Abstract
Tramadol is a veterinary analgesic for dogs. In this study, the steady-state pharmacokinetics of a sustained-release (SR) tablet (Tramagetic OD®) and immediate-release capsules (IR) were compared. In a crossover design, six dogs received five doses of IR 50 mg four times a day (qid), or two doses of SR 200 mg once a day (sid). Eight blood samples were collected per dog, per formulation, up to 6 and 24 h after the last dose, respectively. Serum concentrations of tramadol and its metabolites were measured with LC-MS/MS. Metabolite M1 levels were below the lower limit of quantification (LLOQ) in all samples. The non-compartmental analysis of the time–concentration data showed a later Tmax with the SR formulation (median 6.00 h (3.00–9.00)) and a lower Cmax/D (median 7.74 µg/L/mg/kg (0.09–25.3)) compared to the IR formulation (median Tmax 1.75 h (0.75–2.00) and median Cmax/D 11.1 µg/L/mg/kg (4.8–70.4)). AUCtau/D after SR administration was 55.5 h × kg × µg/L/mg (0–174.1) compared to 29.8 h × kg × µg/L/mg (12.2–140.8) after IR administration. The terminal elimination half-lives were 2.38 h (1.77–6.22) and 1.70 h (0.95–2.11) for the SR and IR formulations, respectively. Strong conclusions cannot be drawn from this study because of the high percentage of samples that were below LLOQ and the great interindividual variability, but these results suggest that Tramagetic OD can be administered less frequently in dogs.
1. Introduction
Tramadol is a widely used analgesic in dogs. It has a complex, multimodal mechanism of action: it is a weak µ-opioid agonist, but it also inhibits the reuptake of serotonin and noradrenaline, and it has an inhibiting effect on muscarinergic receptors [1]. Tramadol is metabolized by different cytochrome P450 (CYP) enzymes into various metabolites, of which O-desmethyltramadol (M1), N-desmethyltramadol (M2) and N,O-desmethyltramadol (M5) are described the best. In dogs, CYP 2D15 metabolizes tramadol to M1, and CYP 2B11 and CYP 3A12 metabolize tramadol to M2. M5 is formed from M1 and M2, mainly by CYP 2C21 [2,3]. M1 is an active metabolite and the most important metabolite with regard to analgesic effect. It is reported to have a >200-fold higher affinity for the µ-opioid receptor compared to tramadol [4,5]. However, in contrast to tramadol, M1 does not influence noradrenaline and serotonin reuptake. In vitro, M5 also shows µ-agonism, but its contribution to the analgesic effect is expected to be negligible because it does not pass the blood–brain barrier. M2 is thought to be an inactive metabolite [6].
There are concerns about the analgesic efficacy of tramadol in dogs because they hardly produce M1 [6,7,8]. However, tramadol might still be of benefit in dogs, possibly as part of a multimodal therapy [9].
Tramadol is available for dogs as immediate-release (IR) tablets. These tablets need to be administered 3–4 times a day, which is a schedule that is hard for the owner to follow.
Several studies in humans have shown that compliance is improved by reducing the dosing frequency [10,11,12]. Animal owners also seem to find it difficult to accurately follow dosing regimens, even for the short-term administration of medication [13,14]. For compliance, once-daily dosing (sid) is preferred above more frequent dosing.
Dosing frequency can be reduced by using tablets with sustained-release (SR) properties. Besides the advantage of less frequent dosing, SR formulations also result in less fluctuations in serum concentrations, giving a more consistent effect with less side-effects. For dogs, there are only immediate-release (IR) formulations are on the marke. However, there are various oral SR formulations with tramadol available for humans. Using the SR tablets in dogs could facilitate a dosing regimen that is more tractable for owners, resulting in better compliance and better analgesic efficacy, without inducing breakthrough pains.
To dose SR tablets correctly, the pharmacokinetics (PK), such as the rate of absorption and bioavailability, of these formulations in dogs must be known. It is difficult to predict these parameters in dogs based on human data because of the differences in anatomy and physiology between these species. [15]. Pharmacokinetic studies in dogs are therefore needed.
