glycerol, and 1 mM EDTA). The resulting nuclei were disrupted by 1 min sonication (60% amplitude, 1 min cycles with 10 s on and 20 s off) using a sonicator Vibra Cell VCX 130 device (Sonics & Materials, Inc., Newtown, CT, USA). Finally, samples were centrifuged at 13,000× g for 15 min, and nuclear proteins were collected from the supernatant for quantification by the Bradford method using the Bio-Rad protein assay kit (Bio-Rad Labs., Richmond, CA, USA) [].
2.4. Mass Spectrometry Analysis
First, 2 µg of protein extracts were solubilized with 8 M urea in a proportion of 1:1, reduced with 5 mM DTT, alkylated with 14 mM iodoacetamide (IAA), treated with 5 mM DTT for 15 min in the dark, and digested with trypsin (Promega, Madison, WI, USA) in the proportion 1:50 for 16 h at 37 °C. The samples containing the peptides were desalted with STAGE tips with C18 discs (3M, Neuss, Germany). Peptides were eluted in 0.1% formic acid. The peptides were separated and analyzed by high-resolution LC–MS/MS using the Mass Spectrometer Q-ToF Maxis 3G (Bruker, Billerica, MA, USA) coupled to a Nano LC Acquity (Waters Corporation, Milford, MA, USA) at the Central Analytical Instrumentation Facility of the Institute of Chemistry of the University of São Paulo. The CaptiveSpray ion source was used in spectrometry, and the electrospray voltage was adjusted to 2 kV, with the source temperature at 150 °C. Peptide separation was performed in two steps, with accumulation of peptides in a nanoAcquity UPLC® 2G-V/MTrap Symmetry® C18 column (180 µm × 20 mm, 5 µm) for 3 min at a flow of 7 µL/min in 0.1% formic acid, followed by an analytical separation using a nanoAcquity UPLC® BEH130 (100 µm × 100 mm, 1.7 µm), with an acetonitrile gradient of 2–85% in 0.1% formic acid as the mobile phase at a flow of 0.3 µL/min for 85 min.
2.5. Data Analysis
Proteins were identified using MaxQuant software v.1.6.17.0 [] available on our Galaxy server platform (http://143.107.55.132/galaxy/, accessed on 20 September 2021) against the reference proteome database of C. reinhardtii v5.5 annotation (17,741 proteins), retrieved from the Phytozome 12 [] platform. The maximum error tolerance accepted was 6 ppm for the precursor ion search (MS search) and 0.5 Da for the fragment search (MS/MS search), while a maximum of two missed cleavages were accepted, with a maximum of 1% false discovery rate (FDR) applied for the identification of peptides and proteins. For FDR calculation, we applied a reverse proteome database as decoy, which was created automatically from the reference proteome database by reversing the sequence of the amino acids of the peptides generated from in silico digestion of the reference proteome. Cysteine carbamidomethylation was set as a fixed modification, and methionine oxidation was set as a variable modification.
Statistical analyses were carried out using Perseus software (version 1.6.14.0) []. Protein hits were accepted if they were quantified in at least two of the three experimental replicates performed and were present in at least one experimental condition. Proteins only identified by site and reverse entries were excluded from further analysis. Significantly differentially expressed proteins were calculated using a two-sample Student’s t-test with a Benjamini–Hochberg FDR threshold of 0.05 as a post hoc test [].
Enrichment analysis of the protein annotation terms from Gene Ontology (GO; www.geneontology.org, accessed on 22 September 2021) was applied for the data mining of the proteomics data. Significantly overrepresented GO terms for the selected list of proteins identified were retrieved using the BiNGO tool [] with a hypergeometric test and p-value correction using the Benjamini–Hochberg method. The visualization of the network of enriched GO terms was performed using the AutoAnnotate application of Cytoscape [,]. The node color (orange–red gradient) indicates the scale of the corrected p-values [].
Protein subcellular localization was predicted using the SUBA prediction tool (http://suba.live/, accessed on 22 September 2021) [] with an Arabidopsis TAIR annotation for homologs of C. reinhardtii proteins []. Furthermore, prediction of nuclear location was performed using the NucPred prediction server (https://nucpred.bioinfo.se/nucpred/, accessed on 22 september of 2021) [] and PredAlgo Subcellular localization prediction tool (https://giavap-genomes.ibpc.fr/predalgo/, accessed on 22 September 2021) [].
2.6. Microscopy
To evaluate nuclei integrity and enrichment, an aliquot of purified nuclei was stained with DAPI (Sigma-Aldrich, St. Louis, MO, USA) and observed with a fluorescence microscope Dmi8 (Leica Camera, Wetzlar, Germany) and confocal microscope CTR6 LED (Leica Camera, Germany).
2.7. Prediction of Transcription Factor Target Genes
Transcription factors and transcription regulators found in the nuclear proteome were identified according to the annotation available at the PlnTFDB database (http://plntfdb.bio.uni-potsdam.de/, accessed on 1 October 2021) []. Furthermore, we performed the identification of their homologs in Arabidopsis thaliana (Araport11 annotation []) using BLASTP (https://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on 1 October 2021). The sequence of known DNA cis-element-binding motifs recognized by these Arabidopsis TF homologous sequences and annotated in the Plant Transcription factor Database v4.0 (http://planttfdb.cbi.pku.edu.cn/, accessed on 1 October 2021) [] were then searched in the promoter regions (≤1500 bp upstream of the transcription start site) of the Chlamydomonas genes (v5.6 genome, Phytozome13) using the MAST tool (http://memesuite.org/tools/mast, accessed on 1 October 2021) []. Sequence hits with an e-value ≤1 were considered for further analysis as potential target genes.
