
Kinases Inhibitors as New Therapeutic Opportunities in Cutaneous T-Cell Lymphoma



Abstract
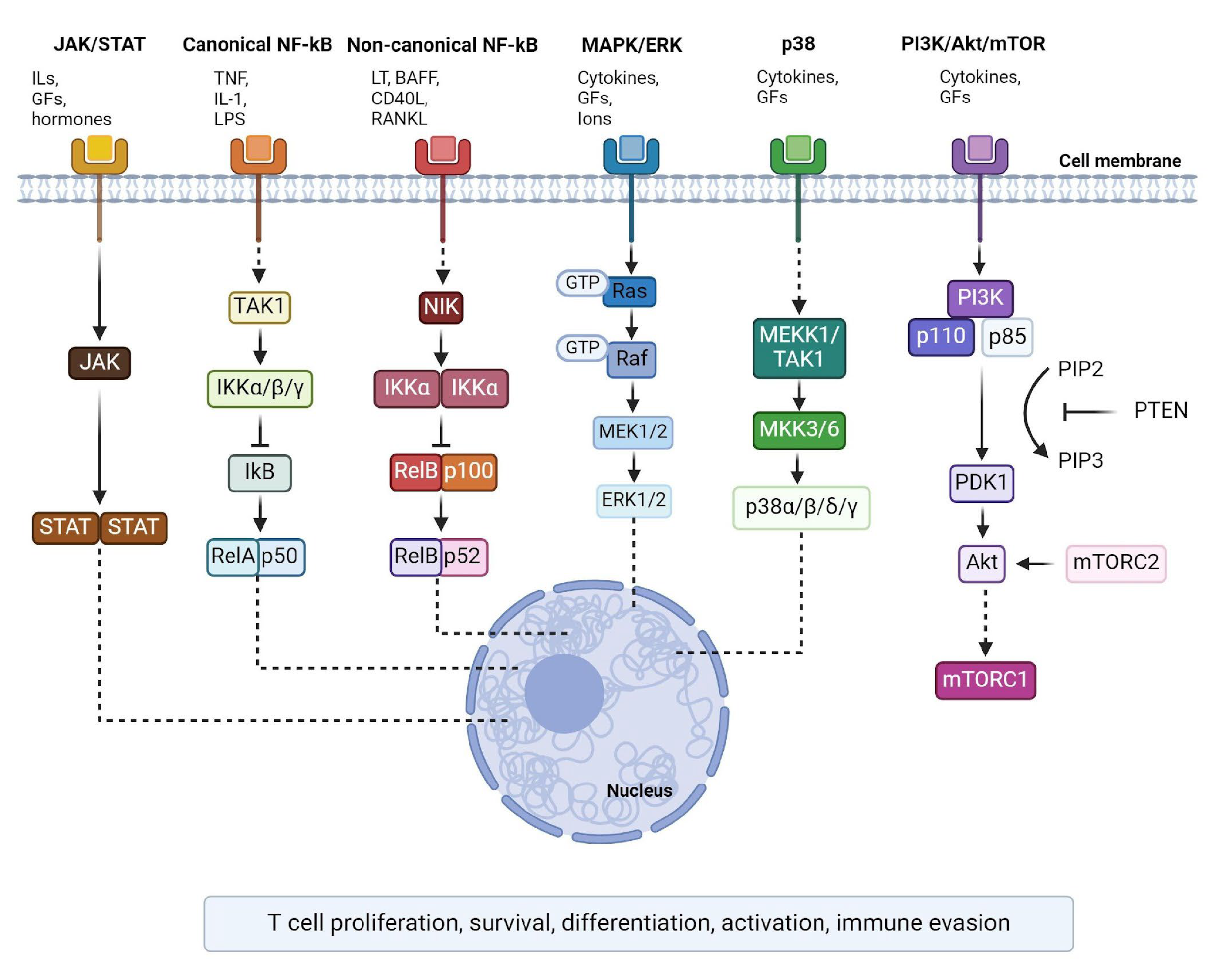
:1. Introduction
2. JAK/STAT Signalling
2.1. JAK Inhibitors
2.2. JAK Inhibitors Effect on Cell Proliferation and Apoptosis in CTCL In Vitro Models
2.3. JAK Inhibitors Interfere with Pro-Tumorigenic Cellular Signalling In Vitro and In Vivo CTCL Models
2.4. JAK Inhibitors Block Primary Tumour Formation and the Metastatic Cascade in CTCL
2.5. Clinical Trials and Case Reports of JAK Inhibitors in CTCL
2.6. JAK Inhibition to Overcome Staphylococcus Aureus-Induced Drug Resistance
3. NF-kB Signalling
3.1. IKK Inhibitors
3.2. TAK1 Inhibitor in CTCL
4. MAPK Signalling
4.1. ERK Signalling
4.2. MAPK/ERK Inhibitors Effect on Cell Proliferation and Apoptosis in CTCL In Vitro Models
4.3. MAPK/ERK Inhibitors Effect on CTCL Microenvironment
4.4. Clinical Trials of MAPK/ERK Inhibitors in CTCL
5. p38 Signalling
5.1. p38 Inhibitors Effect on Differentiation and Apoptosis in CTCL In Vitro Models
5.2. Clinical Trials of p38 Inhibitors in CTCL
6. PI3K/Akt/mTOR Signalling
6.1. PI3K/Akt/mTOR Inhibitors in CTCL In Vitro and In Vivo Models
6.2. Clinical Trials Targeting PI3K/Akt/mTOR Signalling Pathway in CTCL
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rendón-Serna, N.; Correa-Londoño, L.A.; Velásquez-Lopera, M.M.; Bermudez-Muñoz, M. Cell signaling in cutaneous T-cell lymphoma microenvironment: Promising targets for molecular-specific treatment. Int. J. Dermatol. 2021, 60, 1462–1480. [Google Scholar] [CrossRef] [PubMed]
- Vahabi, S.M.; Bahramian, S.; Esmaeili, F.; Danaei, B.; Kalantari, Y.; Fazeli, P.; Sadeghi, S.; Hajizadeh, N.; Assaf, C.; Etesami, I. JAK Inhibitors in Cutaneous T-Cell Lymphoma: Friend or Foe? A Systematic Review of the Published Literature. Cancers 2024, 16, 861. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Vermeer, M.H.; Scarisbrick, J.J.; Kim, Y.H.; Stonesifer, C.; Tensen, C.P.; Geskin, L.J.; Quaglino, P.; Ramelyte, E. Cutaneous T cell lymphoma. Nat. Rev. Dis. Primers 2021, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Hague, C.; Farquharson, N.; Menasce, L.; Parry, E.; Cowan, R. Cutaneous T-cell lymphoma: Diagnosing subtypes and the challenges. Br. J. Hosp. Med. 2022, 83, 1–7. [Google Scholar] [CrossRef]
- Pavlidis, A.; Piperi, C.; Papadavid, E. Novel therapeutic approaches for cutaneous T cell lymphomas. Expert. Rev. Clin. Immunol. 2021, 17, 629–641. [Google Scholar] [CrossRef]
- De Masson, A.; Beylot-Barry, M.; Ram-Wolff, C.; Mear, J.-B.; Dalle, S.; d’Incan, M.; Ingen-Housz-Oro, S.; Orvain, C.; Abraham, J.; Dereure, O.; et al. Allogeneic transplantation in advanced cutaneous T-cell lymphomas (CUTALLO): A propensity score matched controlled prospective study. Lancet 2023, 401, 1941–1950. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Sig Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Groner, B.; Manstein, V. von Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Mol. Cell. Endocrinol. 2017, 451, 1–14. [Google Scholar] [CrossRef]
