Sirtuins and Immuno-Metabolism of Sepsis



Abstract
:1. Introduction
2. Immunologic Dysfunction in Sepsis
3. Metabolic and Bioenergetic Changes during Sepsis
3.1. Microenvironment Contributions
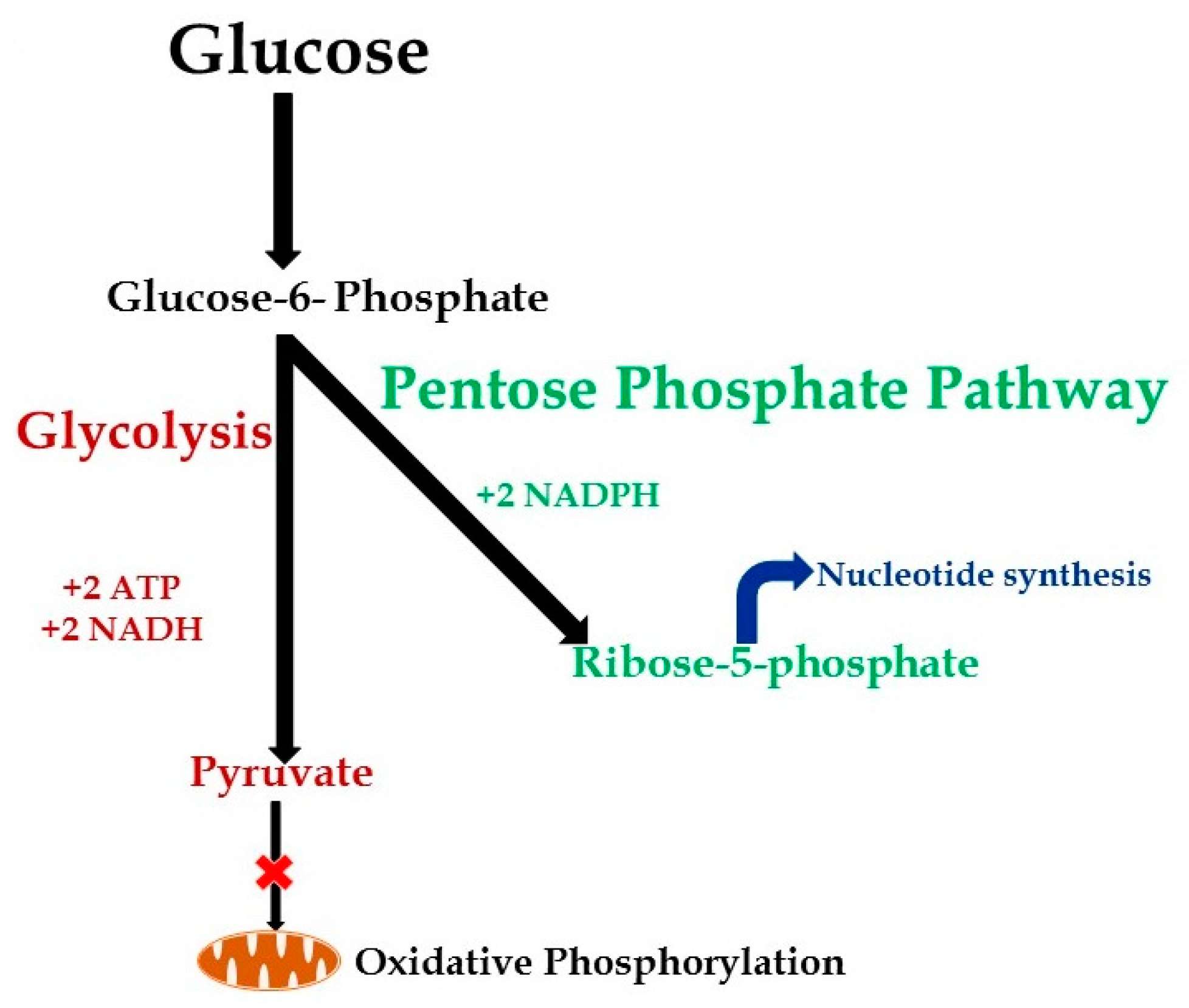
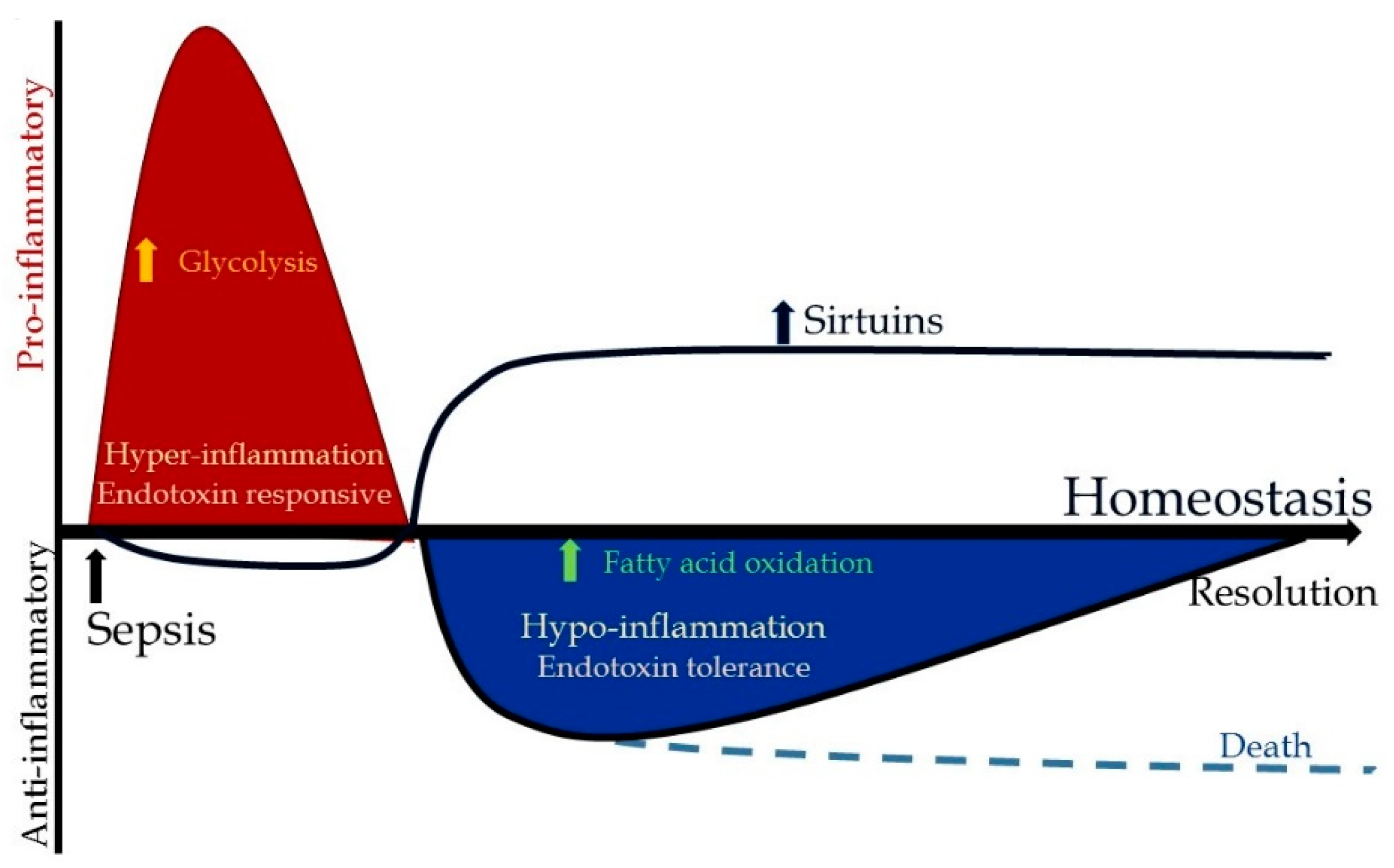
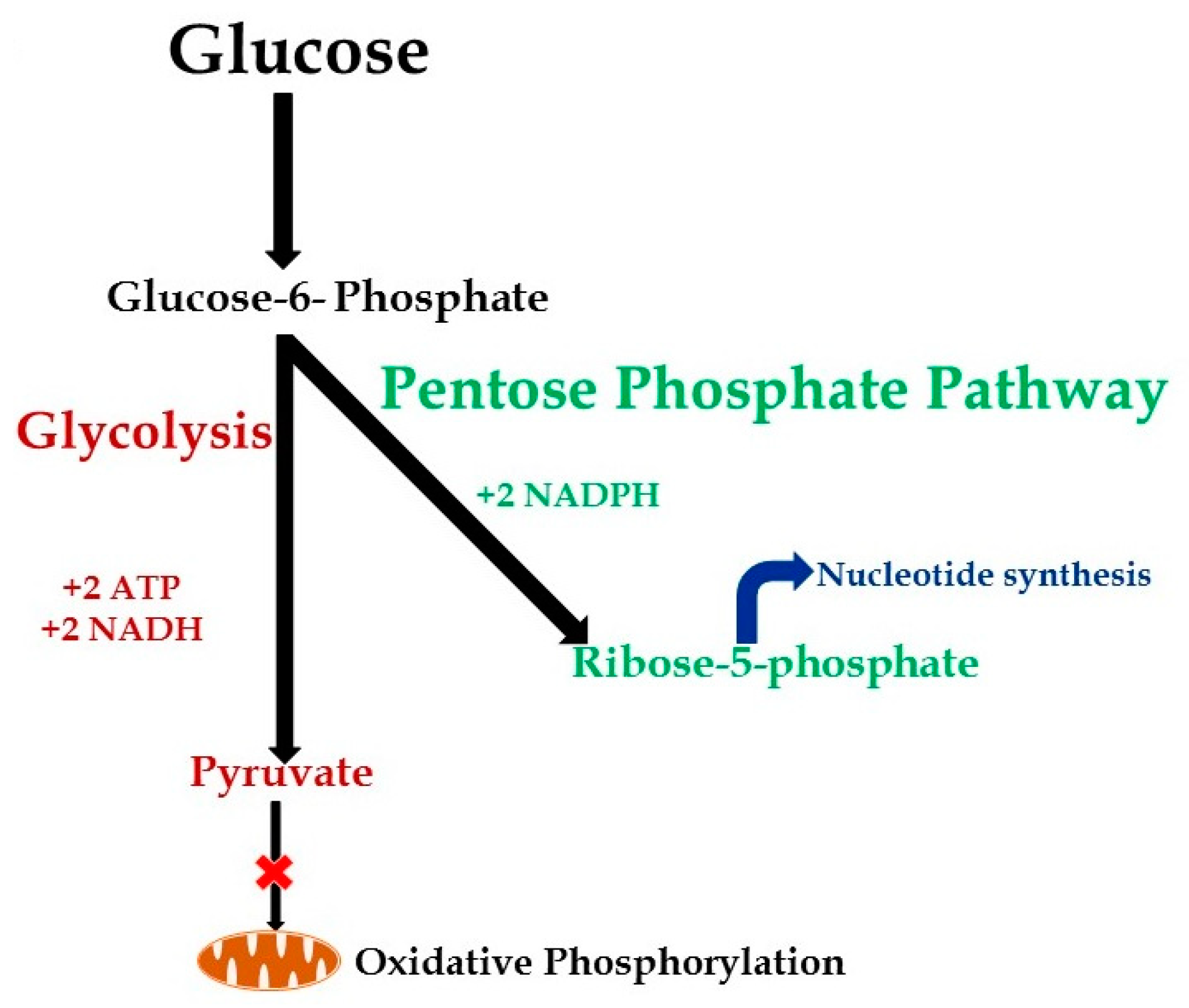
3.2. Metabolic Changes during Hyper-Inflammation
3.3. Metabolic Changes during Hypo-Inflammation
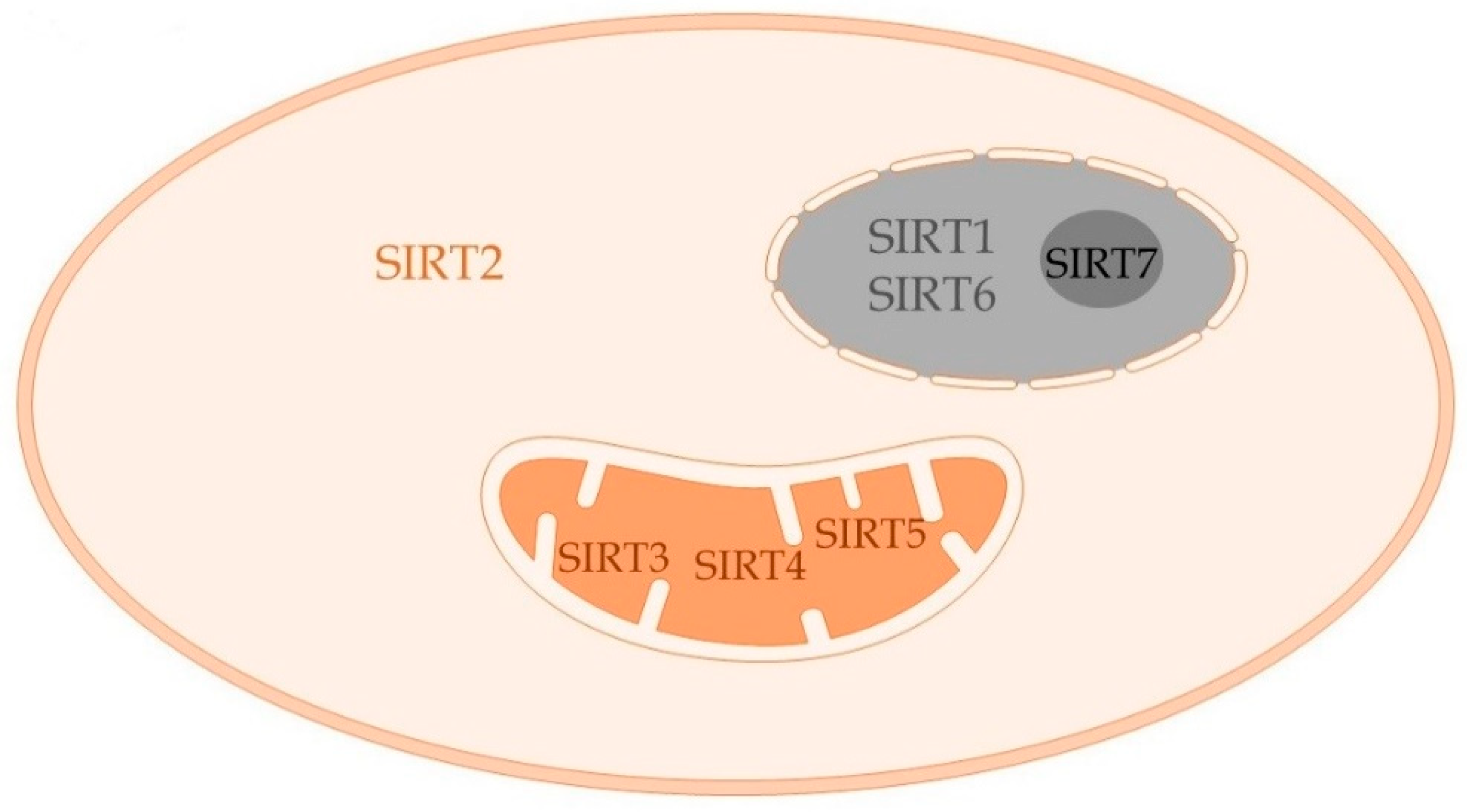
4. Sirtuins and Sepsis Immuno-Metabolism
4.1. SIRT1
4.2. SIRT2
4.3. SIRT3
4.4. SIRT4
4.5. SIRT6
4.6. SIRT5 and SIRT7
5. Sirtuin Modulation as Potential Therapeutic Targets?
5.1. Sirtuin Modulation during Hyper-Inflammation
5.2. Sirtuin Modulation during Hypo-Inflammation
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Sir2 | Silent mating-type information regulator 2 |
| SIRT | Sirtuin |
| ADP | Adenosine diphosphate |
| ATP | Adenosine triphosphate |
| NAD | Nicotinamide adenine dinucleotide |
| NAMPT | Nicotinamide phosphoribosyltransferase |
| GLUT | Glucose transporter |
| PDC | Pyruvate dehydrogenase complex |
| Acetyl-CoA | Acetyl coenzyme A |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| ROS | Reactive oxygen species |
| TCA | Tricarboxylic acid |
| CPT1 | Carnitine palmitoyltransferase 1 |
| STAT6 | Signal transducer and activator of transcription 6 |
| PPAR | Peroxisome proliferator-activated receptor |
| PGC | Peroxisome proliferator-activated receptor gamma coactivator |
| FOXO1 | Forkhead box protein O1 |
| HIF | Hypoxia inducible factor |
| LD | linear dichroism |
| UCP1 | Uncoupling protein 1 |
| PDH | Pyruvate dehydrogenase |
| ACLY | ATP-citrate lyase |
| PEPCK1 | Phosphoenolpyruvate carboxykinase 1 |
| OXPHOS | Oxidative phosphorylation |
| NFĸB | Nuclear factor kappa B |
| TNF | Tumor necrosis factor |
| H3K9 | Histone 3; lysine 9 |
| CD36 | Cluster of differentiation 36 |
| AMPK | AMP-activated protein kinase |
References
- Torio, C.M.; Andrews, R.M. National inpatient hospital costs: The most expensive conditions by payer, 2011: Statistical brief #160. In Healthcare Cost and Utilization Project (HCUP) Statistical Briefs; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2006. [Google Scholar]
- Reinhart, K.; Daniels, R.; Machado, F.R. The burden of sepsis: A call to action in support of world sepsis day 2013. Rev. Brasil. Terapia Intensiva 2013, 25, 3–5. [Google Scholar] [CrossRef]
- Boomer, J.S.; To, K.; Chang, K.C.; Takasu, O.; Osborne, D.F.; Walton, A.H.; Bricker, T.L.; Jarman, S.D., 2nd; Kreisel, D.; Krupnick, A.S.; et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 2011, 306, 2594–2605. [Google Scholar] [CrossRef] [PubMed]
- Otto, G.P.; Sossdorf, M.; Claus, R.A.; Rodel, J.; Menge, K.; Reinhart, K.; Bauer, M.; Riedemann, N.C. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit. Care 2011, 15, R183. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.C.; Cook, D.J.; Christou, N.V.; Bernard, G.R.; Sprung, C.L.; Sibbald, W.J. Multiple organ dysfunction score: A reliable descriptor of a complex clinical outcome. Crit. Care Med. 1995, 23, 1638–1652. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.C. After xigris, researchers look to new targets to combat sepsis. Nat. Med. 2012, 18, 1001. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.F.; Vachharajani, V.T.; Yoza, B.K.; McCall, C.E. Nad+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J. Biol. Chem. 2012, 287, 25758–25769. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; Scicluna, B.P.; Arts, R.J.; Gresnigt, M.S.; Lachmandas, E.; Giamarellos-Bourboulis, E.J.; Kox, M.; Manjeri, G.R.; Wagenaars, J.A.; Cremer, O.L.; et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat. Immunol. 