3. Results
3.1. The Impact of 0.1 M NaCl on Growth and Lipid Accumulation in C. reinhardtii
C. reinhardtii has been postulated as a prominent organism for biomass production and, thus, a potential source for sustainable energy production. Importantly, during adaptation to some adverse conditions, prominently salt stress, the ability of C. reinhardtii to produce biomass is usually reduced. To assess this, a comparison between cultures with the supplementation of 0.1 M NaCl (treatment) and without adding NaCl (control) was performed. Cell growth and the concentration of neutral lipids were analyzed over time (0, 24, 48, and 72 h) (Figure 1).
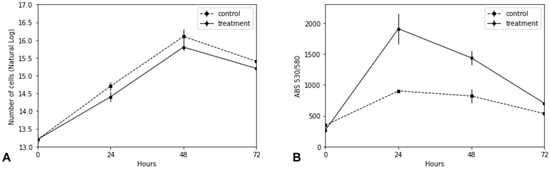
Figure 1.
C. reinhardtii growth and lipid production under saline stress with 0.1 M NaCl. (A) Logarithmic transformed growth curve of non-stressed (dotted line) and salt (0.1 M NaCl)-stressed (solid curve) C. reinhardtii cultures. (B) Relative quantification of neutral lipids of non-stressed (dotted line) and salt-stressed (0.1 M NaCl)- (solid curve). Neutral lipid accumulation was performed in aliquots of 106 cells from C. reinhardtii cultures and measured by fluorescence at 530/580 nm after Nile Red staining. The X-axis indicates the time in hours.
As shown in Figure 1, the treatment of cell cultures with 0.1 M NaCl only marginally affected the growth rate of C. reinhardtii, while it strongly stimulated the accumulation of neutral lipids, especially in the first 48 h of cultivation under salt treatment. After this time, the rate of lipid production sharply decreased, reaching similar levels to that of non-stressed cells. Thus, our study indicates that slight deviations from physiological conditions are sufficient to trigger increased lipid production without affecting cell growth. Although it is reported that C. reinhardtii can survive up to 0.2 M NaCl [,], this condition is reported to cause growth arrest and loss of biomass and, thus, is not well suited for bioenergy production purposes. Therefore, we aimed to identify possible target nuclear regulatory proteins and their target genes that may be associated with increased lipid biosynthesis without affecting cell growth performance, revealing the identity of novel targets for microalgae transcriptional engineering.
3.2. Proteomics Analysis
Having determined that 0.1 M NaCl salt stress does not impact growth but boosts intracellular neutral lipid synthesis, we performed the nuclear proteomics analysis to unravel stress-related factors. During the time-course experiment, cells were harvested after 24 h of exposure to stress conditions for nucleus isolation and protein extraction (please see Section 2 for details). Firstly, the integrity of isolated nuclei was analyzed using 4′,6-diamidino-2-phenylindole (DAPI). The fluorescence micrograph (Figure S1) shows uniform spheres with an average diameter of approximately 3 µm with no visible contamination with other cellular compartments.
3.3. Protein Identification and Subcellular Localization
Nuclear proteins extracted from C. reinhardtii were then analyzed using nano-LC–MS/MS. During data analysis, we initially identified a total of 472 proteins. These data were processed to remove contaminant entries and to filter the list of proteins identified by the number of missing values. We then only considered in further analysis the proteins with at least two valid label-free quantification values across all three replicates in at least one experimental condition, which narrowed our dataset to 323 proteins. Multi-scatter plots showing Pearson correlation maps were used to assess the data quality (Figure S2). In Table S1, we summarize the GO functional categories and expression levels of all 323 proteins identified.
3.4. Enrichment of Nuclei Sample
An important aspect of subcellular proteome analysis is the purity and enrichment of the sample, since it can include protein contamination from other organelles, especially the chloroplast. The prediction of the subcellular localization of the identified proteins and assessment of the presence of potential contaminants were performed using three prediction tools for subcellular location: SUBA [], NucPred [], and PredAlgo [].
At first, to estimate the enrichment of nuclear samples with nuclear proteins, we compared the total proteome and nuclear proteome of cell cultures under the same conditions of 0.1 M NaCl for 24 h (Figure 2A). SUBA location was used to predict the subcellular location of proteins using as input their homologous sequences from Arabidopsis thaliana available at the Arabidopsis Information Resource (TAIR) database (https://www.arabidopsis.org/, accessed on 22 September 2021). In our analysis, we found an increment superior to 50% of annotated nuclear proteins, when compared to the observed 10% of annotated nuclear proteins identified in the whole-cell proteome and the 68% of annotated nuclear proteins identified in the nuclear proteome (Figure 2B).
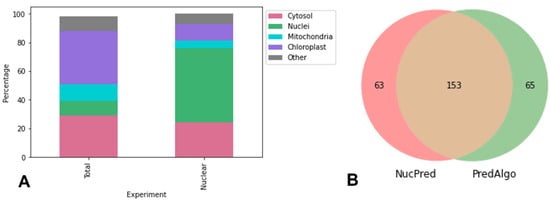
Figure 2.
Prediction of subcellular localization of identified proteins. (A) Percentage of categories of subcellular localization prediction for nuclear and whole-cell proteins performed with SUBA4 (Arabidopsis Subcellular Database). (B) Venn diagram of groups of nuclear-predicted proteins identified by PredAlgo (green circle) (https://giavapgenomes.ibpc.fr/predalgo/) and by NucPred (https://nucpred.bioinfo.se/cgi-bin/single.cgi) (red circle) both accessed on 22 September 2021. The overlapping region represents proteins that were identified by both predictors as nuclear proteins.
PredAlgo and NucPred prediction servers were used to estimate the presence of nuclear proteins in the nuclear proteome analyzed (Figure 2B). The combination of PredAlgo and NucPred predictions revealed that 153 (47%) out of 323 proteins analyzed were predicted to be nuclear localized. PredAlgo analysis revealed that 216 (67%) proteins were indeed nuclear-related proteins, while 62 proteins (19% of total proteins) were associated with the chloroplast, 21 proteins (6.5% of total proteins) were associated with the mitochondria, 10 proteins (3% of total proteins) were classified as extracellular proteins, and the remaining proteins were associated with other cellular compartments.
