
Development of Interface-Specific Two-Dimensional Vibrational–Electronic (i2D-VE) Spectroscopy for Vibronic Couplings at Interfaces



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
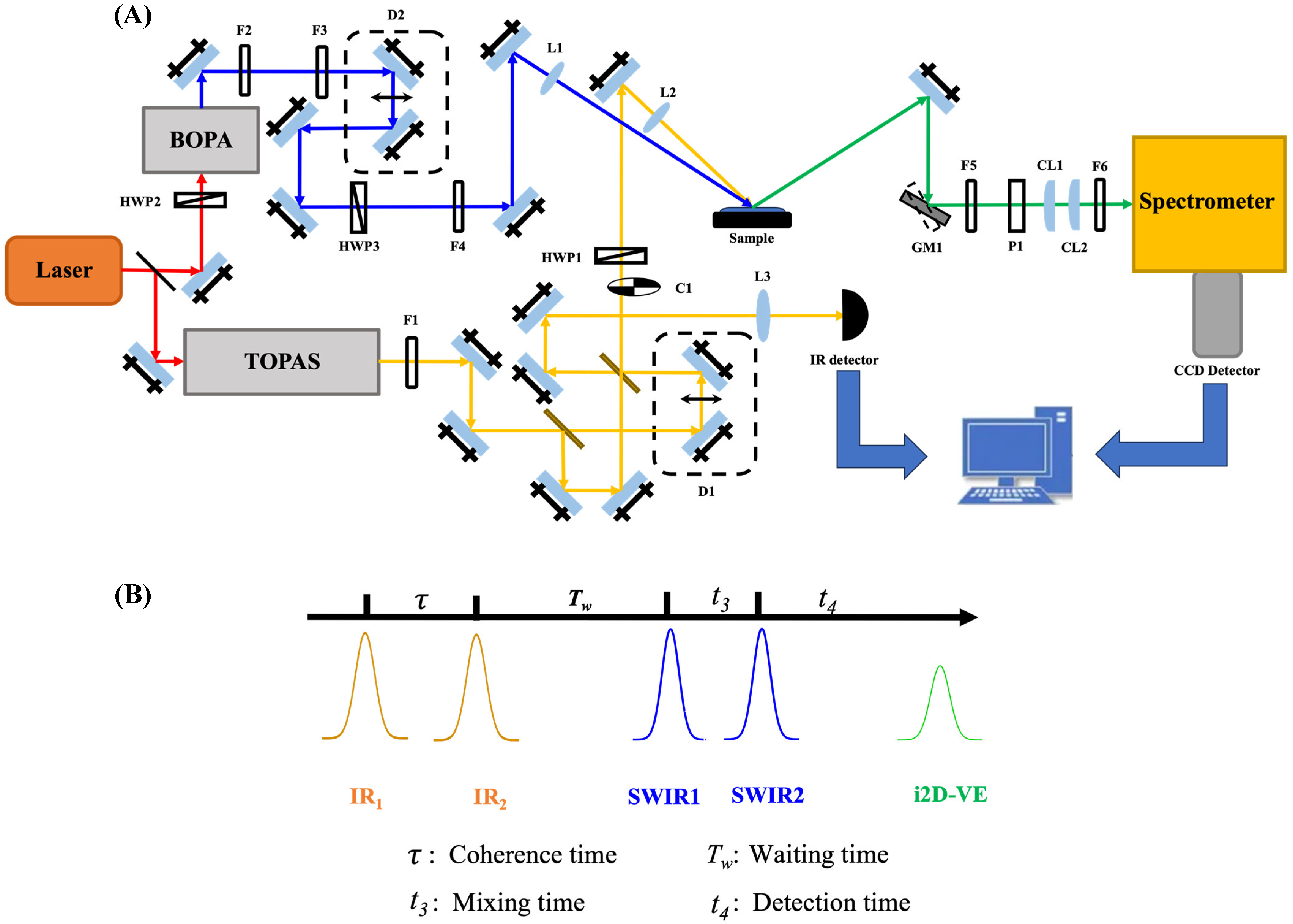
2. Materials and Methods
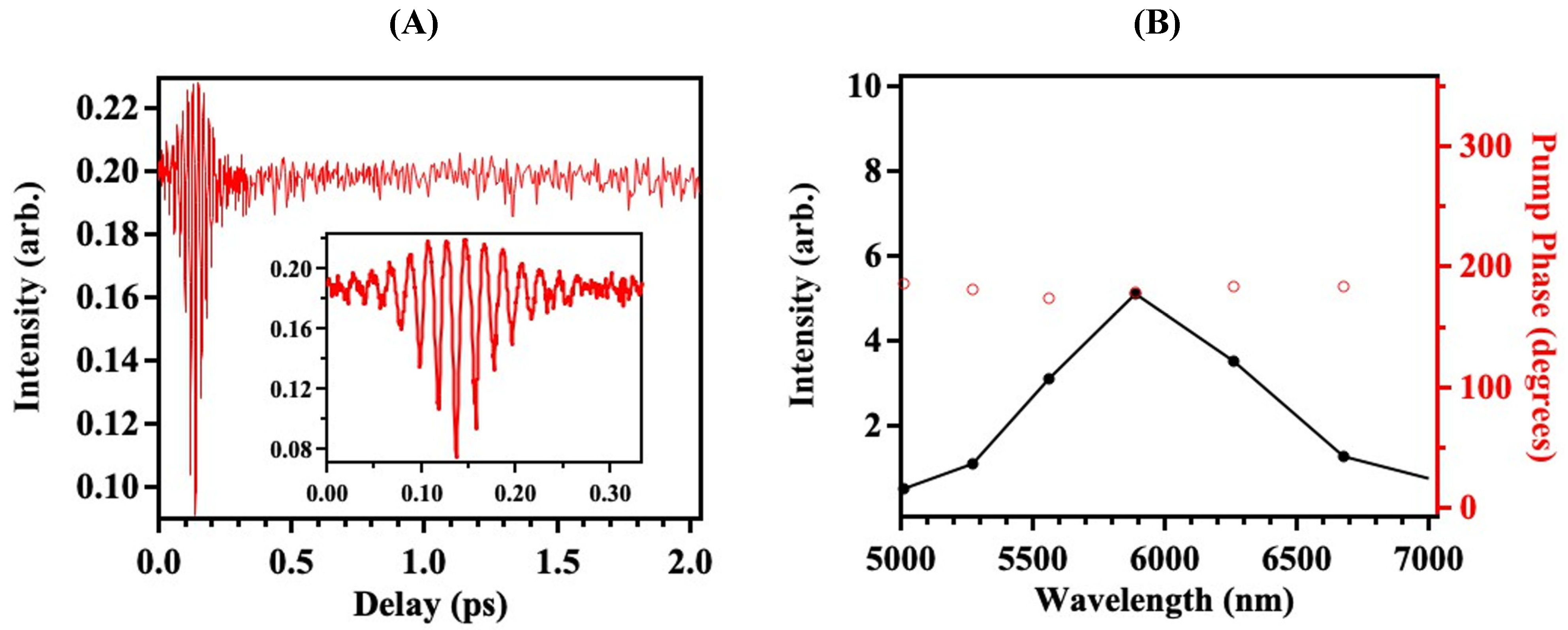
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mukamel, S.; Tanimura, Y.; Hamm, P. Coherent Multidimensional Optical Spectroscopy. Acc. Chem. Res. 2009, 42, 1207–1209. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.C. Coherent multidimensional vibrational spectroscopy. Int. Rev. Phys. Chem. 2002, 21, 185–255. [Google Scholar] [CrossRef]
- Cundiff, S.T.; Mukamel, S. Optical multidimensional coherent spectroscopy. Phys. Today 2013, 66, 44–49. [Google Scholar] [CrossRef]
- Bruder, L.; Bangert, U.; Binz, M.; Uhl, D.; Stienkemeier, F. Coherent multidimensional spectroscopy in the gas phase. J. Phys. B At. Mol. Opt. Phys. 2019, 52, 183501. [Google Scholar] [CrossRef]
- Petti, M.K.; Lomont, J.P.; Maj, M.; Zanni, M.T. Two-Dimensional Spectroscopy Is Being Used to Address Core Scientific Questions in Biology and Materials Science. J. Phys. Chem. B 2018, 122, 1771–1780. [Google Scholar] [CrossRef]
- Baiz, C.; Bredenbeck, J.; Cho, M.; Jansen, T.; Krummel, A.; Roberts, S. Celebrating 25 years of 2D IR spectroscopy. J. Chem. Phys. 2024, 160, 010401. [Google Scholar] [CrossRef]
- Biswas, S.; Kim, J.; Zhang, X.; Scholes, G.D. Coherent Two-Dimensional and Broadband Electronic Spectroscopies. Chem. Rev. 2022, 122, 4257–4321. [Google Scholar] [CrossRef]
- Wright, J.C. Multiresonant Coherent Multidimensional Spectroscopy. Annu. Rev. Phys. Chem. 2011, 62, 209–230. [Google Scholar] [CrossRef]
- Schultz, J.D.; Yuly, J.L.; Arsenault, E.A.; Parker, K.; Chowdhury, S.N.; Dani, R.; Kundu, S.; Nuomin, H.; Zhang, Z.; Valdiviezo, J.; et al. Coherence in Chemistry: Foundations and Frontiers. Chem. Rev. 2024, 124, 11641–11766. [Google Scholar] [CrossRef]
- Buchanan, L.E.; Dunkelberger, E.B.; Tran, H.Q.; Cheng, P.-N.; Chiu, C.-C.; Cao, P.; Raleigh, D.P.; de Pablo, J.J.; Nowick, J.S.; Zanni, M.T. Mechanism of IAPP amyloid fibril formation involves an intermediate with a transient β-sheet. Proc. Natl. Acad. Sci. USA 2013, 110, 19285–19290. [Google Scholar] [CrossRef]