2016, 17, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.F.; Vachharajani, V.; Millet, P.; Bharadwaj, M.S.; Molina, A.J.; McCall, C.E. Sequential actions of sirt1-relb-sirt3 coordinate nuclear-mitochondrial communication during immunometabolic adaptation to acute inflammation and sepsis. J. Biol. Chem. 2015, 290, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Sirtuins, aging, and metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Natoli, G. When sirtuins and nf-kappab collide. Cell 2009, 136, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Vachharajani, V.T.; Liu, T.; Wang, X.; Hoth, J.J.; Yoza, B.K.; McCall, C.E. Sirtuins link inflammation and metabolism. J. Immunol. Res. 2016, 2016, 8167273. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Buechler, N.L.; Martin, A.; Wells, J.; Yoza, B.; McCall, C.E.; Vachharajani, V. Sirtuin-2 regulates sepsis inflammation in ob/ob mice. PLoS ONE 2016, 11, e0160431. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.F.; Yoza, B.K.; El Gazzar, M.; Vachharajani, V.T.; McCall, C.E. Nad+-dependent sirt1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J. Biol. Chem. 2011, 286, 9856–9864. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E. The many faces of sirtuins: Coupling of nad metabolism, sirtuins and lifespan. Nat. Med. 2014, 20, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Coopersmith, C.M.; McDunn, J.E.; Ferguson, T.A. The sepsis seesaw: Tilting toward immunosuppression. Nat. Med. 2009, 15, 496–497. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Karl, I.E. The pathophysiology and treatment of sepsis. N. Engl. J. Med. 2003, 348, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Vachharajani, V.; Liu, T.; McCall, C.E. Epigenetic coordination of acute systemic inflammation: Potential therapeutic targets. Expert Rev. Clin. Immunol. 2014, 10, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Vachharajani, V.T.; Fu Liu, T.; Brown, C.M.; Wang, X.; Buechler, N.L.; Wells, J.D.; Yoza, B.K.; McCall, C.E. Sirt1 inhibition during the hypoinflammatory phenotype of sepsis enhances immunity and improves outcome. J. Leukocyte Biol. 2014, 96, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Arts, R.J.; Gresnigt, M.S.; Joosten, L.A.; Netea, M.G. Cellular metabolism of myeloid cells in sepsis. J. Leukocyte Biol. 2017, 101, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Vitko, N.P.; Spahich, N.A.; Richardson, A.R. Glycolytic dependency of high-level nitric oxide resistance and virulence in staphylococcus aureus. mBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Loftus, R.M.; Finlay, D.K. Immunometabolism: Cellular metabolism turns immune regulator. J. Biol. Chem. 2016, 291, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Maguire, T.G.; Alwine, J.C. Human cytomegalovirus activates glucose transporter 4 expression to increase glucose uptake during infection. J. Virol. 2011, 85, 1573–1580. [Google Scholar] [CrossRef] [PubMed]
- Thai, M.; Graham, N.A.; Braas, D.; Nehil, M.; Komisopoulou, E.; Kurdistani, S.K.; McCormick, F.; Graeber, T.G.; Christofk, H.R. Adenovirus e4orf1-induced myc activation promotes host cell anabolic glucose metabolism and virus replication. Cell Metab. 2014, 19, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; El Gazzar, M.; Yoza, B.K.; McCall, C.E. The nf-kappab factor relb and histone h3 lysine methyltransferase g9a directly interact to generate epigenetic silencing in endotoxin tolerance. J. Biol. Chem. 2009, 284, 27857–27865. [Google Scholar] [CrossRef] [PubMed]
- Yoza, B.K.; McCall, C.E. Facultative heterochromatin formation at the il-1 beta promoter in lps tolerance and sepsis. Cytokine 2011, 53, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Souza-Fonseca-Guimaraes, F.; Parlato, M.; Fitting, C.; Cavaillon, J.M.; Adib-Conquy, M. Nk cell tolerance to tlr agonists mediated by regulatory t cells after polymicrobial sepsis. J. Immunol. 2012, 188, 5850–5858. [Google Scholar] [CrossRef] [PubMed]
- Lotz, M.; Gutle, D.; Walther, S.; Menard, S.; Bogdan, C.; Hornef, M.W. Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells. J. Exp. Med. 2006, 203, 973–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, A.; Mannam, P. Warburg revisited: Lessons for innate immunity and sepsis. Front. Physiol. 2015, 6, 70. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Gawehn, K.; Geissler, A.W. [metabolism of leukocytes]. Z. Naturforsch. Teil B Chem. Biochem. Biophys. Biol. 1958, 13B, 515–516. [Google Scholar]