- Liau, N.P.D.; Laktyushin, A.; Lucet, I.S.; Murphy, J.M.; Yao, S.; Whitlock, E.; Callaghan, K.; Nicola, N.A.; Kershaw, N.J.; Babon, J.J. The molecular basis of JAK/STAT inhibition by SOCS1. Nat. Commun. 2018, 9, 1558. [Google Scholar] [CrossRef]
- Patil, K.; Kuttikrishnan, S.; Khan, A.Q.; Ahmad, F.; Alam, M.; Buddenkotte, J.; Ahmad, A.; Steinhoff, M.; Uddin, S. Molecular pathogenesis of Cutaneous T cell Lymphoma: Role of chemokines, cytokines, and dysregulated signaling pathways. Semin. Cancer Biol. 2022, 86, 382–399. [Google Scholar] [CrossRef]
- Karagianni, F.; Piperi, C.; Mpakou, V.; Spathis, A.; Foukas, P.G.; Dalamaga, M.; Pappa, V.; Papadavid, E. Ruxolitinib with resminostat exert synergistic antitumor effects in Cutaneous T-cell Lymphoma. PLoS ONE 2021, 16, e0248298. [Google Scholar] [CrossRef] [PubMed]
- Shawky, A.M.; Almalki, F.A.; Abdalla, A.N.; Abdelazeem, A.H.; Gouda, A.M. A Comprehensive Overview of Globally Approved JAK Inhibitors. Pharmaceutics 2022, 14, 1001. [Google Scholar] [CrossRef]
- Aihie, O.; Dyer, J. JAK Inhibitors: A New Weapon in the Skin Care Providers’ Arsenal. Mo. Med. 2023, 120, 45–48. [Google Scholar] [PubMed]
- Huang, M.; Armstrong, A. Janus-kinase inhibitors in dermatology: A review of their use in psoriasis, vitiligo, systemic lupus erythematosus, hidradenitis suppurativa, dermatomyositis, lichen planus, lichen planopilaris, sarcoidosis and graft-versus-host disease. Indian J. Dermatol. Venereol. Leprol. 2023, 90, 30–40. [Google Scholar] [CrossRef]
- Müller, S.; Maintz, L.; Bieber, T. Treatment of atopic dermatitis: Recently approved drugs and advanced clinical development programs. Allergy 2024, 79, 1501–1515. [Google Scholar] [CrossRef] [PubMed]
- McGirt, L.Y.; Jia, P.; Baerenwald, D.A.; Duszynski, R.J.; Dahlman, K.B.; Zic, J.A.; Zwerner, J.P.; Hucks, D.; Dave, U.; Zhao, Z.; et al. Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood 2015, 126, 508–519. [Google Scholar] [CrossRef]
- Karagianni, F.; Piperi, C.; Casar, B.; de la Fuente-Vivas, D.; García-Gómez, R.; Lampadaki, K.; Pappa, V.; Papadavid, E. Combination of Resminostat with Ruxolitinib Exerts Antitumor Effects in the Chick Embryo Chorioallantoic Membrane Model for Cutaneous T Cell Lymphoma. Cancers 2022, 14, 1070. [Google Scholar] [CrossRef]
- Yumeen, S.; Mirza, F.N.; Lewis, J.M.; King, A.L.O.; Kim, S.R.; Carlson, K.R.; Umlauf, S.R.; Surovtseva, Y.V.; Foss, F.M.; Girardi, M. JAK inhibition synergistically potentiates BCL2, BET, HDAC, and proteasome inhibition in advanced CTCL. Blood Adv. 2020, 4, 2213–2226. [Google Scholar] [CrossRef]