- Mehlenbacher, R.D.; McDonough, T.J.; Grechko, M.; Wu, M.-Y.; Arnold, M.S.; Zanni, M.T. Energy transfer pathways in semiconducting carbon nanotubes revealed using two-dimensional white-light spectroscopy. Nat. Commun. 2015, 6, 6732. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, N.S.; Cheng, Y.-C.; Fleming, G.R. Two-Dimensional Electronic Spectroscopy of Molecular Aggregates. Acc. Chem. Res. 2009, 42, 1352–1363. [Google Scholar] [CrossRef] [PubMed]
- Harel, E.; Fidler, A.F.; Engel, G.S. Real-time mapping of electronic structure with single-shot two-dimensional electronic spectroscopy. Proc. Natl. Acad. Sci. USA 2010, 107, 16444–16447. [Google Scholar] [CrossRef]
- Kim, Y.S.; Hochstrasser, R.M. Applications of 2D IR Spectroscopy to Peptides, Proteins, and Hydrogen-Bond Dynamics. J. Phys. Chem. B 2009, 113, 8231–8251. [Google Scholar] [CrossRef]
- Zheng, J.; Kwak, K.; Fayer, M.D. Ultrafast 2D IR Vibrational Echo Spectroscopy. Acc. Chem. Res. 2007, 40, 75–83. [Google Scholar] [CrossRef]
- Fernández-Terán, R.; Ruf, J.; Hamm, P. Vibrational Couplings in Hydridocarbonyl Complexes: A 2D-IR Perspective. Inorg. Chem. 2020, 59, 7721–7726. [Google Scholar] [CrossRef]
- Hybl, J.D.; Albrecht, A.W.; Gallagher Faeder, S.M.; Jonas, D.M. Two-dimensional electronic spectroscopy. Chem. Phys. Lett. 1998, 297, 307–313. [Google Scholar] [CrossRef]
- Gallagher Faeder, S.M.; Jonas, D.M. Two-Dimensional Electronic Correlation and Relaxation Spectra: Theory and Model Calculations. J. Phys. Chem. A 1999, 103, 10489–10505. [Google Scholar] [CrossRef]
- Hybl, J.D.; Albrecht Ferro, A.; Jonas, D.M. Two-dimensional Fourier transform electronic spectroscopy. J. Chem. Phys. 2001, 115, 6606–6622. [Google Scholar] [CrossRef]
- Brixner, T.; Mančal, T.; Stiopkin, I.V.; Fleming, G.R. Phase-stabilized two-dimensional electronic spectroscopy. J. Chem. Phys. 2004, 121, 4221–4236. [Google Scholar] [CrossRef]
- Schlau-Cohen, G.S.; Ishizaki, A.; Fleming, G.R. Two-dimensional electronic spectroscopy and photosynthesis: Fundamentals and applications to photosynthetic light-harvesting. Chem. Phys. 2011, 386, 1–22. [Google Scholar] [CrossRef]