- Borregaard, N.; Herlin, T. Energy metabolism of human neutrophils during phagocytosis. J. Clin. Investig. 1982, 70, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Boxer, L.A.; Baehner, R.L.; Davis, J. The effect of 2-deoxyglucose on guinea pig polymorphonuclear leukocyte phagocytosis. J. Cell. Physiol. 1977, 91, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Michl, J.; Ohlbaum, D.J.; Silverstein, S.C. 2-deoxyglucose selectively inhibits fc and complement receptor-mediated phagocytosis in mouse peritoneal macrophages ii. Dissociation of the inhibitory effects of 2-deoxyglucose on phagocytosis and atp generation. J. Exp. Med. 1976, 144, 1484–1493. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, R.J.; Sbarra, A.J. Phagocytosis inhibition and reversal. Ii. Possible role of pyruvate as an alternative source of energy for particle uptake by guinea-pig leukocytes. Biochim. Biophys. Acta 1966, 127, 159–171. [Google Scholar] [CrossRef]
- Sbarra, A.J.; Karnovsky, M.L. The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J. Biol. Chem. 1959, 234, 1355–1362. [Google Scholar] [PubMed]
- Venet, F.; Demaret, J.; Blaise, B.J.; Rouget, C.; Girardot, T.; Idealisoa, E.; Rimmele, T.; Mallet, F.; Lepape, A.; Textoris, J.; et al. Il-7 restores t lymphocyte immunometabolic failure in septic shock patients through mtor activation. J. Immunol. 2017, 199, 1606–1615. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory cd4+ t cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [PubMed]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: Glucose transporter 1 (glut1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guthrie, L.A.; McPhail, L.C.; Henson, P.M.; Johnston, R.B., Jr. Priming of neutrophils for enhanced release of oxygen metabolites by bacterial lipopolysaccharide. Evidence for increased activity of the superoxide-producing enzyme. J. Exp. Med. 1984, 160, 1656–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, I.; Horvath, M.; Lanyi, A.; Petheo, G.L.; Geiszt, M. Reactive oxygen species-mediated bacterial killing by b lymphocytes. J. Leukocyte Biol. 2015, 97, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Aldini, R.; Marangoni, A.; Guardigli, M.; Sambri, V.; Giacani, L.; Montagnani, M.; Roda, A.; Cevenini, R. Chemiluminescence detection of reactive oxygen species in isolated kupffer cells during phagocytosis of treponema pallidum. Comp. Hepatol. 2004, 3 (Suppl. 1), S41. [Google Scholar] [CrossRef] [PubMed]
- Vernon, P.J.; Tang, D. Eat-me: Autophagy, phagocytosis, and reactive oxygen species signaling. Antioxid. Redox Signal. 2013, 18, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Spolarics, Z.; Bautista, A.P.; Spitzer, J.J. Primed pentose cycle activity supports production and elimination of superoxide anion in kupffer cells from rats treated with endotoxin in vivo. Biochim. Biophys. Acta 1993, 1179, 134–140. [Google Scholar] [CrossRef]
- Azevedo, E.P.; Rochael, N.C.; Guimaraes-Costa, A.B.; de Souza-Vieira, T.S.; Ganilho, J.; Saraiva, E.M.; Palhano, F.L.; Foguel, D. A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril- and phorbol 12-myristate 13-acetate-induced neutrophil extracellular trap (net) formation. J. Biol. Chem. 2015, 290, 22174–22183. [Google Scholar] [CrossRef] [PubMed]
- Rostami-Far, Z.; Ghadiri, K.; Rostami-Far, M.; Shaveisi-Zadeh, F.; Amiri, A.; Rahimian Zarif, B. Glucose-6-phosphate dehydrogenase deficiency (g6pd) as a risk factor of male neonatal sepsis. J. Med. Life 2016, 9, 34–38. [Google Scholar] [PubMed]
- Wilmanski, J.; Villanueva, E.; Deitch, E.A.; Spolarics, Z. Glucose-6-phosphate dehydrogenase deficiency and the inflammatory response to endotoxin and polymicrobial sepsis. Crit. Care Med. 2007, 35, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Spolarics, Z.; Siddiqi, M.; Siegel, J.H.; Garcia, Z.C.; Stein, D.S.; Ong, H.; Livingston, D.H.; Denny, T.; Deitch, E.A. Increased incidence of sepsis and altered monocyte functions in severely injured type A-glucose-6-phosphate dehydrogenase-deficient African American trauma patients. Crit. Care Med. 2001, 29, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.R.; DeChatelet, L.R.; McCall, C.E.; LaVia, M.F.; Spurr, C.L.; Baehner, R.L. Leucocyte g.-6-p.D. Deficiency. Lancet 1970, 1, 110. [Google Scholar] [CrossRef]