3.5. Differentially Expressed and Exclusive Nuclear Proteins
For the identification of differentially expressed proteins (DEs), we performed a Student’s t-test (p < 0.05) analysis, with further Benjamini–Hochberg post hoc test correction. We found 61 proteins differentially expressed during exposure to 0.1 M NaCl out of the 323 proteins identified, as summarized in Table 1, which also includes proteins unique in control or treatment samples.

Table 1.
Differentially expressed nuclear proteins in response to 0.1 M salt stress.
We conducted Gene Ontology (GO) term enrichment analysis for all 323 proteins and the 61 DEs to annotate their molecular function and to describe the possible biological role of these proteins using the GO categories biological process (BP), molecular function (MF), and cellular component (CC) with analysis of the distribution of proteins identified into their corresponding GO annotations. The results of GO annotation of the DEs and unique proteins indicated that their annotations were mostly related to the categories of biosynthetic metabolic process (ribosomal proteins in translation section in Table 1) and complex nucleosome assembly (Histone H1-Cre13.g567450.t1.2 and Cre06.g275900.t1.2) (Table 1 and Figure 3). In the CC category, the DEs were mainly enriched in the nucleus annotation category, as expected, and, in terms of MF, the DEs were mainly associated with nucleobase-containing compound kinase activity, represented by Cre16.g654300.t1.2 and Cre01.g029750.t1.1 (Figure S3).
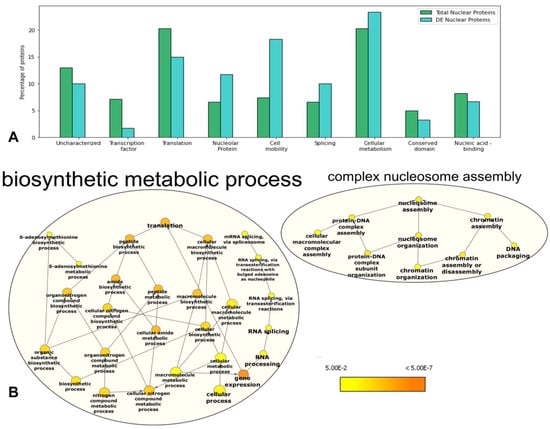
Figure 3.
Functional classification of overrepresented Gene Ontology (GO) terms in the dataset of total and differentially expressed proteins. (A) Bar plot showing a comparison of the percentage distribution of the GO terms categories retrieved for the datasets of total and DEs proteins. (B) Networks of the significantly enriched biological process GO terms in the analysis of the differentially expressed proteins using BiNGO. Illustration of downregulated gene GO enrichment category of biological process (BP). The color gradient refers to the scale of enrichment significance, where orange indicates the highest significance of enrichment, and yellow indicates the minimum significance of term enrichment above the significance cutoff (FDR-corrected = 0.05). The node size represents the GO hierarchy level. The significance threshold of the hypergeometric distribution of the functional annotation categories was set as p < 0.05.
The most significant GO categories annotated for the dataset of nuclear proteomics and nuclear DEs were cellular metabolism and translation. The nuclear proteome list of DEs was annotated to the GO categories of translation, nucleolar proteins, cell mobility, and cellular metabolism. Previous studies of the nuclear proteome of C. reinhardtii also showed the presence of flagellar proteins in the nuclear fraction []. These proteins are expected to be found in the nuclear proteome since they have an association with nuclear transport and may even play a role in nuclear function []. However, these annotated flagellar proteins were absent in our results from cells cultivated in 0.1 M NaCl treatment. This result may be connected to the paralysis or loss of flagella, which is one of the first cellular responses to stress [,].
3.6. Prediction of Nuclear Transcription Factor Target Genes in C. reinhardtii under Salt Stress
We took all 323 proteins present in the nuclear proteome and searched for known transcription factors (TFs) according to the annotation available at the PlnTFDB database (http://plntfdb.bio.uni-potsdam.de/, accessed on 1 October 2021. We identified 19 C. reinhardtii TFs, and their amino-acid sequences were identified and used to further search for their homologous sequences in A. thaliana. Their respective cis-element recognition motifs were then searched in PlantPan. Starting with 323 proteins in C. reinhardtii, we found 16 A. thaliana TFs homologs, all of which featured at least one reported cis-element recognition motif. With the motifs retrieved, we performed a MEME/MAST analysis to find putative target motifs in the promoter regions of Chlamydomonas genes. Overall, we identified 733 unique targets associated with eight of the TFs identified (Table S2).
4. Discussion
4.1. Proteomic Analysis of C. reinhardtii Nuclear Proteins in Salt Stress
In this report, we presented the characterization of the nuclear proteome of C. reinhardtii under salt stress via supplementation of cell culture with 0.1 M NaCl for 24 h, which did not affect the cell growth but stimulated an increase in neutral lipid accumulation.
An increase in lipids was previously reported in salt stress conditions with 0.2 M NaCl [,]. Under salt stress (0.1 M NaCl), cells were reported to increase 50-fold their triacylglycerol (TAG) content in the C. reinhardtii wild-type CC124 strain []. However, the regulatory aspects of the cellular response to salt stress are not yet completely understood.
In our analysis, we identified 323 proteins, with 68% predicted to be nuclear using the SUBA database. We found that 19% of the proteins present in our sample were annotated to the chloroplast, which is somehow expected for protein contaminants from the chloroplast, the largest organelle that occupies almost half of the cell volume in C. reinhardtii. Therefore, even without observing visible contamination of chloroplast fragments in our fluorescence microscopy analysis of the isolated nuclei, we believe that some of the identified proteins may have been derived from organelles other than the nucleus. PredAlgo and NucPred analyses revealed a great enrichment of nuclear proteins (about 58%) in our nuclear proteome with just a few protein contaminants from other cellular compartments. Notably, the presence of such contaminants did not impact further analysis, similar to the results observed in previous publications [,]. Nevertheless, proteins present in both nuclear and cellular fractions (Figure 2B) may also be cytoplasmic proteins that may exert their function in the nucleus at some point during the cellular response to salt stress. Further investigation is needed to elucidate their functional role.