There are several studies available on the PK of tramadol in dogs, but only one study of an SR tablet [5]. This was a pilot study in which one tramadol SR tablet (Contramal 100 mg) was administered once to six dogs weighing between 20 and 27 kg, resulting in a dose of 3.7–5.0 mg/kg. In this study, the Tmax in dogs was 2–4 h, compared to 4.9 h when the same formulation was administered to humans. The absorption therefore appears to be faster in dogs compared to humans.
Our study aimed to compare the steady-state pharmacokinetics between a different tramadol SR tablet (Tramagetic Once Daily®) and IR capsules in dogs.
2. Materials and Methods
The study was performed at the Utrecht University Department for Companion Animals (Utrecht, the Netherlands). It was approved by the Animal Experiments Committee of the university, as required under Dutch legislation (approval number DEC 2008.III.05.049). Six university-owned female research dogs (four Beagles and two cross-breed Beagle-Bedlington terriers, 13.3–16.9 kg, aged 4–7 years) received one IR tramadol capsule of 50 mg every six hours (qid) on a total of five times (Pharmachemie BV, Haarlem, the Netherlands) (3.0–3.8 mg/kg), or an SR tramadol tablet 200 mg every 24 h (sid) (Tramagetic® Once-Daily tablet from Nycomed BV, Hoofddorp, The Netherlands) (11.8–15.0 mg/kg) twice. Dogs were randomly assigned to receive either the IR capsules or the SR tablet first. All dogs received the alternate medication after a wash-out period of 48 h. Immediately after the last administration, blood samples (2.5 mL) were collected via jugular veni-puncture at 0, 30, 45, 60 and 90 min and 2, 4 and 6 h after the IR capsule and at 30, 60, 90 min and 3, 6, 9, 15 and 24 h after the SR tablet.
Serum concentrations of tramadol and its metabolites M1, M2, and M5 were measured by the Department of Clinical Pharmacy and Pharmacology of University Medical Centre Groningen (Groningen, The Netherlands) using a validated LC-MS method. For sample preparation a volume of 100 μL serum was mixed with 750 μL precipitation reagent. This reagent consisted of a mixture of methanol and acetonitrile (4:21, v/v) and the internal standard cyanoimipramine. After centrifuging the samples at 10,000 g for 5 min, 5 μL of the supernatant was injected on the LC-MS/MS. A Thermo Finnigan Surveyor® LC-system consisting of a quaternary LC Pump, an autosampler set at 10 °C and a column compartment, was coupled to a TSQ Quantum triple quadruplose mass spectrometer (Thermo Finnigan). A Thermo Electron Hypurity Aquasta column (50 × 2.1 mm, 5 μm), maintained at a temperature of 23 °C, was used for chromatographic separation. The mobile phase contained an aqueous buffer (consisting of ammonium acetate p.a. 0.25 g/L, glacial acetic acid 1.75 mL/L and trifluoroacetic anhydride 0.1 mL/L water; pH 3.5), purified water and acetonitrile (LC-MS grade) with a flow rate of 0.3 mL/min. With a constant part of 5% of the aqueous buffer, a gradient between water and acetonitrile was run for 3.6 min. The concentration gradient of acetonitrile went from 0% the first minute, to 30–95% from 1–2.5 min; the concentration stayed at 95% for 0.3 min, after which it was turned back to 0%. The mass spectrometer operated in positive mode using single-ion mass detection (SIM). Electrospray ionization was performed with an ion source spray voltage of 3500 V. The sheath and auxiliary gas pressure were set to 35 arbitrary units (Arb.) and 10 Arb., respectively, and the vaporizer temperature and the capillary temperature were set at 75 °C and 350 °C, respectively.
Table 1 summarizes the validation data of the analytical method of tramadol and its metabolites. The validation process had been performed in human sera before. Therefore, a cross-validation of dog and human sera was performed, and the results met the preset requirements: for all concentrations, including LLOQ, both bias and CV were <15%.

Table 1.
Summary of the validation information of the analysis of tramadol and the metabolites.