- Pérez, C.; González-Rincón, J.; Onaindia, A.; Almaráz, C.; García-Díaz, N.; Pisonero, H.; Curiel-Olmo, S.; Gómez, S.; Cereceda, L.; Madureira, R.; et al. Mutated JAK kinases and deregulated STAT activity are potential therapeutic targets in cutaneous T-cell lymphoma. Haematologica 2015, 100, e450–e453. [Google Scholar] [CrossRef]
- Karagianni, F.; Saraki, K.; Papadaki, M.; Mpakou, V.; Piperi, C.; Pappa, V.; Papadavid, E. 010—In vitro effect of Jak and HDAC inhibitors in cutaneous T-cell lymphoma. Eur. J. Cancer 2019, 119, S4. [Google Scholar] [CrossRef]
- Zhu, M.; Liu, Y.; Lei, P.; Shi, X.; Tang, W.; Huang, X.; Pan, X.; Wang, C.; Ma, W. ND-16: A Novel Compound for Inhibiting the Growth of Cutaneous T Cell Lymphoma by Targeting JAK2. Curr. Cancer Drug Targets 2022, 22, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, F.; Bertran, J.; López-Arribillaga, E.; González, J.; Menéndez, S.; Sánchez, I.; Colomo, L.; Iglesias, M.; Garrido, M.; Santamaría-Babí, L.F.; et al. Novel phosphorylated TAK1 species with functional impact on NF-κB and β-catenin signaling in human Cutaneous T-cell lymphoma. Leukemia 2018, 32, 2211–2223. [Google Scholar] [CrossRef]
- Seto, A.G.; Beatty, X.; Lynch, J.M.; Hermreck, M.; Tetzlaff, M.; Duvic, M.; Jackson, A.L. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br. J. Haematol. 2018, 183, 428–444. [Google Scholar] [CrossRef]
- Kießling, M.K.; Nicolay, J.P.; Schlör, T.; Klemke, C.-D.; Süss, D.; Krammer, P.H.; Gülow, K. NRAS mutations in cutaneous T cell lymphoma (CTCL) sensitize tumors towards treatment with the multikinase inhibitor Sorafenib. Oncotarget 2017, 8, 45687–45697. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.R.; Robey, R.W.; Luchenko, V.L.; Zhan, Z.; Piekarz, R.L.; Gillet, J.-P.; Kossenkov, A.V.; Wilkerson, J.; Showe, L.C.; Gottesman, M.M.; et al. MAPK pathway activation leads to Bim loss and histone deacetylase inhibitor resistance: Rationale to combine romidepsin with an MEK inhibitor. Blood 2013, 121, 4115–4125. [Google Scholar] [CrossRef]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/β-Catenin and NF-κB Signaling Pathway during Inflammation. Front. Immunol. 2016, 7, 378. [Google Scholar] [CrossRef] [PubMed]
- Haller, V.; Nahidino, P.; Forster, M.; Laufer, S.A. An updated patent review of p38 MAP kinase inhibitors (2014-2019). Expert. Opin. Ther. Pat. 2020, 30, 453–466. [Google Scholar] [CrossRef]
- Witzig, T.E.; Reeder, C.; Han, J.J.; LaPlant, B.; Stenson, M.; Tun, H.W.; Macon, W.; Ansell, S.M.; Habermann, T.M.; Inwards, D.J.; et al. The mTORC1 inhibitor everolimus has antitumor activity in vitro and produces tumor responses in patients with relapsed T-cell lymphoma. Blood 2015, 126, 328–335. [Google Scholar] [CrossRef]