- Girardot, T.; Rimmele, T.; Venet, F.; Monneret, G. Apoptosis-induced lymphopenia in sepsis and other severe injuries. Apoptosis 2017, 22, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Tinsley, K.W.; Swanson, P.E.; Grayson, M.H.; Osborne, D.F.; Wagner, T.H.; Cobb, J.P.; Coopersmith, C.; Karl, I.E. Depletion of dendritic cells, but not macrophages, in patients with sepsis. J. Immunol. 2002, 168, 2493–2500. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. T cells and their immunometabolism: A novel way to understanding sepsis immunopathogenesis and future therapeutics. Eur. J. Cell Biol. 2018, 97, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Martins, P.S.; Kallas, E.G.; Neto, M.C.; Dalboni, M.A.; Blecher, S.; Salomao, R. Upregulation of reactive oxygen species generation and phagocytosis, and increased apoptosis in human neutrophils during severe sepsis and septic shock. Shock 2003, 20, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.F.; Sun, L.; Gao, H.; Shi, K.X.; Rittirsch, D.; Sarma, V.J.; Zetoune, F.S.; Ward, P.A. In vivo regulation of neutrophil apoptosis by c5a during sepsis. J. Leukocyte Biol. 2006, 80, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Taneja, R.; Parodo, J.; Jia, S.H.; Kapus, A.; Rotstein, O.D.; Marshall, J.C. Delayed neutrophil apoptosis in sepsis is associated with maintenance of mitochondrial transmembrane potential and reduced caspase-9 activity. Crit. Care Med. 2004, 32, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Singer, M. Cellular dysfunction in sepsis. Clin. Chest Med. 2008, 29, 655–660, viii-ix. [Google Scholar] [CrossRef] [PubMed]
- Anantha, R.V.; Mazzuca, D.M.; Xu, S.X.; Porcelli, S.A.; Fraser, D.D.; Martin, C.M.; Welch, I.; Mele, T.; Haeryfar, S.M.; McCormick, J.K. T helper type 2-polarized invariant natural killer t cells reduce disease severity in acute intra-abdominal sepsis. Clin. Exp. Immunol. 2014, 178, 292–309. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Brewington, R.; Chatterji, M.; Zoubine, M.; Kinasewitz, G.T.; Peer, G.T.; Chang, A.C.; Taylor, F.B., Jr.; Shnyra, A. Infection-induced modulation of m1 and m2 phenotypes in circulating monocytes: Role in immune monitoring and early prognosis of sepsis. Shock 2004, 22, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Suzuki, Y.; Inokuchi, S.; Inoue, S. Sepsis induces incomplete m2 phenotype polarization in peritoneal exudate cells in mice. J. Intensive Care 2016, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- West, M.A.; Heagy, W. Endotoxin tolerance: A review. Crit. Care Med. 2002, 30, S64–S73. [Google Scholar] [CrossRef] [PubMed]
- Yoza, B.; LaRue, K.; McCall, C. Molecular mechanisms responsible for endotoxin tolerance. Prog. Clin. Biol. Res. 1998, 397, 209–215. [Google Scholar] [PubMed]
- Fukumoto, K.; Pierro, A.; Zammit, V.A.; Spitz, L.; Eaton, S. Tyrosine nitration of carnitine palmitoyl transferase i during endotoxaemia in suckling rats. Biochim. Biophys. Acta 2004, 1683, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and pgc-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.J.; Tsalik, E.L.; van Velkinburgh, J.C.; Glickman, S.W.; Rice, B.J.; Wang, C.; Chen, B.; Carin, L.; Suarez, A.; Mohney, R.P.; et al. An integrated clinico-metabolomic model improves prediction of death in sepsis. Sci. Transl. Med. 2013, 5, 195ra195. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.J.; Tipper, J.L.; Bruse, S.; Baron, R.M.; Tsalik, E.L.; Huntley, J.; Rogers, A.J.; Jaramillo, R.J.; O’Donnell, D.; Mega, W.M.; et al. Integrative “omic” analysis of experimental bacteremia identifies a metabolic signature that distinguishes human sepsis from systemic inflammatory response syndromes. Am. J. Respir. Crit. Care Med. 2014, 190, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Voll, R.E.; Herrmann, M.; Roth, E.A.; Stach, C.; Kalden, J.R.; Girkontaite, I. Immunosuppressive effects of apoptotic cells. Nature 1997, 390, 350–351. [Google Scholar] [CrossRef] [PubMed]