A noncompartmental analysis (NCA) profiles was performed of the serum concentration–time using the commercially available software program Phoenix® WinNonLin from Certara USA, Inc. (Princeton, NJ, USA), version 8.3. The concentrations below LLOQ were replaced by LLOQ/2, i.e., 2.5 µg/L before the NCA was executed. Replacing LLOQ by LLOQ/2 is a common way of handling these data in the NCA [16,17,18,19]. Leaving these out would have resulted in an overestimation of some of the parameter values, especially of the area under the concentration–time curve (AUC). The maximum concentration (Cmax) and its corresponding time (Tmax) were observed directly from the individual serum concentration–time profiles.
3. Results
The plasma concentrations of tramadol, M2 and M5 for each dog can be found in Supplementary Materials (Tables S1–S7). M1 concentrations were below the LLOQ in all samples. Of the 48 (8 time points of 6 dogs) measurements after IR administration, 12 (25%) were below the LLOQ for tramadol (5 µg/L). All measured concentrations of M2 and M5 were above the LLOQ. For the SR formulation, 22 (46%) of the 48 measurements for tramadol and one measurement for both M2 and M5 were below the LLOQ. One dog had tramadol levels below the LLOQ at all time points after administration of the SR tablets, accounting for 8 of the 22 LLOQ measurements of tramadol.
The mean serum concentration–time curves of tramadol, M2 and M5 for both formulations are shown in Figure 1. Serum concentration–time curves of the individual dogs are available in Supplementary Materials (Figures S1–S3).
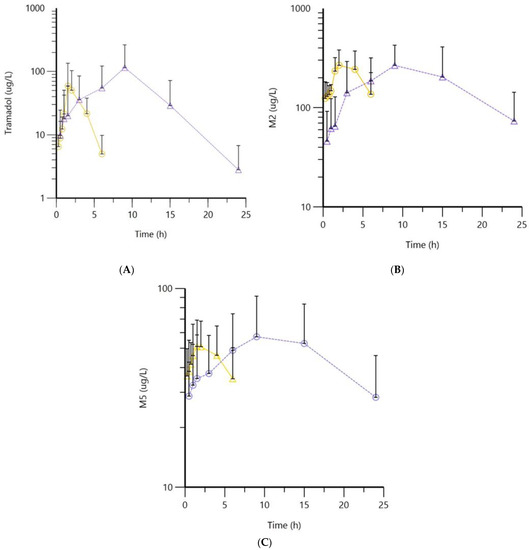
Figure 1.
Mean (triangles show the mean concentrations after administration of the IR formulation and circles the mean concentration after administration of the SR formulation) and standard deviation of tramadol (A), M2 (B) and M5 (C) serum concentrations over time, after administration of IR (yellow lines) and SR (purple lines) formulations. In these graphs, all measured values are included, as well as values below LLOQ.
The pharmacokinetic parameters of tramadol and its metabolites, M2 and M5, are summarized in Table 2. The ratio of the AUC overdose, given in mg/kg (AUC/D), was calculated using the AUC from the time of administration of the last dose up to the last measured concentration (AUClast). Because the time to the last measurement was the same as the dosing interval, AUClast is the same as AUCtau. The terminal elimination half-life (T1/2) was calculated using the time points falling along a constant slope. These time points were selected by visual inspection of the semi-log of the concentration–time profiles for the individual dogs.
Table 2.
Pharmacokinetic values of tramadol, M2 and M5 based on NCA; values below LLOQ are replaced by LLOQ/2 (i.e., 2.5 µg/L).
4. Discussion
A simple dosing regimen improves owner compliance, resulting in a better clinical outcome for the canine patient. SR tablets may help to simplify dosing regimens by reducing dose frequency. Tramagetic Once-Daily (OD) tablets were developed to be administered once a day to humans. This study shows that in dogs, for the same daily dose, the overall exposure to tramadol (expressed as AUCtau/D) is higher after the administration of Tramagetic OD compared to treatment with IR capsules. The slower rate of tramadol release from these SR tablets in dogs may enable less frequent dosing compared to that possible with the IR capsules.