- Zanni, M.T.; Ge, N.-H.; Kim, Y.S.; Hochstrasser, R.M. Two-dimensional IR spectroscopy can be designed to eliminate the diagonal peaks and expose only the crosspeaks needed for structure determination. Proc. Natl. Acad. Sci. USA 2001, 98, 11265–11270. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.; Demirdöven, N.; Tokmakoff, A. Coherent 2D IR Spectroscopy: Molecular Structure and Dynamics in Solution. J. Phys. Chem. A 2003, 107, 5258–5279. [Google Scholar] [CrossRef]
- Kwak, K.; Park, S.; Finkelstein, I.J.; Fayer, M.D. Frequency-frequency correlation functions and apodization in two-dimensional infrared vibrational echo spectroscopy: A new approach. J. Chem. Phys. 2007, 127, 124503. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kwak, K.; Fayer, M.D. Ultrafast 2D-IR vibrational echo spectroscopy: A probe of molecular dynamics. Laser Phys. Lett. 2007, 4, 704. [Google Scholar] [CrossRef]
- Hochstrasser, R.M. Two-dimensional spectroscopy at infrared and optical frequencies. Proc. Natl. Acad. Sci. USA 2007, 104, 14190–14196. [Google Scholar] [CrossRef]
- Weiner, A.M.; De Silvestri, S.; Ippen, E.P. Three-pulse scattering for femtosecond dephasing studies: Theory and experiment. J. Opt. Soc. Am. B 1985, 2, 654–662. [Google Scholar] [CrossRef]
- Hamm, P.; Lim, M.; Hochstrasser, R.M. Structure of the Amide I Band of Peptides Measured by Femtosecond Nonlinear-Infrared Spectroscopy. J. Phys. Chem. B 1998, 102, 6123–6138. [Google Scholar] [CrossRef]
- Fuller, F.D.; Ogilvie, J.P. Experimental Implementations of Two-Dimensional Fourier Transform Electronic Spectroscopy. Annu. Rev. Phys. Chem. 2015, 66, 667–690. [Google Scholar] [CrossRef]
- Oliver, T.A.A.; Lewis, N.H.C.; Fleming, G.R. Correlating the motion of electrons and nuclei with two-dimensional electronic–vibrational spectroscopy. Proc. Natl. Acad. Sci. USA 2014, 111, 10061–10066. [Google Scholar] [CrossRef]
- Courtney, T.L.; Fox, Z.W.; Estergreen, L.; Khalil, M. Measuring Coherently Coupled Intramolecular Vibrational and Charge-Transfer Dynamics with Two-Dimensional Vibrational–Electronic Spectroscopy. J. Phys. Chem. Lett. 2015, 6, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Courtney, T.L.; Fox, Z.W.; Slenkamp, K.M.; Khalil, M. Two-dimensional vibrational-electronic spectroscopy. J. Chem. Phys. 2015, 143, 154201. [Google Scholar] [CrossRef] [PubMed]
