Activated Rho GTPases in Cancer—The Beginning of a New Paradigm

Abstract
:1. Introduction
2. Identification of the Classical Rho GTPases
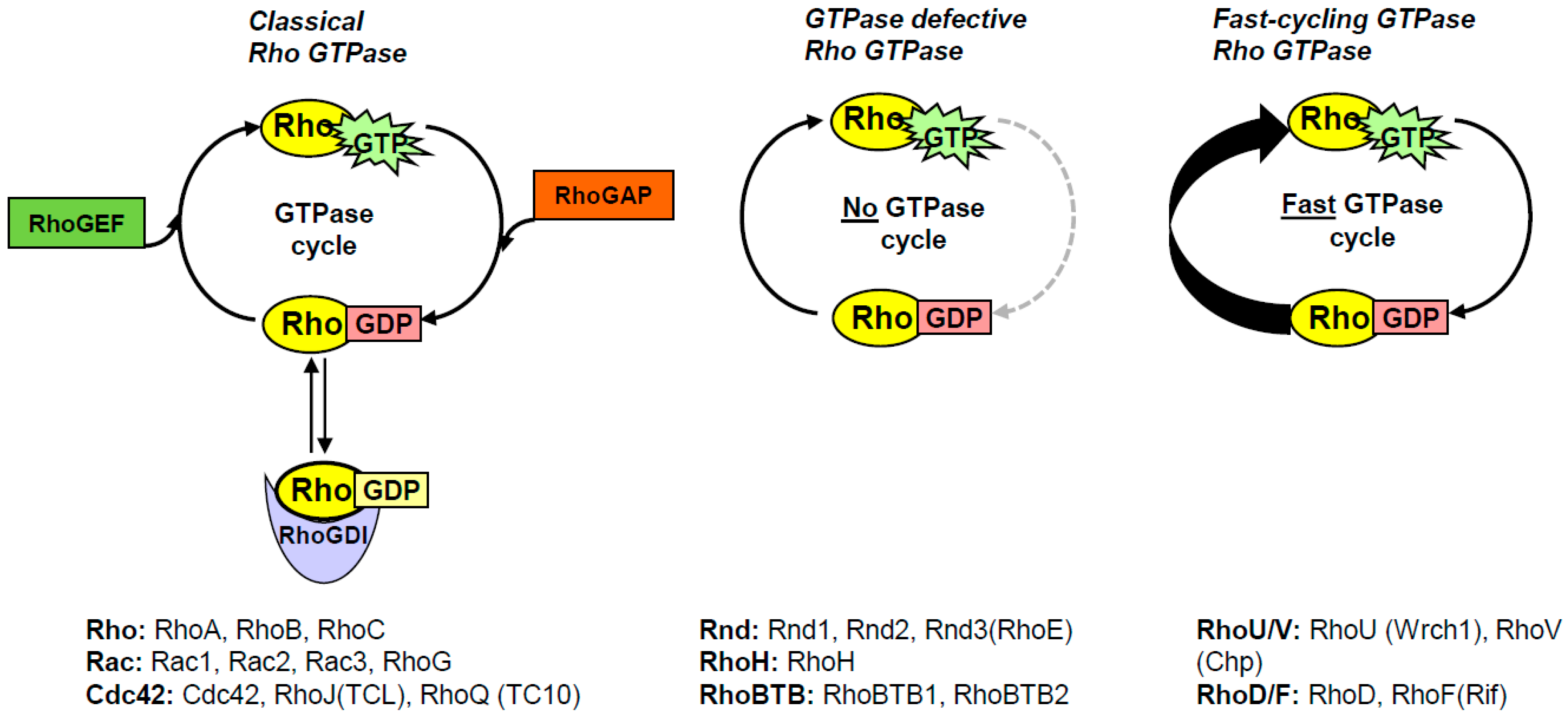
3. Some Basic Facts about Rho GTPases
4. Why Were the Rho GTPases Not Considered to Be Oncogenes?
5. An Oncogenic Splice Variant of Rac1
6. The Concept of Fast-Cycling Small GTPases
7. Cancer-Associated Point Mutations in Rac1
8. Cancer-Associated Point Mutations in Other Rho GTPases
9. Summary
Funding
Conflicts of Interest
References
- Wennerberg, K.; Der, C.J. Rho-family GTPases: It’s not only Rac and Rho (and I like it). J. Cell Sci. 2004, 117 Pt 8, 1301–1312. [Google Scholar] [CrossRef]
- Aspenström, P. Fast-cycling Rho GTPases. Small GTPases 2018, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bustelo, X.R. RHO GTPases in cancer: Known facts, open questions, and therapeutic challenges. Biochem. Soc. Trans. 2018, 46, 741–760. [Google Scholar] [CrossRef] [PubMed]
- Madaule, P.; Axel, R. A novel Ras-related gene family. Cell 1985, 41, 31–40. [Google Scholar] [CrossRef]
- Yeramian, P.; Chardin, P.; Madaule, P.; Tavitian, A. Nucleotide sequence of human rho cDNA clone 12. Nucl. Acids Res. 1987, 15, 1869. [Google Scholar] [CrossRef]
- Chardin, P.; Madaule, P.; Tavitian, A. Coding sequence of human rho cDNAs clone 6 and clone 9. Nucl. Acids Res. 1988, 16, 2717. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.; Brown, M.L.; Fraser, E.D.; Northup, J.K. Purification of the major GTP-binding proteins from human placental membranes. J. Biol. Chem. 1986, 261, 7052–7059. [Google Scholar] [PubMed]
- Didsbury, J.; Weber, R.F.; Bokoch, G.M.; Evans, T.; Snyderman, R. Rac, A novel ras-related family of proteins that are botulinum toxin substrates. J. Biol. Chem. 1989, 264, 16378–16382. [Google Scholar]
- Munemitsu, S.; Innis, M.A.; Clark, R.; McCormick, F.; Ullrich, A.; Polakis, P. Molecular cloning and expression of a G25K cDNA, the human homolog of the yeast cell cyclegene CDC42. Mol. Cell. Biol. 1990, 10, 5977–5982. [Google Scholar] [CrossRef]
- Shinjo, K.; Koland, J.G.; Hart, M.J.; Narasimhan, V.; Johnson, D.I.; Evans, T.; Cerione, R.A. Molecular cloning of the gene for the human placental GTP-binding protein Gp (G25K): Identification of this GTP-binding protein as the human homolog of the yeast cell-division-cycle protein CDC42. Proc. Natl. Acad. Sci. USA 1990, 87, 9853–9857. [Google Scholar] [CrossRef]