- Singer, M. Mitochondrial function in sepsis: Acute phase versus multiple organ failure. Crit. Care Med. 2007, 35, S441–S448. [Google Scholar] [CrossRef] [PubMed]
- Singer, M. Critical illness and flat batteries. Crit. Care 2017, 21, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, M.; De Santis, V.; Vitale, D.; Jeffcoate, W. Multiorgan failure is an adaptive, endocrine-mediated, metabolic response to overwhelming systemic inflammation. Lancet 2004, 364, 545–548. [Google Scholar] [CrossRef]
- Li, T.; Zhang, J.; Feng, J.; Li, Q.; Wu, L.; Ye, Q.; Sun, J.; Lin, Y.; Zhang, M.; Huang, R.; et al. Resveratrol reduces acute lung injury in a lpsinduced sepsis mouse model via activation of sirt1. Mol. Med. Rep. 2013, 7, 1889–1895. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Chen, Y.; Liao, L.; Wu, W. Resveratrol protects huvecs from oxidized-ldl induced oxidative damage by autophagy upregulation via the ampk/sirt1 pathway. Cardiovasc. Drugs Ther. 2013, 27, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Kim, D.W.; Park, J.H.; Kim, S.J.; Lee, C.H.; Yong, J.I.; Ryu, E.J.; Cho, S.B.; Yeo, H.J.; Hyeon, J.; et al. Pep-1-sirt2 inhibits inflammatory response and oxidative stress-induced cell death via expression of antioxidant enzymes in murine macrophages. Free Radic. Biol. Med. 2013, 63, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Feldman, J.L.; Dittenhafer-Reed, K.E.; Kudo, N.; Thelen, J.N.; Ito, A.; Yoshida, M.; Denu, J.M. Kinetic and structural basis for acyl-group selectivity and nad(+) dependence in sirtuin-catalyzed deacylation. Biochemistry 2015, 54, 3037–3050. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The nad(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Imai, S. The nad world: A new systemic regulatory network for metabolism and aging—Sirt1, systemic nad biosynthesis, and their importance. Cell Biochem. Biophys. 2009, 53, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Buechler, N.L.; Yoza, B.K.; McCall, C.E.; Vachharajani, V.T. Resveratrol attenuates microvascular inflammation in sepsis via sirt-1-induced modulation of adhesion molecules in ob/ob mice. Obesity 2015, 23, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Lo Sasso, G.; Menzies, K.J.; Mottis, A.; Piersigilli, A.; Perino, A.; Yamamoto, H.; Schoonjans, K.; Auwerx, J. Sirt2 deficiency modulates macrophage polarization and susceptibility to experimental colitis. PLoS ONE 2014, 9, e103573. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Zhang, J.; Ling, Y.; McCall, C.E.; Liu, T.F. Mitochondrial sirtuin 4 resolves immune tolerance in monocytes by rebalancing glycolysis and glucose oxidation homeostasis. Front. Immunol. 2018, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Huang, C.; Huang, Y.; Hong, L.; Wang, H.; Zhou, Z.; Qiu, Y. Sirt4 suppresses inflammatory responses in human umbilical vein endothelial cells. Cardiovasc. Toxicol. 2015, 15, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; McVey, M.; Guarente, L. The sir2/3/4 complex and sir2 alone promote longevity in saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Sirtuins in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Li, X. Sirt1 and energy metabolism. Acta Biochim. Biophys. Sin. 2013, 45, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of nf-kappab-dependent transcription and cell survival by the sirt1 deacetylase. Embo J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Bordone, L.; Guarente, L. Calorie restriction, sirt1 and metabolism: Understanding longevity. Nat. Rev. Mol. Cell Biol. 2005, 6, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Jiang, L.Q.; Deshmukh, A.S.; Mataki, C.; Coste, A.; Lagouge, M.; Zierath, J.R.; Auwerx, J. Interdependence of ampk and