The nucleus is a dynamic organelle which changes its proteome in response to intracellular and environmental stimuli []. The characterization of proteins modulated in response to salt stress conditions can provide novel information about mechanisms of the lipid accumulation and adaptation to salt stress at the molecular level. The function of most proteins found in the nuclear proteome seems to be related to cellular metabolism (nitrogen/organic cyclic compound metabolic process) and translation. In the set of DE proteins, we found proteins related to translation, nucleolar proteins, cell mobility, and cellular metabolism downregulated or unique in the non-stressed sample (control), indicating target proteins that may be involved in the regulation of ribosome assembly under abiotic stress. At 24 h under salt stress, even though the cells did not show significant alteration of cell growth rate, it seems that cells already respond by modulating the expression of proteins of the cellular machinery of protein synthesis.
Among the differentially expressed proteins identified, we highlight the several ribosomal and nucleolar proteins observed. Although their presence is expected in a nuclear isolate, their differential expression indicates a drastic change in the ribosomal protein profile, which is known to influence translational efficiency [,]. This has been reported to have a regulatory role in other plant models, especially in stress response mechanisms [,,,]. In Chlamydomonas, specifically, drastic changes in ribosomal protein expression have been observed to accompany transcriptome changes in other phenotypes such as nutrient deprivation responses [,,,] suggesting the possibility for similar regulatory mechanisms for salt stress and other stress-related responses.
This translation-level regulatory response is further supported by the identification of a range of differentially expressed pre-mRNA splicing factors. However, alternative splicing is unlikely to fully explain these findings, as only roughly 10.1% of Chlamydomonas’s protein-coding loci have annotated alternative transcripts considering the latest genome annotation [,] compared to ~30% for flowering plants []. Alternatively, the changes in pre-mRNA processing introduced by these differential proteins could be related to noncoding RNAs (ncRNAs), which have been found to act in the regulation of certain stress response behaviors and chloroplast metabolism in Chlamydomonas [,,,]. Along with the changes observed in ribosomal proteins, these nuclear proteome findings highlight the early regulatory shift into a sustained, long-term salt stress response phenotype, markedly characterized by alterations in ribosome-related mechanisms and protein translation rate, even though the results observed for treatment condition did not indicate significant changes in the cell growth performance.
4.2. Transcription Factors
Among the proteins identified on the nuclear proteome, we also observed 15 TFs and seven TRs providing new insights into the regulatory cascades that establish the medium- and long-term response of C. reinhardtii to salt stress beyond the 24 h time point analyzed here. These regulatory proteins feature limited experimental characterization in the literature, as do most Chlamydomonas TFs and TRs [,]; therefore, their confirmed presence in the nuclear proteome under salt stress is deeply impactful. In particular, we bring attention to the five differentially expressed TFs (Table 2), split among the Top and MYB families. Members of the latter, in particular, are well known in the literature for their roles in various biotic and abiotic stress responses in other plants [,,]. The two downregulated MYB TFs we found, Cre03.g176651.t1.1 and Cre03.g197350.t1.2, were previously identified as histone H2A deubiquitinase MYSM1 and cell division cycle 5 (CDC5) [,], respectively. As a histone deubiquitinase, MYSM1 acts in transcriptional regulation through chromatin modifications, but its functional role is poorly characterized in plants as very few plant species have been found to possess MYSM1-like proteins [,]. Thus, the identification of the differential expression of C. reinhardtii MYSM1 under salt stress may be vital to understanding its role and what processes it may regulate. On the other hand, CDC5 is well characterized in land plants and eukaryotes in general as a crucial regulator of the cell cycle []. More interestingly, Chlamydomonas CDC5 mutants have been shown to accumulate neutral lipids [] in a manner similar to what we observed under salt stress, further corroborating this protein’s crucial role we propose herein.
Table 2.
C. reinhardtii transcription factor and transcriptional regulator homologs identified in A. thaliana.
Furthermore, the TRs with the highest number of identified target genes (Cre06.g261450.t1.2, Cre16.g672300.t1.2, and Cre17.g702650.t1.1) were all mapped to a single A. thaliana TR homologous sequence (AT4G11080), with high conservation of sequences on and around their high-mobility group box (HMG_box, PF00505, http://pfam.xfam.org/, accessed on 2 October 2021) motifs. This motif is known to interact with DNA, in both sequence-specific and nonspecific ways, and, in some cases, mediate chromatin remodeling processes [,]. Furthermore, the HMG box-containing proteins which are found in plants are divided into four main groups: (1) HMGB proteins; (2) structure-specific recognition protein 1 (SSRP1) homologs; (3) proteins containing both an HMG-box domain and an AT-rich interaction domain (ARID); (4) proteins containing three HMG-box domains (3 × HMG-box). The Arabidopsis protein in question, AT4G11080, was recently identified as one of the two 3 × HMG-box proteins in this species and found to interact with mitotic and meiotic chromosomes [].
While Chlamydomonas does not possess, to our knowledge, any 3 × HMG-box protein in its genome, the three TRs identified in our proteomics analysis show the relatively high conservation in and around their HMG-box domains, as well as at the N-terminus (data not shown). This leads us to believe that the reported recognition motif for AT4G11080 might be reasonably conserved in Chlamydomonas, providing the most promising insight into the biological role of the four algae TRs, which have very limited functional characterization. Analyzing the 585 target genes identified for these TFs, we observed an enrichment of GO annotation terms related to kinase activity and protein modification, which may be indicative of their participation in signal transduction processes necessary to sustain over time the metabolic changes observed that activate lipid accumulation while minimally affecting cell growth (Figure S4). Further functional characterization of these target genes is necessary, enabling more detailed insights into stress-related signaling cascades in Chlamydomonas and novel targets for biotechnological applications.