In our pharmacokinetic analysis, all values below the LLOQ are included by replacing them with LLOQ/2 (i.e., 2.5 µg/L). Handling values below the LLOQ can highly influence the results of the analysis. Especially the AUC is highly affected by how one deals with these LLOQ data [16]. The influence is most pronounced in datasets with a large percentage of data below the LLOQ coupled with other low concentrations [17]. In our study, 25% of the tramadol values were below the LLOQ after IR capsules and 46% after SR tablets. Replacing those values with LLOQ/2 is a common way to handle these data in NCA. Several studies in recent years have compared different methods of handling LLOQ in NCA [16,17,18,19]. These studies all refer to the methods about handling data below LLOQ in pharmacokinetic modeling, described by Beal [20], and it has since been shown that handling data that are below the LLOQ as fixed-point censored data (called method 3, M3) gives the best and least biased results in pharmacokinetic modeling, especially when a high percentage (>30%) of the data are below the LLOQ [21]. The method of replacing data below LLOQ by LLOQ/2 is only recommended in pharmacokinetic modeling when less than 5% of the data are below LLOQ [21]. We compared the results of AUCtau/D using M3 and LLOQ/2 because of the high percentages of data below LLOQ. AUCtau/D was 0.44% and 1.54% higher in the IR and SR formulations, respectively, when using the LLOQ/2 method compared to the M3 method. We therefore chose the simpler method.
For the comparison of the exposure to tramadol between an IR and SR formulation, we used AUCtau/D. For both formulations, the samples used to measure concentrations started being taken 24 h after the first administration (this was directly after the fifth administration of the IR formulation and the second administration of the SR formulation). As the half-life of tramadol after IR has a range of 0.95–2.11 h in our study, the samples are taken at a steady state for this formulation. The half-life after SR ranges from 1.77 to 6.22 h in our study. The sampling of the dog with a half-life of 6.22 h might not have been in a set of steady-state conditions. As the other dogs had a shorter half-life (maximum of 4.00 h), the sampling was performed at a steady state in these dogs as well, which justifies the use of AUCtau/D after SR formulation too. The median AUCtau/D for the IR capsules was 46% lower compared to the SR tablets (29.8 h×kg×µg/L/mg and 55.5 h×kg×µg/L, respectively). These results suggest a higher oral bioavailability of tramadol administered as an SR formulation.
It should, however, be remembered that the AUC might be overestimated. The percentage extrapolated AUC0-inf is 35.2% with the SR tablet. If the percentage is more than 20%, this implies that the sampling period was not long enough to estimate AUC reliably. This is a known pitfall, especially in SR formulations [22]. Furthermore, SR formulations often show flip-flop pharmacokinetics. Flip-flop pharmacokinetics are expected in slow-release formulations as the active component of these tablets is meant to be absorbed slowly, resulting in a slower absorption rate than the elimination rate and a flatter decline during the elimination phase until the compound is fully absorbed. This is reflected in a larger AUC, leading to an overestimation of the bioavailability. Although physico-chemical properties can also result in flip-flop pharmacokinetics [23], with tramadol, this is not expected, as it has a high solubility and high permeability (Biopharmaceutics Classification System (BCS) class 1). The absence of flip-flop characteristics in the IR formulation also indicates that the flip-flop pharmacokinetics of the SR tablets are a result of the formulation.
This assumption is confirmed if we compare our results with the results of the PK study of the slow-release tramadol tablet, Contramal, in dogs [5]. This study reported a higher AUC/D: 82.6, versus that of 55.5 in our study. The half-life of Contramal in dogs was calculated by Giorgi to be 1.4–2.2 h, which lies between the half-lives we found for the IR formulation (0.95–2.11 h) and Tramagetic OD (1.77–6.22 h). Tmax of Contramal (2–4 h) is also shorter than that seen with Tramagetic OD (3–9 h). The differences in half-life and Tmax can be explained by the following formulation: Contramal tablets are based solely on a hydrophilic matrix; however, in Tramagetic OD, the hydrophilic matrix is combined with castor oil to delay the release of tramadol even more.
Looking at the median Cmax and Clast of the parent tramadol of both formulations (35.3 and 5.7 µg/L for IR, respectively, and 107.5 and 3.0 µg/L for SR, respectively), it is expected that once a day Tramagetic OD 200 mg will result in similar analgesic efficacy as tramadol IR capsules 50 mg four times a day. This is due to comparable drug levels during the major part of the dosing interval. However, the minimal serum levels considered analgesic in human are 56–590 µg/L for tramadol [24,25,26,27] and 36–84 µg/L for M1 [25,26]. Considering these thresholds, neither the IR capsules nor the Tramagetic OD will provide analgesic efficacy in dogs in the given dosing regimens. This is consistent with the conclusion of a recent review on the efficacy of tramadol as a post-operative analgesic in dogs [28].