- Gaynor, J.D.; Khalil, M. Signatures of vibronic coupling in two-dimensional electronic-vibrational and vibrational-electronic spectroscopies. J. Chem. Phys. 2017, 147, 094202. [Google Scholar] [CrossRef] [PubMed]
- Gaynor, J.D.; Weakly, R.B.; Khalil, M. Multimode two-dimensional vibronic spectroscopy. I. Orientational response and polarization-selectivity. J. Chem. Phys. 2021, 154, 184201. [Google Scholar] [CrossRef]
- Weakly, R.B.; Gaynor, J.D.; Khalil, M. Multimode two-dimensional vibronic spectroscopy. II. Simulating and extracting vibronic coupling parameters from polarization-selective spectra. J. Chem. Phys. 2021, 154, 184202. [Google Scholar] [CrossRef]
- Deng, G.-H.; Qian, Y.; Zhang, T.; Han, J.; Chen, H.; Rao, Y. Two-dimensional electronic–vibrational sum frequency spectroscopy for interactions of electronic and nuclear motions at interfaces. Proc. Natl. Acad. Sci. USA 2021, 118, e2100608118. [Google Scholar] [CrossRef]
- Huang-Fu, Z.-C.; Qian, Y.; Zhang, T.; Deng, G.-H.; Brown, J.B.; Fisher, H.; Schmidt, S.; Chen, H.; Rao, Y. Orientational Coupling of Molecules at Interfaces Revealed by Two-Dimensional Electronic–Vibrational Sum Frequency Generation (2D-EVSFG). JACS Au 2023, 3, 1413–1423. [Google Scholar] [CrossRef]
- Huang-Fu, Z.-C.; Qian, Y.; Deng, G.-H.; Zhang, T.; Schmidt, S.; Brown, J.; Rao, Y. Development of Two-Dimensional Electronic-Vibrational Sum Frequency Generation (2D-EVSFG) for Vibronic and Solvent Couplings of Molecules at Interfaces and Surfaces. ACS Phys. Chem. Au 2023, 3, 374–385. [Google Scholar] [CrossRef]
- Huang-Fu, Z.-C.; Tkachenko, N.V.; Qian, Y.; Zhang, T.; Brown, J.B.; Harutyunyan, A.; Chen, G.; Rao, Y. Conical Intersections at Interfaces Revealed by Phase-Cycling Interface-Specific Two-Dimensional Electronic Spectroscopy (i2D-ES). J. Am. Chem. Soc. 2024, 146, 20996–21007. [Google Scholar] [CrossRef]
- Zhang, T.; Huang-Fu, Z.-C.; Qian, Y.; Gao, H.; Brown, J.B.; Rao, Y. Photoinduced Surface Electric Fields and Surface Population Dynamics of GaP(100) Photoelectrodes. J. Phys. Chem. C 2022, 126, 6531–6541. [Google Scholar] [CrossRef]
- Zhang, T.; Qian, Y.; Gao, H.; Huang-Fu, Z.-C.; Brown, J.B.; Rao, Y. Surface States for Photoelectrodes of Gallium Phosphide (GaP) with Surface-Specific Electronic Spectra and Phase Measurements. J. Phys. Chem. C 2022, 126, 6761–6772. [Google Scholar] [CrossRef]