5. Conclusions
In this study, we demonstrated that mild osmotic abiotic stress triggered enhanced lipid biosynthesis in C. reinhardtii, while not impairing cell growth. Triggering such a physiological status is highly desirable for biomass production toward sustainable lipid accumulation and, thus, bioenergy production. In addition, we provided important insights into the nuclear proteome of this biomass-producing phenotype of C. reinhardtii. Interestingly, we identified 61 downregulated DEs proteins related to functions such as translation and nucleosome assembly under salt stress. We anticipate that our results correlating the alterations of abundance for some nuclear proteins during the short-term acclimation of cells to salt stress and the increase in neutral lipid production will contribute to studies on bioenergy production from C. reinhardtii, highlighting the potential of salt stress as a viable industrial approach for inducing lipid accumulation. However, further studies must be performed to identify and characterize the function of some target differential regulated proteins to address their regulatory role in this type of stress response.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/phycology2030015/s1: Figure S1. Analysis of the structural integrity of the isolated nucleus of C. reinhardtii under 0.1 M NaCl salt stress; Figure S2. Reproducibility between replicates of proteome datasets. Figure S3. Functional classification of overrepresented Gene Ontology (GO) annotation terms in the dataset of differentially expressed (DEs) nuclear proteins under salt stress (0.1 M NaCl) according to BiNGO (Cytoscape software) tool; Figure S4. Functional classification of overrepresented Gene Ontology (GO) terms in the dataset of putative targets of Cre17.g702650.t1.1 according to BiNGO (Cytoscape software) tool; Table S1. All proteins identified in the nuclear proteome of Chlamydomonas reinhardtii in salt stress; Table S2. C. reinhardtii targets for the transcription factor and transcriptional regulator homologs identified in A. thaliana; Table S3. ProteinGroups file of protein identification without data preprocessing.
Author Contributions
Conceptualization, L.d.O.M., F.N.d.M. and F.V.W.; methodology, L.d.O.M. and F.N.d.M.; formal analysis, L.d.O.M., F.N.d.M. and F.V.W.; investigation, L.d.O.M., F.N.d.M. and F.V.W.; resources, F.V.W.; writing—original draft preparation, L.d.O.M. and F.N.d.M.; writing—review and editing, L.d.O.M. and F.V.W.; visualization, L.d.O.M. and F.N.d.M.; supervision, F.V.W.; project administration, F.V.W.; funding acquisition, F.V.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the São Paulo Research Foundation (grant #2016/06601-4, São Paulo Research Foundation (FAPESP)). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium via the PRIDE partner repository (https://www.ebi.ac.uk/pride/, accessed on 16 June 2022 with the dataset identifier PXD034581. The complete file of proteinGroups identified by mass spectrometry containing the protein identification and expression data without data preprocessing or data filtering is available in Table S3.
Acknowledgments
We thank F.D.B Almeida from the Biosciences Institute (IB) and F.C. Meotti from the Institute of Chemistry (IQ), both from São Paulo University (USP), for providing the fluorescence microscope used to visualize the nuclei and the fluorescence plate reader used to visualize the Nile red fluorescence, respectively. The authors thank the Sciences PhD school in Biochemistry from the Institute of Chemistry (IQ) from São Paulo University (USP) for support. The authors thank D. Yamamoto and S. M. de Oliveira for technical assistance. The authors thank the Central Analytical Instrumentation Facility of the Institute of Chemistry of the University of São Paulo for technical support and mass spectrometry analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hoegh-Guldberg, O.; Bruno, J.F. The Impact of Climate Change on the World’s Marine Ecosystems. Science 2010, 328, 1523–1528. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.C.; Yung, Y.L.; Lacis, A.A.; Mo, T.; Hansc, J.E. Greenhouse Effects Due to Man-Mad Perturbations of Trace Gases: Anthropogenic Gases May Alter Our Climate by Plugging an Atmospheric Window for Escaping Thermal Radiation. Science 1976, 194, 4266. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.S.; Kropat, J.; Liu, B.; Shaw, J.; Warakanont, J. TAG, You’re It! Chlamydomonas as a Reference Organism for Understanding Algal Triacylglycerol Accumulation. Curr. Opin. Biotechnol. 2012, 23, 352–363. [Google Scholar] [PubMed]
- Ryan Georgianna, D.; Mayfield, S.P. Exploiting Diversity and Synthetic Biology for the Production of Algal Biofuels. Nature 2012, 488, 329–335. [Google Scholar] [CrossRef] [PubMed]