A limitation of this study is the high percentage of tramadol concentrations below LLOQ. There was also a lot of interindividual variability among the concentrations. In other PK studies in which human SR tablets with different compounds are administered to dogs (5–11 dogs per study), the interindividual variability was also high [29,30,31,32,33,34]. This could be due to variable dissolution of the formulations in the dog. The high variability in our study specifically could also be explained by the CYP metabolism of tramadol. Dogs might have different activities of the CYP enzymes involved, resulting in different variations in concentrations of tramadol and its metabolites.
More animals and higher doses would improve the characterization of the PK of tramadol in dogs. However, with higher doses, adverse effects could occur. The purpose of this study was limited to the comparison of steady-state PK of the two oral formulations of tramadol. Better estimates of the PK parameters could have been attained if blood samples were also taken after the first dose of both oral formulations and after intravenous administration.
We conclude that Tramagetic OD shows sustained release of tramadol in dogs. More data are needed to determine what dose and dosing frequency is comparable to that of the IR capsules. Nevertheless, the great interindividual variability, especially seen with the SR formulation, will result in variability in serum concentration and analgesic effect. This reinforces the importance of evaluating the analgesic effect in an individual patient.
Although tramadol might not be the ideal analgesic for all dogs and types of pain, these results provide insight into the possible use of human SR medicines in dogs.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/futurepharmacol2040040/s1, Figures S1–S3: Serum concentration–time profiles of tramadol, M2 and M5 for the individual dogs, Tables S1–S7: raw data of tramadol, M2 and M5.
Author Contributions
Conceptualization, E.W.; methodology, I.v.G. and I.A.; software, E.W. and R.G.; validation, M.S.; formal analysis, E.W., R.G. and M.S.; writing—original draft preparation, E.W.; writing—review and editing, I.v.G., I.A., M.S. and R.G.; supervision, R.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. The software license for Phoenix® was provided by Certara USA, Inc. (Princeton, NJ, USA) as part of their Centers of Excellence program.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Utrecht, and approved by the Animal Ethics Committee of Utrecht University (DEC 2008.III.05.049, 1 May 2008).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available as supplementary files, see also supplementary materials.
Acknowledgments
We thank Manon Noordhoff and Anke Loeffen for their work during their internship on this project. We also thank Remco Koster and Ben Greijdanus from the UMCG for developing and validating the analysis and analyzing the samples. Additionally, we thank Mireille Wessels from the UMCG for her help in finding and reconstructing all the details on the method of analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Raffa, R.B.; Friderichs, E.; Reimann, W.; Shank, R.P.; Codd, E.E.; Vaught, J.L. Opioid and nonopioid components independently contribute to the mechanism of action of tramadol, an ‘atypical’ opioid analgesic. J. Pharmacol. Exp. Ther. 1992, 260, 275–285. [Google Scholar] [PubMed]