- Horwitz, S.M.; Koch, R.; Porcu, P.; Oki, Y.; Moskowitz, A.; Perez, M.; Myskowski, P.; Officer, A.; Jaffe, J.D.; Morrow, S.N.; et al. Activity of the PI3K-δ,γ inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood 2018, 131, 888–898. [Google Scholar] [CrossRef]
- Bazewicz, C.; Verardi, N.; Akilov, O. Utility of Low-Dose Duvelisib for Advanced Mycosis Fungoides: A Single-Institution Study. Oncologist 2024, 29, 272–274. [Google Scholar] [CrossRef]
- Dai, J.; Duvic, M. Cutaneous T-Cell Lymphoma: Current and Emerging Therapies. Cancer Netw. 2023, 37, 55–62. [Google Scholar]
- Vadivel, C.K.; Willerslev-Olsen, A.; Namini, M.R.J.; Zeng, Z.; Yan, L.; Danielsen, M.; Gluud, M.; Pallesen, E.M.H.; Wojewoda, K.; Osmancevic, A.; et al. Staphylococcus aureus induces drug resistance in cancer T cells in Sézary syndrome. Blood 2024, 143, 1496–1512. [Google Scholar] [CrossRef]
- Chang, T.-P.; Vancurova, I. NFκB function and regulation in cutaneous T-cell lymphoma. Am. J. Cancer Res. 2013, 3, 433–445. [Google Scholar] [PubMed]
- Li, Y.; Zhao, B.; Peng, J.; Tang, H.; Wang, S.; Peng, S.; Ye, F.; Wang, J.; Ouyang, K.; Li, J.; et al. Inhibition of NF-κB signaling unveils novel strategies to overcome drug resistance in cancers. Drug Resist. Updates 2024, 73, 101042. [Google Scholar] [CrossRef]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H.; et al. Role of the NFκB-signaling pathway in cancer. OncoTargets Ther. 2018, 11, 2063–2073. [Google Scholar] [CrossRef]
- Paul, A.; Edwards, J.; Pepper, C.; Mackay, S. Inhibitory-κB Kinase (IKK) α and Nuclear Factor-κB (NFκB)-Inducing Kinase (NIK) as Anti-Cancer Drug Targets. Cells 2018, 7, 176. [Google Scholar] [CrossRef]
- Zinzani, P.L.; Musuraca, G.; Tani, M.; Stefoni, V.; Marchi, E.; Fina, M.; Pellegrini, C.; Alinari, L.; Derenzini, E.; de Vivo, A.; et al. Phase II Trial of Proteasome Inhibitor Bortezomib in Patients with Relapsed or Refractory Cutaneous T-Cell Lymphoma. J. Clin. Oncol. 2007, 25, 4293–4297. [Google Scholar] [CrossRef] [PubMed]
- Prescott, J.A.; Cook, S.J. Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors. Cells 2018, 7, 115. [Google Scholar] [CrossRef]
- Wuerzberger-Davis, S.M.; Miyamoto, S. TAK-ling IKK Activation: “Ub” the Judge. Sci. Signal. 2010, 3, pe3. [Google Scholar] [CrossRef]
- Takaesu, G.; Surabhi, R.M.; Park, K.-J.; Ninomiya-Tsuji, J.; Matsumoto, K.; Gaynor, R.B. TAK1 is Critical for IκB Kinase-mediated Activation of the NF-κB Pathway. J. Mol. Biol. 2003, 326, 105–115. [Google Scholar] [CrossRef]