- García, J.L.; de Vicente, M.; Galán, B. Microalgae, Old Sustainable Food and Fashion Nutraceuticals. Microb. Biotechnol. 2017, 10, 1017–1024. [Google Scholar] [CrossRef]
- Fan, J.; Zheng, L. Acclimation to NaCl and Light Stress of Heterotrophic Chlamydomonas reinhardtii for Lipid Accumulation. J. Biosci. Bioeng. 2017, 124, 302–308. [Google Scholar] [CrossRef]
- Lin, Q.; Zhuo, W.H.; Wang, X.W.; Chen, C.P.; Gao, Y.H.; Liang, J.R. Effects of Fundamental Nutrient Stresses on the Lipid Accumulation Profiles in Two Diatom Species Thalassiosira weissflogii and Chaetoceros muelleri. Bioprocess Biosyst. Eng. 2018, 41, 1213–1224. [Google Scholar] [CrossRef]
- He, Q.; Yang, H.; Wu, L.; Hu, C. Effect of Light Intensity on Physiological Changes, Carbon Allocation and Neutral Lipid Accumulation in Oleaginous Microalgae. Bioresour. Technol. 2015, 191, 219–228. [Google Scholar] [CrossRef]
- Fan, J.; Cui, Y.; Wan, M.; Wang, W.; Li, Y. Lipid Accumulation and Biosynthesis Genes Response of the Oleaginous Chlorella pyrenoidosa under Three Nutrition Stressors. Biotechnol. Biofuels 2014, 7, 17. [Google Scholar] [CrossRef]
- Hu, Q.; Sommerfeld, M.; Jarvis, E.; Ghirardi, M.; Posewitz, M.; Seibert, M.; Darzins, A. Microalgal Triacylglycerols as Feedstocks for Biofuel Production: Perspectives and Advances. Plant J. 2008, 54, 621–639. [Google Scholar] [CrossRef]
- Winck, F.V.; Páez Melo, D.O.; González Barrios, A.F. Carbon Acquisition and Accumulation in Microalgae Chlamydomonas: Insights from “Omics” Approaches. J. Proteomics 2013, 94, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Mettler, T.; Mühlhaus, T.; Hemme, D.; Schöttler, M.-A.; Rupprecht, J.; Idoine, A.; Veyel, D.; Pal, S.K.; Yaneva-Roder, L.; Winck, F.V.; et al. Systems Analysis of the Response of Photosynthesis, Metabolism, and Growth to an Increase in Irradiance in the Photosynthetic Model Organism Chlamydomonas reinhardtii. Plant Cell 2014, 26, 2310–2350. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Maréchal-Drouard, L.; et al. The Chlamydomonas Genome Reveals the Evolution of Key Animal and Plant Functions. Science 2007, 318, 245–250. [Google Scholar] [CrossRef]
- May, P.; Wienkoop, S.; Kempa, S.; Usadel, B.; Christian, N.; Rupprecht, J.; Weiss, J.; Recuenco-Munoz, L.; Ebenhöh, O.; Weckwerth, W.; et al. Metabolomics- and Proteomics-Assisted Genome Annotation and Analysis of the Draft Metabolic Network of Chlamydomonas reinhardtii. Genetics 2008, 179, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Mastrobuoni, G.; Irgang, S.; Pietzke, M.; Aßmus, H.E.; Wenzel, M.; Schulze, W.X.; Kempa, S. Proteome Dynamics and Early Salt Stress Response of the Photosynthetic Organism Chlamydomonas reinhardtii. BMC Genom. 2012, 13, 215. [Google Scholar] [CrossRef]
- Hounslow, E.; Vijay Kapoore, R.; Vaidyanathan, S.; Gilmour, D.J.; Wright, P.C. The Search for a Lipid Trigger: The Effect of Salt Stress on the Lipid Profile of the Model Microalgal Species Chlamydomonas reinhardtii for Biofuels Production. Curr. Biotechnol. 2016, 5, 305–313. [Google Scholar] [CrossRef]
- Wang, N.; Qian, Z.; Luo, M.; Fan, S.; Zhang, X.; Zhang, L. Identification of Salt Stress Responding Genes Using Transcriptome Analysis in Green Alga Chlamydomonas reinhardtii. Int. J. Mol. Sci. 2018, 19, 3359. [Google Scholar] [CrossRef]
- Arias, C.; Obudulu, O.; Zhao, X.; Ansolia, P.; Zhang, X.; Paul, S.; Bygdell, J.; Pirmoradian, M.; Zubarev, R.A.; Samuelsson, G.; et al. Nuclear Proteome Analysis of Chlamydomonas with Response to CO2 Limitation. Algal Res. 2020, 46, 101765. [Google Scholar] [CrossRef]
- Winck, F.V.; Riaño-Pachón, D.M.; Sommer, F.; Rupprecht, J.; Mueller-Roeber, B. The Nuclear Proteome of the Green Alga Chlamydomonas reinhardtii. PROTEOMICS 2012, 12, 95–100. [Google Scholar] [CrossRef]
- Gorman, D.S.; Levine, R.P. Cytochrome f and Plastocyanin: Their Sequence in the Photosynthetic Electron Transport Chain of Chlamydomonas reinhardi. Proc. Natl. Acad. Sci. USA 1965, 54, 1665–1669. [Google Scholar] [CrossRef]
- Kou, Z.; Bei, S.; Sun, J.; Pan, J. Fluorescent Measurement of Lipid Content in the Model Organism Chlamydomonas reinhardtii. J. Appl. Phycol. 2013, 25, 1633–1641. [Google Scholar] [CrossRef]
- Winck, F.V.; Kwasniewski, M.; Wienkoop, S.; Mueller-Roeber, B. AN OPTIMIZED METHOD FOR THE ISOLATION OF NUCLEI FROM CHLAMYDOMONAS REINHARDTII (CHLOROPHYCEAE)1. J. Phycol. 2011, 47, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-Wide Label-Free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A Comparative Platform for Green Plant Genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape Plugin to Assess Overrepresentation of Gene Ontology Categories in Biological Networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef]
- Kucera, M.; Isserlin, R.; Arkhangorodsky, A.; Bader, G.D. AutoAnnotate: A Cytoscape App for Summarizing Networks with Semantic Annotations. F1000Research 2016, 5, 1717. [Google Scholar] [CrossRef]