sirt1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010, 11, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Gomes, P.; Outeiro, T.F.; Cavadas, C. Emerging role of sirtuin 2 in the regulation of mammalian metabolism. Trends Pharmacol. Sci. 2015, 36, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Nguyen, M.; Qin, F.X.; Tong, Q. Sirt2 deacetylates foxo3a in response to oxidative stress and caloric restriction. Aging Cell 2007, 6, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, M.M.; Tomkinson, E.M.; Nobles, J.; Wizeman, J.W.; Amore, A.M.; Quinti, L.; Chopra, V.; Hersch, S.M.; Kazantsev, A.G. The sirtuin 2 microtubule deacetylase is an abundant neuronal protein that accumulates in the aging cns. Hum. Mol. Genet. 2011, 20, 3986–3996. [Google Scholar] [CrossRef] [PubMed]
- Jing, E.; Gesta, S.; Kahn, C.R. Sirt2 regulates adipocyte differentiation through foxo1 acetylation/deacetylation. Cell Metab. 2007, 6, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, J.; Danzer, C.; Simka, T.; Ukropec, J.; Walter, K.M.; Kumpf, S.; Mirtschink, P.; Ukropcova, B.; Gasperikova, D.; Pedrazzini, T.; et al. Dietary obesity-associated hif1alpha activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the sirt2-nad+ system. Genes Dev. 2012, 26, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Hiratsuka, M.; Osaki, M.; Yamada, H.; Kishimoto, I.; Yamaguchi, S.; Nakano, S.; Katoh, M.; Ito, H.; Oshimura, M. Sirt2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene 2007, 26, 945–957. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Verdin, E. Interphase nucleo-cytoplasmic shuttling and localization of sirt2 during mitosis. PLoS ONE 2007, 2, e784. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Vassilopoulos, A.; Wang, R.H.; Lahusen, T.; Xiao, Z.; Xu, X.; Li, C.; Veenstra, T.D.; Li, B.; Yu, H.; et al. Sirt2 maintains genome integrity and suppresses tumorigenesis through regulating apc/c activity. Cancer Cell 2011, 20, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Lain, S.; Hollick, J.J.; Campbell, J.; Staples, O.D.; Higgins, M.; Aoubala, M.; McCarthy, A.; Appleyard, V.; Murray, K.E.; Baker, L.; et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 2008, 13, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Chopra, V.; Quinti, L.; Kim, J.; Vollor, L.; Narayanan, K.L.; Edgerly, C.; Cipicchio, P.M.; Lauver, M.A.; Choi, S.H.; Silverman, R.B.; et al. The sirtuin 2 inhibitor ak-7 is neuroprotective in huntington’s disease mouse models. Cell Rep. 2012, 2, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Bobrowska, A.; Donmez, G.; Weiss, A.; Guarente, L.; Bates, G. Sirt2 ablation has no effect on tubulin acetylation in brain, cholesterol biosynthesis or the progression of huntington’s disease phenotypes in vivo. PLoS ONE 2012, 7, e34805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.H.; Ozden, O.; Liu, G.; Song, H.Y.; Zhu, Y.; Yan, Y.; Zou, X.; Kang, H.J.; Jiang, H.; Principe, D.R.; et al. Sirt2-mediated deacetylation and tetramerization of pyruvate kinase directs glycolysis and tumor growth. Cancer Res. 2016, 76, 3802–3812. [Google Scholar] [CrossRef] [PubMed]
- Shirai, T.; Nazarewicz, R.R.; Wallis, B.B.; Yanes, R.E.; Watanabe, R.; Hilhorst, M.; Tian, L.; Harrison, D.G.; Giacomini, J.C.; Assimes, T.L.; et al. The glycolytic enzyme pkm2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J. Exp. Med. 2016, 213, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Alves-Filho, J.C.; Palsson-McDermott, E.M. Pyruvate kinase m2: A potential target for regulating inflammation. Front. Immunol. 2016, 7, 145. [Google Scholar] [CrossRef] [PubMed]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.; et al. Pyruvate kinase m2 regulates hif-1alpha activity and il-1beta induction and is a critical determinant of the warburg effect in lps-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Buechler, N.; Long, D.L.; Furdui, C.M.; Yoza, B.K.; McCall, C.E.; Vachharajani, V. Cysteine thiol oxidation on SIRT2 regulates inflammation in obese mice with sepsis. Inflammation, in press.