- Perez Jimenez, T.E.; Mealey, K.L.; Grubb, T.L.; Greene, S.A.; Court, M.H. Tramadol metabolism to O-desmethyl tramadol (M1) and N-desmethyl tramadol (M2) by dog liver microsomes: Species comparison and identification of responsible canine cytochrome P-450s (CYPs). Drug Metab. Dispos. 2016, 44, 1963–1972, Erratum in Drug Metab Dispos. 2017, 45, 706. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Perez Jimenez, T.E.; Mealey, K.L.; Schnider, D.; Grubb, T.L.; Greene, S.A.; Court, M.H. Identification of canine cytochrome P-450s (CYPs) metabolizing the tramadol (+)-M1 and (+)-M2 metabolites to the tramadol (+)-M5 metabolite in dog liver microsomes. J. Vet. Pharmacol. Ther. 2018, 41, 815–824. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kukanich, B.; Papich, M.G. Pharmacokinetics and antinociceptive effects of oral tramadol hydrochloride administration in Greyhounds. Am. J. Vet. Res. 2011, 72, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, M.; Saccomanni, G.; Lebkowska-Wieruszewska, B.; Kowalski, C. Pharmacokinetic evaluation of tramadol and its major metabolites after single oral sustained tablet administration in the dog: A pilot study. Vet. J. 2009, 180, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Kögel, B.; Terlinden, R.; Schneider, J. Characterisation of tramadol, morphine and tapentadol in an acute pain model in Beagle dogs. Vet. Anaesth. Analg. 2014, 41, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, M.; Del Carlo, S.; Saccomanni, G.; Łebkowska-Wieruszewska, B.; Kowalski, C.J. Pharmacokinetic and urine profile of tramadol and its major metabolites following oral immediate release capsules administration in dogs. Vet. Res. Commun. 2009, 33, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Schütter, A.F.; Tünsmeyer, J.; Kästner, S.B.R. Influence of tramadol on acute thermal and mechanical cutaneous nociception in dogs. Vet. Anaesth. Analg. 2017, 44, 309–316. [Google Scholar] [CrossRef] [PubMed]
- American Society of Anesthesiologists Task Force on Acute Pain Management. Practice guidelines for acute pain management in the perioperative setting: An updated report by the American Society of Anesthesiologists Task Force on Acute Pain Management. Anesthesiology 2012, 116, 248–273. [Google Scholar] [CrossRef] [PubMed]
- Eisen, S.A.; Miller, D.K.; Woodward, R.S.; Spitznagel, E.; Przybeck, T.R. The effect of prescribed daily dose frequency on patient medication compliance. Arch. Intern Med. 1990, 150, 1881–1884. [Google Scholar] [CrossRef] [PubMed]
- Leenen, F.H.; Wilson, T.W.; Bolli, P.; Larochelle, P.; Myers, M.; Handa, S.P.; Boileau, G.; Tanner, J. Patterns of compliance with once versus twice daily antihypertensive drug therapy in primary care: A randomized clinical trial using electronic monitoring. Can. J. Cardiol. 1997, 13, 914–920. [Google Scholar] [PubMed]
- Petrilla, A.A.; Benner, J.S.; Battleman, D.S.; Tierce, J.C.; Hazard, E.H. Evidence-based interventions to improve patient compliance with antihypertensive and lipid-lowering medications. Int. J. Clin. Pract. 2005, 59, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Wareham, K.J.; Brennan, M.L.; Dean, R.S. Systematic review of the factors affecting cat and dog owner compliance with pharmaceutical treatment recommendations. Vet. Rec. 2019, 184, 154. [Google Scholar] [CrossRef] [PubMed]
- Barter, L.S.; Watson, A.D.; Maddison, J.E. Owner compliance with short term antimicrobial medication in dogs. Aust. Vet. J. 1996, 74, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Toutain, P.L.; Ferran, A.; Bousquet-Mélou, A. Species differences in pharmacokinetics and pharmacodynamics. Handb. Exp. Pharmacol. 2010, 199, 19–48. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.H.; Upton, R.N.; Foster, D.J.R. Comparison of non-compartmental and mixed effect modelling methods for establishing bioequivalence for the case of two compartment kinetics and censored concentrations. J. Pharmacokinet. Pharmacodyn. 2017, 44, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Barnett, H.Y.; Geys, H.; Jacobs, T.; Jaki, T. Methods for Non-Compartmental Pharmacokinetic Analysis With Observations Below the Limit of Quantification. Stat. Biopharm. Res. 2021, 13, 59–70. [Google Scholar] [CrossRef]
- Hing, J.P.; Woolfrey, S.G.; Greenslade, D.; Wright, P.M. Analysis of toxicokinetic data using NONMEM: Impact of quantification limit and replacement strategies for censored data. J. Pharmacokinet. Pharmacodyn. 2001, 28, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R. Methods for Handling Concentration Values Below the Limit of Quantification in PK Studies. In PhUSE US Connect 2018; Kent Innovation Centre: Broadstairs, UK, 2018; pp. 1–9. [Google Scholar]