- Bellei, B.; Cota, C.; Amantea, A.; Muscardin, L.; Picardo, M. Association of p53 Arg72Pro polymorphism and β-catenin accumulation in mycosis fungoides. Br. J. Dermatol. 2006, 155, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Scarneo, S.; Hughes, P.; Freeze, R.; Yang, K.; Totzke, J.; Haystead, T. Development and Efficacy of an Orally Bioavailable Selective TAK1 Inhibitor for the Treatment of Inflammatory Arthritis. ACS Chem. Biol. 2022, 17, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Almeida, A.C.; Abate, F.; Khiabanian, H.; Martinez-Escala, E.; Guitart, J.; Tensen, C.P.; Vermeer, M.H.; Rabadan, R.; Ferrando, A.; Palomero, T. The mutational landscape of cutaneous T cell lymphoma and Sézary syndrome. Nat. Genet. 2015, 47, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK signalling pathway and tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Brown, M.D.; Sacks, D.B. Compartmentalised MAPK Pathways. In Protein-Protein Interactions as New Drug Targets; Klussmann, E., Scott, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 205–235. ISBN 978-3-540-72843-6. [Google Scholar]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing Effects of ERK and JNK-p38 MAP Kinases on Apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef]
- Gibson, J.F.; Foss, F.; Cooper, D.; Seropian, S.; Irizarry, D.; Barbarotta, L.; Lansigan, F. Pilot study of sorafenib in relapsed or refractory peripheral and cutaneous T-cell lymphoma. Br. J. Haematol. 2014, 167, 141–144. [Google Scholar] [CrossRef]
- Zhang, X.H.; Chen, C.-H.; Li, H.; Hsiang, J.; Wu, X.; Hu, W.; Horne, D.; Nam, S.; Shively, J.; Rosen, S.T. Targeting the non-ATP-binding pocket of the MAP kinase p38γ mediates a novel mechanism of cytotoxicity in cutaneous T-cell lymphoma (CTCL). FEBS Lett. 2021, 595, 2570–2592. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Yong, H.-Y.; Koh, M.-S.; Moon, A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert. Opin. Investig. Drugs 2009, 18, 1893–1905. [Google Scholar] [CrossRef]
- Manfè, V.; Biskup, E.; Rosbjerg, A.; Kamstrup, M.; Skov, A.G.; Lerche, C.M.; Lauenborg, B.T.; Ødum, N.; Gniadecki, R. miR-122 Regulates p53/Akt Signalling and the Chemotherapy-Induced Apoptosis in Cutaneous T-Cell Lymphoma. PLoS ONE 2012, 7, e29541. [Google Scholar] [CrossRef]

{kind=link}
| Signalling Pathway | Kinase Target | Drug | Tests and Results on CTCL |
|---|---|---|---|
| JAK/STAT | JAK1/2 | Ruxolitinib | Positive results from in vitro, in vivo and clinical studies. |
| JAK3 JAK2 JAK1/2/3 SYK JAK1 | Tofacitinib ND-16 Cerdulatinib Upadacitinib |
In vitro study: cell cycle arrest and increased apoptosis in H9 cell line [21]. Clinical trial (phase II): ORR 35%, with highest ORR in MF patients [2]. Case reports: complete response in two MF patients [2]. | |
| NF-kB | TAK1 | 5Z-7-Oxozeaenol | In vitro study: reduced proliferation and apoptosis induction in HH and SeAx cells. CTCL-derived tumour growth reduction [22]. |
| IKKβ | HS-276 BAY65-8072 | No results on CTCL. Suggested. In vitro study: reduced HH, MyLa, and SeAx cell growth [22]. | |