- Jiang, W.; Wang, S.; Xiao, M.; Lin, Y.; Zhou, L.; Lei, Q.; Xiong, Y.; Guan, K.L.; Zhao, S. Acetylation regulates gluconeogenesis by promoting pepck1 degradation via recruiting the ubr5 ubiquitin ligase. Mol. Cell 2011, 43, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.R.; Tang, F.J.; Zhang, X.; Wang, H. Suppression of nf-kappab activation by pdlim2 restrains hepatic lipogenesis and inflammation in high fat diet induced mice. Biochem. Biophys. Res. Commun. 2018, 503, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Srere, P.A. The citrate cleavage enzyme. I. Distribution and purification. J. Biol. Chem. 1959, 234, 2544–2547. [Google Scholar] [PubMed]
- Lin, R.; Tao, R.; Gao, X.; Li, T.; Zhou, X.; Guan, K.L.; Xiong, Y.; Lei, Q.Y. Acetylation stabilizes atp-citrate lyase to promote lipid biosynthesis and tumor growth. Mol. Cell 2013, 51, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Sun, B.; Jiang, C.; Hong, H.; Zheng, Y. Sirt2 suppresses inflammatory responses in collagen-induced arthritis. Biochem. Biophys. Res. Commun. 2013, 441, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Jung, Y.J.; Kim, D.; Nguyen-Thanh, T.; Kang, K.P.; Lee, S.; Park, S.K.; Kim, W. Sirt2 ameliorates lipopolysaccharide-induced inflammation in macrophages. Biochem. Biophys. Res. Commun. 2014, 450, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Buechler, N.; Wang, X.; Yoza, B.K.; McCall, C.E.; Vachharajani, V. Sirtuin 2 regulates microvascular inflammation during sepsis. J. Immunol. Res. 2017, 2017, 2648946. [Google Scholar] [CrossRef] [PubMed]
- Ciarlo, E.; Heinonen, T.; Theroude, C.; Herderschee, J.; Mombelli, M.; Lugrin, J.; Pfefferle, M.; Tyrrell, B.; Lensch, S.; Acha-Orbea, H.; et al. Sirtuin 2 deficiency increases bacterial phagocytosis by macrophages and protects from chronic staphylococcal infection. Front. Immunol. 2017, 8, 1037. [Google Scholar] [CrossRef] [PubMed]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian sir2 homolog sirt3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Y.; Zhang, L.; Sui, M.X.; Zhu, Y.H.; Zeng, L. Protective effects of sirtuin 3 in a murine model of sepsis-induced acute kidney injury. Sci. Rep. 2016, 6, 33201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurd, B.J.; Holloway, G.P.; Yoshida, Y.; Bonen, A. In mammalian muscle, sirt3 is present in mitochondria and not in the nucleus; and sirt3 is upregulated by chronic muscle contraction in an adenosine monophosphate-activated protein kinase-independent manner. Metab. Clin. Exp. 2012, 61, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Iwahara, T.; Bonasio, R.; Narendra, V.; Reinberg, D. Sirt3 functions in the nucleus in the control of stress-related gene expression. Mol. Cell. Biol. 2012, 32, 5022–5034. [Google Scholar] [CrossRef] [PubMed]
- Bellizzi, D.; Rose, G.; Cavalcante, P.; Covello, G.; Dato, S.; De Rango, F.; Greco, V.; Maggiolini, M.; Feraco, E.; Mari, V.; et al. A novel vntr enhancer within the sirt3 gene, a human homologue of sir2, is associated with survival at oldest ages. Genomics 2005, 85, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Gao, B.; Li, N.; Wang, J.; Qiu, C.; Zhang, G.; Liu, M.; Zhang, R.; Li, C.; Ji, G.; et al. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: Role of foxo3a-parkin-mediated mitophagy. Biochim. Biophys. Acta 2017, 1863, 1973–1983. [Google Scholar] [CrossRef] [PubMed]
- Lantier, L.; Williams, A.S.; Williams, I.M.; Yang, K.K.; Bracy, D.P.; Goelzer, M.; James, F.D.; Gius, D.; Wasserman, D.H. Sirt3 is crucial for maintaining skeletal muscle insulin action and protects against severe insulin resistance in high-fat-fed mice. Diabetes 2015, 64, 3081–3092. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Ciarlo, E.; Heinonen, T.; Lugrin, J.; Acha-Orbea, H.; Le Roy, D.; Auwerx, J.; Roger, T. Sirtuin 3 deficiency does not alter host defenses against bacterial and fungal infections. Sci. Rep. 2017, 7, 3853. [Google Scholar] [CrossRef] [PubMed]
- Sosulski, M.L.; Gongora, R.; Feghali-Bostwick, C.; Lasky, J.A.; Sanchez, C.G. Sirtuin 3 deregulation promotes pulmonary fibrosis. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2017, 72, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Wang, F.; Stieren, E.; Tong, Q. Sirt3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 2005, 280, 13560–13567. [Google Scholar] [CrossRef] [PubMed]
- Jing, E.; O’Neill, B.T.; Rardin, M.J.; Kleinridders, A.; Ilkeyeva, O.R.; Ussar, S.; Bain, J.R.; Lee, K.Y.; Verdin, E.M.; Newgard, C.B.; et al. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 2013, 62, 3404–3417. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Ma, H.; Zhang, X.; Tu, F.; Wang, X.; Ha, T.; Fan, M.; Liu, L.; Xu, J.; Yu, K.; et al. Enhanced glycolytic metabolism contributes to cardiac dysfunction in polymicrobial sepsis. J. Infect. Dis. 2017, 215, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; He, X.; Tuo, Q.H.; Liao, D.F.; Zhang, G.Q.; Chen, J.X. Lps causes pericyte loss and microvascular dysfunction via disruption of sirt3/angiopoietins/tie-2 and hif-2alpha/notch3 pathways. Sci. Rep. 2016, 6, 20931. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. Sirt4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, J.J.; Guo, L.Y.; Li, P.; Zhao, Z.; Zhou, H.; Di, L.J. Molecular link between glucose and glutamine consumption in cancer cells mediated by ctbp and sirt4. Oncogenesis 2018, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Mathias, R.A.; Greco, T.M.; Oberstein, A.; Budayeva, H.G.; Chakrabarti, R.; Rowland, E.A.; Kang, Y.; Shenk, T.; Cristea, I.M. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 2014, 159, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Laurent, G.; German, N.J.; Saha, A.K.; de Boer, V.C.; Davies, M.; Koves, T.R.; Dephoure, N.; Fischer, F.; Boanca, G.; Vaitheesvaran, B.; et al. Sirt4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl coa decarboxylase. Mol. Cell 2013, 50, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Michishita, E.; McCord, R.A.; Boxer, L.D.; Barber, M.F.; Hong, T.; Gozani, O.; Chua, K.F. Cell cycle-dependent deacetylation of telomeric histone h3 lysine k56 by human sirt6. Cell Cycle 2009, 8, 2664–2666. [Google Scholar] [CrossRef] [PubMed]
- Michishita, E.; McCord, R.A.; Berber, E.; Kioi, M.; Padilla-Nash, H.; Damian, M.; Cheung, P.; Kusumoto, R.; Kawahara, T.L.; Barrett, J.C.; et al. Sirt6 is a histone h3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 2008, 452, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human sirt proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The histone deacetylase sirt6 regulates glucose homeostasis via hif1alpha. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Shakespear, M.R.; Iyer, A.; Cheng, C.Y.; Das Gupta, K.; Singhal, A.; Fairlie, D.P.; Sweet, M.J. Lysine deacetylases and regulated glycolysis in macrophages. Trends Immunol. 2018, 39, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.L.; Michishita, E.; Adler, A.S.; Damian, M.; Berber, E.; Lin, M.; McCord, R.A.; Ongaigui, K.C.; Boxer, L.D.; Chang, H.Y.; et al. Sirt6 links histone h3 lysine 9 deacetylation to nf-kappab-dependent gene expression and organismal life span. Cell 2009, 136, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Ka, S.O.; Lee, S.M.; Lee, S.I.; Park, J.W.; Park, B.H. Overexpression of sirtuin 6 suppresses inflammatory responses and bone destruction in mice with collagen-induced arthritis. Arth. Rheumatism 2013, 65, 1776–1785. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, S.; Chang, K.; Sakai, M.; Shimizu, N.; Yamada, M.; Tanaka, T.; Nakazawa, H.; Ichinose, F.; Yamada, Y.; Ishigami, A.; et al. Inflammatory stimuli induce inhibitory s-nitrosylation of the deacetylase sirt1 to increase acetylation and activation of p53 and p65. Sci. Signal. 2014, 7, ra106. [Google Scholar] [CrossRef] [PubMed]