- Beal, S.L. Ways to fit a PK model with some data below the quantification limit. J. Pharmacokinet. Pharmacodyn. 2001, 28, 481–504, Erratum in J. Pharmacokinet. Pharmacodyn. 2002, 29, 309. [Google Scholar] [CrossRef] [PubMed]
- Irby, D.J.; Ibrahim, M.E.; Dauki, A.M.; Badawi, M.A.; Illamola, S.M.; Chen, M.; Wang, Y.; Liu, X.; Phelps, M.A.; Mould, D.R. Approaches to handling missing or “problematic” pharmacology data: Pharmacokinetics. CPT Pharmacometrics Syst. Pharmacol. 2021, 10, 291–308. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yáñez, J.A.; Remsberg, C.M.; Sayre, C.L.; Forrest, M.L.; Davies, N.M. Flip-flop pharmacokinetics--delivering a reversal of disposition: Challenges and opportunities during drug development. Ther. Deliv. 2011, 2, 643–672. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Garrison, K.L.; Sahin, S.; Benet, L.Z. Few Drugs Display Flip-Flop Pharmacokinetics and These Are Primarily Associated with Classes 3 and 4 of the BDDCS. J. Pharm. Sci. 2015, 104, 3229–3235. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lintz, W.; Barth, H.; Osterloh, G.; Schmidt-Böthelt, E. Bioavailability of enteral tramadol formulations. 1st communication: Capsules. Arzneimittelforschung 1986, 36, 1278–1283. [Google Scholar] [PubMed]
- Lehmann, K.A.; Kratzenberg, U.; Schroeder-Bark, B.; Horrichs-Haermeyer, G. Postoperative patient-controlled analgesia with tramadol: Analgesic efficacy and minimum effective concentrations. Clin. J. Pain. 1990, 6, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Grond, S.; Meuser, T.; Uragg, H.; Stahlberg, H.J.; Lehmann, K.A. Serum concentrations of tramadol enantiomers during patient-controlled analgesia. Br. J. Clin. Pharmacol. 1999, 48, 254–257. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sarbu, A.; Radulescu, F.; Robertson, S.; Bouchard, S. Onset of analgesic effect and plasma levels of controlled-release tramadol (Tramadol Contramid once-a-day) 200-mg tablets in patients with acute low back pain. J. Opioid Manag. 2008, 4, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Donati, P.A.; Tarragona, L.; Franco, J.V.A.; Kreil, V.; Fravega, R.; Diaz, A.; Verdier, N.; Otero, P.E. Efficacy of tramadol for postoperative pain management in dogs: Systematic review and meta-analysis. Vet. Anaesth. Analg. 2021, 48, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Kendall, J.; Papich, M.G. Posaconazole pharmacokinetics after administration of an intravenous solution, oral suspension, and delayed-release tablet to dogs. Am. J. Vet. Res. 2015, 76, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Boozer, L.B.; Platt, S.R.; Haley, A.C.; Linville, A.V.; Kent, M.; Barron, L.E.; Nie, B.; Arnold, R.D. Pharmacokinetic evaluation of immediate- and extended-release formulations of levetiracetam in dogs. Am. J. Vet. Res. 2015, 76, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Thomason, J.D.; Boothe, D.; KuKanich, B.; Rapoport, G. Pharmacokinetic evaluation of a sustained-release compounded procainamide preparation after 24-h (acute) administration in normal dogs. J. Vet. Cardiol. 2019, 24, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Bach, J.E.; Kukanich, B.; Papich, M.G.; McKiernan, B.C. Evaluation of the bioavailability and pharmacokinetics of two extended-release theophylline formulations in dogs. J. Am. Vet. Med. Assoc. 2004, 224, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Cavett, C.L.; Li, Z.; McKiernan, B.C.; Reinhart, J.M. Pharmacokinetics of a modified, compounded theophylline product in dogs. J. Vet. Pharmacol. Ther. 2019, 42, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, J.M.; Perkowski, C.; Lester, C.; Campos, V.; Kadotani, S.; Li, Z.; McKiernan, B.C.; Fries, R. Multidose pharmacokinetics and safety of a modified, compounded theophylline product in dogs. J. Vet. Pharmacol. Ther. 2021, 44, 902–909. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).