| MAPK/ERK | ERK1/2 Multikinase Inhibitor JNK MEK | Cobomarsen (miR-155 inhibitor) Sorafenib Ruxolitinib Selumetinib (AZD6244) | In vitro study: reduces ERK1/2 phosphorylation in HuT-102 MF cell line, decreases cell proliferation and induces apoptosis. In vivo, the tumour size decreased and showed no adverse effects in one patient [23]. In vitro study: inhibits p-MEK and p-ERK successfully in CTCL cell lines like HuT-78, SeAx, MyLa, and HH by producing apoptosis and an anti-proliferative effect [24]. Clinical trial (phase I): 12 patients with refractory/relapsed CTCL, 11% of patients had a complete remission (CR) of the cancer. Better results in combination with Vorinostat (HDACi) by causing apoptosis [24]. In vitro study: combination with Resminostat (HDACi) alters p-JNK levels and inhibits MAPK activation in MyLa and SeAx cell lines.
|
| P38 | p38γ | CSH71 | In vitro study: apoptosis in HuT-78 cell line [26]. |
| p38γ,δ p38α | Imidazo [1,2-a]pyridine UM-60 ATI-450 | No results on CTCL. Suggested [27]. In vitro study. Not approved drug [27]. No results on CTCL. Suggested [27]. | |
| PI3K/Akt/mTOR | PI3K/Akt p-Akt mTOR PI3Kγ,δ | Idelalisib ND-16 Ruxolitinib Everolimus Duvelisib | In vitro study: combination with Cobomarsen produced depletion of both Akt and ERK1/2 phosphorylation [1]. In vitro study: depletion of key signalling pathways in H9 cells and regulation of cell cycle proteins [21]. In vivo study: combination with Resminostat (HDACi) inhibited p-p38, p-Akt and p-ERK using MyLa and SeAx cell lines in derived xenografted tumours [17]. Clinical trial (phase II): ORR 44% in relapsed CTCL patients with minimal cytotoxic response (taste disorder) [28]. Clinical trial (phase I): ORR 32% with several adverse effects with high doses [29]. While at low doses, it has an ORR of 71% and causes fatigue, nausea and transaminitis [30]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valero-Diaz, S.; Amato, C.; Casar, B. Kinases Inhibitors as New Therapeutic Opportunities in Cutaneous T-Cell Lymphoma. Kinases Phosphatases 2024, 2, 255-267. https://doi.org/10.3390/kinasesphosphatases2030016
Valero-Diaz S, Amato C, Casar B. Kinases Inhibitors as New Therapeutic Opportunities in Cutaneous T-Cell Lymphoma. Kinases and Phosphatases. 2024; 2(3):255-267. https://doi.org/10.3390/kinasesphosphatases2030016
Chicago/Turabian StyleValero-Diaz, Sara, Camilla Amato, and Berta Casar. 2024. "Kinases Inhibitors as New Therapeutic Opportunities in Cutaneous T-Cell Lymphoma" Kinases and Phosphatases 2, no. 3: 255-267. https://doi.org/10.3390/kinasesphosphatases2030016
APA StyleValero-Diaz, S., Amato, C., & Casar, B. (2024). Kinases Inhibitors as New Therapeutic Opportunities in Cutaneous T-Cell Lymphoma. Kinases and Phosphatases, 2(3), 255-267. https://doi.org/10.3390/kinasesphosphatases2030016