- Bitter, R.; Mohiuddin, T.; Nawrocki, M. LabView: Advanced Programming Techniques, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar] [CrossRef]
- Hamm, P.; Zanni, M. Concepts and Methods of 2D Infrared Spectroscopy; Cambridge University Press: Cambridge, UK, 2011. [Google Scholar] [CrossRef]
- Shim, S.-H.; Zanni, M.T. How to turn your pump–probe instrument into a multidimensional spectrometer: 2D IR and Vis spectroscopiesvia pulse shaping. Phys. Chem. Chem. Phys. 2009, 11, 748–761. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, P.; Saari, J.I.; Walsh, B.; Kabir, A.; Crozatier, V.; Forget, N.; Kambhampati, P. Two-Color Two-Dimensional Electronic Spectroscopy Using Dual Acousto-Optic Pulse Shapers for Complete Amplitude, Phase, and Polarization Control of Femtosecond Laser Pulses. J. Phys. Chem. A 2013, 117, 6264–6269. [Google Scholar] [CrossRef]
- Huang-Fu, Z.-C.; Qian, Y.; Zhang, T.; Brown, J.B.; Rao, Y. Development of phase-cycling interface-specific two-dimensional electronic sum frequency generation (2D-ESFG) spectroscopy. J. Chem. Phys. 2024, 161, 114201. [Google Scholar] [CrossRef]
- Helbing, J.; Hamm, P. Compact implementation of Fourier transform two-dimensional IR spectroscopy without phase ambiguity. J. Opt. Soc. Am. B 2011, 28, 171–178. [Google Scholar] [CrossRef]
- Deng, G.-H.; Qian, Y.; Wei, Q.; Zhang, T.; Rao, Y. Interface-Specific Two-Dimensional Electronic Sum Frequency Generation Spectroscopy. J. Phys. Chem. Lett. 2020, 11, 1738–1745. [Google Scholar] [CrossRef]
- Zeng, W.-W.; Luo, T.; Xu, P.; Zhou, C.; Yang, X.; Ren, Z. Vibronic coupling of Rhodamine 6G molecules studied by doubly resonant sum frequency generation spectroscopy with narrowband infrared and broadband visible. J. Chem. Phys. 2024, 160, 024705. [Google Scholar] [CrossRef]
- Laaser, J.E.; Xiong, W.; Zanni, M.T. Time-Domain SFG Spectroscopy Using Mid-IR Pulse Shaping: Practical and Intrinsic Advantages. J. Phys. Chem. B 2011, 115, 2536–2546. [Google Scholar] [CrossRef]
- Huang, J.Y.; Shen, Y.R. Theory of doubly resonant infrared-visible sum-frequency and difference-frequency generation from adsorbed molecules. Phys. Rev. A 1994, 49, 3973–3981. [Google Scholar] [CrossRef]