- Carnielli, C.M.; Winck, F.V.; Paes Leme, A.F. Functional Annotation and Biological Interpretation of Proteomics Data. Biochim. Biophys. Acta BBA-Proteins Proteom. 2015, 1854, 46–54. [Google Scholar] [CrossRef]
- Hooper, C.M.; Castleden, I.R.; Tanz, S.K.; Aryamanesh, N.; Millar, A.H. SUBA4: The Interactive Data Analysis Centre for Arabidopsis Subcellular Protein Locations. Nucleic Acids Res. 2017, 45, D1064–D1074. [Google Scholar] [CrossRef]
- Swarbreck, D.; Wilks, C.; Lamesch, P.; Berardini, T.Z.; Garcia-Hernandez, M.; Foerster, H.; Li, D.; Meyer, T.; Muller, R.; Ploetz, L.; et al. The Arabidopsis Information Resource (TAIR): Gene Structure and Function Annotation. Nucleic Acids Res. 2008, 36, D1009–D1014. [Google Scholar] [CrossRef]
- Brameier, M.; Krings, A.; MacCallum, R.M. NucPred—Predicting Nuclear Localization of Proteins. Bioinformatics 2007, 23, 1159–1160. [Google Scholar] [CrossRef] [PubMed]
- Tardif, M.; Atteia, A.; Specht, M.; Cogne, G.; Rolland, N.; Brugière, S.; Hippler, M.; Ferro, M.; Bruley, C.; Peltier, G.; et al. Predalgo: A New Subcellular Localization Prediction Tool Dedicated to Green Algae. Mol. Biol. Evol. 2012, 29, 3625–3639. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, P.; Riaño-Pachón, D.M.; Corrêa, L.G.G.; Rensing, S.A.; Kersten, B.; Mueller-Roeber, B. PlnTFDB: Updated Content and New Features of the Plant Transcription Factor Database. Nucleic Acids Res. 2010, 38, D822–D827. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.Y.; Krishnakumar, V.; Chan, A.P.; Thibaud-Nissen, F.; Schobel, S.; Town, C.D. Araport11: A Complete Reannotation of the Arabidopsis thaliana Reference Genome. Plant J. Cell Mol. Biol. 2017, 89, 789–804. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a Central Hub for Transcription Factors and Regulatory Interactions in Plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Gribskov, M. Combining Evidence Using P-Values: Application to Sequence Homology Searches. Bioinforma. Oxf. Engl. 1998, 14, 48–54. [Google Scholar] [CrossRef]
- Hang, L.T.; Mori, K.; Tanaka, Y.; Morikawa, M.; Toyama, T. Enhanced Lipid Productivity of Chlamydomonas reinhardtii with Combination of NaCl and CaCl2 Stresses. Bioprocess Biosyst. Eng. 2020, 43, 971–980. [Google Scholar] [CrossRef]
- McClure-Begley, T.D.; Klymkowsky, M.W. Nuclear Roles for Cilia-Associated Proteins. Cilia 2017, 6, 8. [Google Scholar] [CrossRef]
- Hessen, D.O.; Van Donk, E.; Andersen, T. Growth Responses, P-Uptake and Loss of Flagellae in Chlamydomonas reinhardtii Exposed to UV-B. J. Plankton Res. 1995, 17, 17–27. [Google Scholar] [CrossRef]
- Rosenbaum, J.L.; Moulder, J.E.; Ringo, D.L. FLAGELLAR ELONGATION AND SHORTENING IN CHLAMYDOMONAS: The Use of Cycloheximide and Colchicine to Study the Synthesis and Assembly of Flagellar Proteins. J. Cell Biol. 1969, 41, 600. [Google Scholar] [CrossRef] [PubMed]
- Siaut, M.; Cuiné, S.; Cagnon, C.; Fessler, B.; Nguyen, M.; Carrier, P.; Beyly, A.; Beisson, F.; Triantaphylidès, C.; Li-Beisson, Y.; et al. Oil Accumulation in the Model Green Alga Chlamydomonas reinhardtii: Characterization, Variability between Common Laboratory Strains and Relationship with Starch Reserves. BMC Biotechnol. 2011, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.; Alegre, S.; Nagler, M.; Escandón, M.; Annacondia, M.L.; Weckwerth, W.; Valledor, L.; Cañal, M.J. The Variations in the Nuclear Proteome Reveal New Transcription Factors and Mechanisms Involved in UV Stress Response in Pinus radiata. J. Proteom. 2016, 143, 390–400. [Google Scholar] [CrossRef]
- Petibon, C.; Malik Ghulam, M.; Catala, M.; Abou Elela, S. Regulation of Ribosomal Protein Genes: An Ordered Anarchy. Wiley Interdiscip. Rev. RNA 2021, 12, e1632. [Google Scholar] [CrossRef]
- Tiruneh, B.S.; Kim, B.H.; Gallie, D.R.; Roy, B.; Von Arnim, A.G. The Global Translation Profile in a Ribosomal Protein Mutant Resembles That of an EIF3 Mutant. BMC Biol. 2013, 11, 123. [Google Scholar] [CrossRef]
- Carroll, A.J. The Arabidopsis Cytosolic Ribosomal Proteome: From Form to Function. Front. Plant Sci. 2013, 4, 32. [Google Scholar] [CrossRef]
- Wang, J.; Lan, P.; Gao, H.; Zheng, L.; Li, W.; Schmidt, W. Expression Changes of Ribosomal Proteins in Phosphate- and Iron-Deficient Arabidopsis Roots Predict Stress-Specific Alterations in Ribosome Composition. BMC Genom. 2013, 14, 783. [Google Scholar] [CrossRef]
- Rosado, A.; Li, R.; Van De Ven, W.; Hsu, E.; Raikhel, N.V. Arabidopsis Ribosomal Proteins Control Developmental Programs through Translational Regulation of Auxin Response Factors. Proc. Natl. Acad. Sci. USA 2012, 109, 19537–19544. [Google Scholar] [CrossRef]
- Schippers, J.H.M.; Mueller-Roeber, B. Ribosomal Composition and Control of Leaf Development. Plant Sci. 2010, 179, 307–315. [Google Scholar] [CrossRef]