- Long, D.; Wu, H.; Tsang, A.W.; Poole, L.B.; Yoza, B.K.; Wang, X.; Vachharajani, V.; Furdui, C.M.; McCall, C.E. The oxidative state of cysteine thiol 144 regulates the sirt6 glucose homeostat. Sci. Rep. 2017, 7, 11005. [Google Scholar] [CrossRef] [PubMed]
- Bringman-Rodenbarger, L.R.; Guo, A.H.; Lyssiotis, C.A.; Lombard, D.B. Emerging roles for sirt5 in metabolism and cancer. Antioxid. Redox Signal. 2018, 28, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a nad-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, Y.; Rardin, M.J.; Carrico, C.; He, W.; Sahu, A.K.; Gut, P.; Najjar, R.; Fitch, M.; Hellerstein, M.; Gibson, B.W.; et al. Sirt5 regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target. Mol. Cell 2015, 59, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Peng, C.; Anderson, K.A.; Chhoy, P.; Xie, Z.; Dai, L.; Park, J.; Chen, Y.; Huang, H.; Zhang, Y.; et al. Lysine glutarylation is a protein posttranslational modification regulated by sirt5. Cell Metab. 2014, 19, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Qin, K.; Han, C.; Zhang, H.; Li, T.; Li, N.; Cao, X. Nad+ dependent deacetylase sirtuin 5 rescues the innate inflammatory response of endotoxin tolerant macrophages by promoting acetylation of p65. J. Autoimmun. 2017, 81, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Zou, R.; Shi, W.; Tao, J.; Li, H.; Lin, X.; Yang, S.; Hua, P. Sirt5 and post-translational protein modifications: A potential therapeutic target for myocardial ischemia-reperfusion injury with regard to mitochondrial dynamics and oxidative metabolism. Eur. J. Pharmacol. 2018, 818, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Canestro, C.; Merlini, M.; Bonetti, N.R.; Liberale, L.; Wust, P.; Briand-Schumacher, S.; Klohs, J.; Costantino, S.; Miranda, M.; Schoedon-Geiser, G.; et al. Sirtuin 5 as a novel target to blunt blood-brain barrier damage induced by cerebral ischemia/reperfusion injury. Int. J. Cardiol. 2018, 260, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.S.; Liang, J.J.; Wang, Y.; Zhao, X.J.; Xu, L.; Xu, Y.Y.; Zou, Q.C.; Zhang, J.M.; Tu, C.E.; Cui, Y.G.; et al. Stat3 undergoes acetylation-dependent mitochondrial translocation to regulate pyruvate metabolism. Sci. Rep. 2016, 6, 39517. [Google Scholar] [CrossRef] [PubMed]
- Buler, M.; Aatsinki, S.M.; Izzi, V.; Uusimaa, J.; Hakkola, J. Sirt5 is under the control of pgc-1alpha and ampk and is involved in regulation of mitochondrial energy metabolism. FASEB J. 2014, 28, 3225–3237. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shi, L.; Yang, S.; Yan, R.; Zhang, D.; Yang, J.; He, L.; Li, W.; Yi, X.; Sun, L.; et al. Sirt7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat. Commun. 2016, 7, 12235. [Google Scholar] [CrossRef] [PubMed]
- Blank, M.F.; Grummt, I. The seven faces of sirt7. Transcription 2017, 8, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, B.N.; Thackray, J.K.; Serrano, L. Sirtuins and DNA damage repair: Sirt7 comes to play. Nucleus 2017, 8, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, B.N.; Thackray, J.K.; Simonet, N.G.; Kane-Goldsmith, N.; Martinez-Redondo, P.; Nguyen, T.; Bunting, S.; Vaquero, A.; Tischfield, J.A.; Serrano, L. Sirt7 promotes genome integrity and modulates non-homologous end joining DNA repair. Embo J. 2016, 35, 1488–1503. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; He, M.; Liu, Y.; Paredes, S.; Villanova, L.; Brown, K.; Qiu, X.; Nabavi, N.; Mohrin, M.; Wojnoonski, K.; et al. Sirt7 represses myc activity to suppress er stress and prevent fatty liver disease. Cell Rep. 2013, 5, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Simonet, N.G.; Vaquero, A. Raising the list of sirt7 targets to a new level. Proteomics 2017, 17, 1700137. [Google Scholar] [CrossRef] [PubMed]
- Holthoff, J.H.; Wang, Z.; Seely, K.A.; Gokden, N.; Mayeux, P.R. Resveratrol improves renal microcirculation, protects the tubular epithelium, and prolongs survival in a mouse model of sepsis-induced acute kidney injury. Kidney Int. 2012, 81, 370–378. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Sirtuin | Immune Cell Type | Mechanism of Action |
|---|---|---|
| SIRT1 | Monocytes | Direct NFκB p65 deacetylation and the HIF-1α and PGC-1α pathway [14] |
| Macrophages | Phenotypic shift from activator to suppressor cells [19] | |
| Lymphocytes | Suppression of pro-inflammatory cytokine expression [71] | |
| Endothelial cells | Direct NFκB p65 deacetylation: attenuation of pro-inflammatory adhesion molecule expression [79] | |
| SIRT2 | Macrophages | Direct NFκB p65 deacetylation and polarization to suppressor phenotype: STAT6/GATA3 signaling cascade [80]. Direct NFκB p65 deacetylation: attenuation of pro-inflammatory cytokine expression [13] |
| SIRT3 | Monocytes | Mitochondrial biogenesis and increased oxidative phosphorylation to sustain hypo-inflammation [9] |
| SIRT4 | Monocytes | Resolution of hypo-inflammatory phase; restoration of glucose oxidation via PDC activity and SIRT1 repression [81] |
| Endothelial cells | Attenuation of pro-inflammatory cytokine and adhesion molecule expression via blocking of nuclear translocation of NFκB p65 [82] | |
| SIRT6 | Monocytes | Decreases glucose oxidation and glycolysis during the hypo-inflammatory phase via epigenetic repression of HIF-1α [7] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Buechler, N.L.; Woodruff, A.G.; Long, D.L.; Zabalawi, M.; Yoza, B.K.; McCall, C.E.; Vachharajani, V. Sirtuins and Immuno-Metabolism of Sepsis. Int. J. Mol. Sci. 2018, 19, 2738. https://doi.org/10.3390/ijms19092738
Wang X, Buechler NL, Woodruff AG, Long DL, Zabalawi M, Yoza BK, McCall CE, Vachharajani V. Sirtuins and Immuno-Metabolism of Sepsis. International Journal of Molecular Sciences. 2018; 19(9):2738. https://doi.org/10.3390/ijms19092738
Chicago/Turabian StyleWang, Xianfeng, Nancy L. Buechler, Alan G. Woodruff, David L. Long, Manal Zabalawi, Barbara K. Yoza, Charles E. McCall, and Vidula Vachharajani. 2018. "Sirtuins and Immuno-Metabolism of Sepsis" International Journal of Molecular Sciences 19, no. 9: 2738. https://doi.org/10.3390/ijms19092738
APA StyleWang, X., Buechler, N. L., Woodruff, A. G., Long, D. L., Zabalawi, M., Yoza, B. K., McCall, C. E., & Vachharajani, V. (2018). Sirtuins and Immuno-Metabolism of Sepsis. International Journal of Molecular Sciences, 19(9), 2738. https://doi.org/10.3390/ijms19092738