- Raschke, M.B.; Hayashi, M.; Lin, S.H.; Shen, Y.R. Doubly-resonant sum-frequency generation spectroscopy for surface studies. Chem. Phys. Lett. 2002, 359, 367–372. [Google Scholar] [CrossRef]
- Yang, S.; Noguchi, H.; Uosaki, K. Electronic Structure of the CO/Pt(111) Electrode Interface Probed by Potential-Dependent IR/Visible Double Resonance Sum Frequency Generation Spectroscopy. J. Phys. Chem. C 2015, 119, 26056–26063. [Google Scholar] [CrossRef]
- Sengupta, S.; Bromley Iii, L.; Velarde, L. Aggregated States of Chalcogenorhodamine Dyes on Nanocrystalline Titania Revealed by Doubly Resonant Sum Frequency Spectroscopy. J. Phys. Chem. C 2017, 121, 3424–3436. [Google Scholar] [CrossRef]
- Arsenault, E.A.; Yoneda, Y.; Iwai, M.; Niyogi, K.K.; Fleming, G.R. The role of mixed vibronic Qy-Qx states in green light absorption of light-harvesting complex II. Nat. Commun. 2020, 11, 6011. [Google Scholar] [CrossRef] [PubMed]
- Gaynor, J.D.; Petrone, A.; Li, X.; Khalil, M. Mapping Vibronic Couplings in a Solar Cell Dye with Polarization-Selective Two-Dimensional Electronic–Vibrational Spectroscopy. J. Phys. Chem. Lett. 2018, 9, 6289–6295. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, Y.; Huang-Fu, Z.-C.; Brown, J.B.; Rao, Y. Development of Interface-Specific Two-Dimensional Vibrational–Electronic (i2D-VE) Spectroscopy for Vibronic Couplings at Interfaces. Spectrosc. J. 2025, 3, 1. https://doi.org/10.3390/spectroscj3010001
Qian Y, Huang-Fu Z-C, Brown JB, Rao Y. Development of Interface-Specific Two-Dimensional Vibrational–Electronic (i2D-VE) Spectroscopy for Vibronic Couplings at Interfaces. Spectroscopy Journal. 2025; 3(1):1. https://doi.org/10.3390/spectroscj3010001
Chicago/Turabian StyleQian, Yuqin, Zhi-Chao Huang-Fu, Jesse B. Brown, and Yi Rao. 2025. "Development of Interface-Specific Two-Dimensional Vibrational–Electronic (i2D-VE) Spectroscopy for Vibronic Couplings at Interfaces" Spectroscopy Journal 3, no. 1: 1. https://doi.org/10.3390/spectroscj3010001
APA StyleQian, Y., Huang-Fu, Z.-C., Brown, J. B., & Rao, Y. (2025). Development of Interface-Specific Two-Dimensional Vibrational–Electronic (i2D-VE) Spectroscopy for Vibronic Couplings at Interfaces. Spectroscopy Journal, 3(1), 1. https://doi.org/10.3390/spectroscj3010001