- Takeuchi, T.; Benning, C. Nitrogen-Dependent Coordination of Cell Cycle, Quiescence and TAG Accumulation in Chlamydomonas. Biotechnol. Biofuels 2019, 12, 292. [Google Scholar] [CrossRef] [PubMed]
- Couso, I.; Pérez-Pérez, M.E.; Martínez-Force, E.; Kim, H.S.; He, Y.; Umen, J.G.; Crespo, J.L. Autophagic Flux Is Required for the Synthesis of Triacylglycerols and Ribosomal Protein Turnover in Chlamydomonas. J. Exp. Bot. 2018, 69, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.; Wang, H.; Gargouri, M.; Deshpande, R.R.; Skepper, J.N.; Holguin, F.O.; Juergens, M.T.; Shachar-Hill, Y.; Hicks, L.M.; Gang, D.R. The Response of Chlamydomonas reinhardtii to Nitrogen Deprivation: A Systems Biology Analysis. Plant J. 2015, 81, 611–624. [Google Scholar] [CrossRef]
- Blaby, I.K.; Blaby-Haas, C.E.; Tourasse, N.; Hom, E.F.Y.; Lopez, D.; Aksoy, M.; Grossman, A.; Umen, J.; Dutcher, S.; Porter, M.; et al. The Chlamydomonas Genome Project: A Decade On. Trends Plant Sci. 2014, 19, 672–680. [Google Scholar] [CrossRef]
- Labadorf, A.; Link, A.; Rogers, M.F.; Thomas, J.; Reddy, A.S.N.; Ben-Hur, A. Genome-Wide Analysis of Alternative Splicing in Chlamydomonas reinhardtii. BMC Genom. 2010, 11, 114. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Y.; Chen, M.; Xiao, P.; Hu, C.; Zeng, Z.; Wang, C.; Wang, J.; Hu, Z. Genome-Wide Long Non-Coding RNA Screening, Identification and Characterization in a Model Microorganism Chlamydomonas reinhardtii. Sci. Rep. 2016 61 2016, 6, 34109. [Google Scholar] [CrossRef]
- Lou, S.; Sun, T.; Li, H.; Hu, Z. Mechanisms of MicroRNA-Mediated Gene Regulation in Unicellular Model Alga Chlamydomonas reinhardtii. Biotechnol. Biofuels 2018, 11, 244. [Google Scholar] [CrossRef]
- Goldschmidt-Clermont, M.; Rahire, M.; Rochaix, J.D. Redundant Cis-Acting Determinants of 3′ Processing and RNA Stability in the Chloroplast RbcL MRNA of Chlamydomonas. Plant J. 2008, 53, 566–577. [Google Scholar] [CrossRef]
- Kück, U.; Schmitt, O. The Chloroplast Trans-Splicing RNA–Protein Supercomplex from the Green Alga Chlamydomonas reinhardtii. Cells 2021, 10, 290. [Google Scholar] [CrossRef]
- Riaño-Pachón, D.M.; Corrêa, L.G.G.; Trejos-Espinosa, R.; Mueller-Roeber, B. Green Transcription Factors: A Chlamydomonas Overview. Genetics 2008, 179, 31–39. [Google Scholar] [CrossRef]
- Anderson, M.S.; Muff, T.J.; Georgianna, D.R.; Mayfield, S.P. Towards a Synthetic Nuclear Transcription System in Green Algae: Characterization of Chlamydomonas reinhardtii Nuclear Transcription Factors and Identification of Targeted Promoters. Algal Res. 2017, 22, 47–55. [Google Scholar] [CrossRef]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB Transcription Factor Genes as Regulators for Plant Responses: An Overview. Physiol. Mol. Biol. Plants Int. J. Funct. Plant Biol. 2013, 19, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB Transcription Factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Roy, S. Function of MYB Domain Transcription Factors in Abiotic Stress and Epigenetic Control of Stress Response in Plant Genome. Plant Signal. Behav. 2016, 11, e1117723. [Google Scholar] [CrossRef]
- Yang, C.; Hager, P.W.; Stiller, J.W. The Identification of Putative RNA Polymerase II C-Terminal Domain Associated Proteins in Red and Green Algae. Transcription 2014, 5, e970944. [Google Scholar] [CrossRef][Green Version]
- Torres-Romero, I.; Kong, F.; Légeret, B.; Beisson, F.; Peltier, G.; Li-Beisson, Y. Chlamydomonas Cell Cycle Mutant Crcdc5 Over-Accumulates Starch and Oil. Biochimie 2020, 169, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yang, S.; Yuan, L.; Cui, Y.; Wu, K. Comparative Analysis of SWIRM Domain-Containing Proteins in Plants. Comp. Funct. Genom. 2012, 2012, 310402. [Google Scholar] [CrossRef]
- March, E.; Farrona, S. Plant Deubiquitinases and Their Role in the Control of Gene Expression through Modification of Histones. Front. Plant Sci. 2018, 8, 2274. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, T.; Shinozaki, K. A Cdc5+ Homolog of a Higher Plant, Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 1996, 93, 13371–13376. [Google Scholar] [CrossRef]
- Antosch, M.; Mortensen, S.A.; Grasser, K.D. Plant Proteins Containing High Mobility Group Box DNA-Binding Domains Modulate Different Nuclear Processes. Plant Physiol. 2012, 159, 875–883. [Google Scholar] [CrossRef]
- Launholt, D.; Grasser, K.D. The HMG-Box: A Versatile Protein Domain Occurring in a Wide Variety of DNA-Binding Proteins. Cell. Mol. Life Sci. 2007, 64, 2590–2606. [Google Scholar] [CrossRef]
- Pedersen, D.S.; Coppens, F.; Ma, L.; Antosch, M.; Marktl, B.; Merkle, T.; Beemster, G.T.S.; Houben, A.; Grasser, K.D. The Plant-Specific Family of DNA-Binding Proteins Containing Three HMG-Box Domains Interacts with Mitotic and Meiotic Chromosomes. New Phytol. 2011, 192, 577–589. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).