- Chardin, P.; Boquet, P.; Madaule, P.; Popoff, M.R.; Rubin, E.J.; Gill, D.M. The mammalian G protein rhoC is ADP-ribosylated by Clostridium botulinum exoenzyme C3 and affects actin microfilaments in Vero cells. EMBO J. 1989, 8, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Braun, U.; Habermann, B.; Just, I.; Aktories, K.; Vandekerckhove, J. Purification of the 22 kDa protein substrate of botulinum ADP-ribosyltransferase C3 from porcine brain cytosol and its characterization as a GTP-binding protein highly homologous to the rho gene product. FEBS Lett. 1989, 243, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Nemoto, Y.; Namba, T.; Teru-uchi, T.; Ushikubi, F.; Morii, N.; Narumiya, S. A rho gene product in human blood platelets. I. Identification of the platelet substrate for botulinum C3 ADP-ribosyltransferase as rhoA protein. J. Biol. Chem. 1992, 267, 20916–20920. [Google Scholar] [PubMed]
- Aktories, K. Rho-modifying bacterial protein toxins. Pathog. Dis. 2015, 73, ftv091. [Google Scholar] [CrossRef]
- Paterson, H.F.; Self, A.J.; Garrett, M.D.; Just, I.; Aktories, K.; Hall, A. Microinjection of recombinant p21rho induces rapid changes in cell morphology. J. Cell Biol. 1990, 111, 1001–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridley, A.J.; Hall, A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef]
- Ridley, A.J.; Paterson, H.F.; Johnston, C.L.; Diekmann, D.; Hall, A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 1992, 70, 401–410. [Google Scholar] [CrossRef]
- Nobes, C.D.; Hall, A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef]
- Hall, A. Rho family GTPases. Biochem. Soc. Trans. 2012, 40, 1378–1382. [Google Scholar] [CrossRef] [Green Version]
- Colicelli, J. Human RAS superfamily proteins and related GTPases. Sci. STKE 2004, 2004, re13. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef]
- Tcherkezian, J.; Lamarche-Vane, N. Current knowledge of the large RhoGAP family of proteins. Biol. Cell 2007, 99, 67–86. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.R.; Rossman, K.L.; Der, C.J. Rho guanine nucleotide exchange factors: Regulators of Rho GTPase activity in development and disease. Oncogene 2014, 33, 4021–4035. [Google Scholar] [CrossRef]
- Feig, L.A. Tools of the trade: Use of dominant-inhibitory mutants of Ras-family GTPases. Nat. Cell Biol. 1999, 1, E25–E27. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Mitin, N.; Keller, P.J.; Chenette, E.J.; Madigan, J.P.; Currin, R.O.; Cox, A.D.; Wilson, O.; Kirschmeier, P.; Der, C.J. Rho family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J. Biol. Chem. 2008, 283, 25150–25163. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Shao, S.; Aziz, A.U.R.; Zhang, B.; Wang, H.; Liu, B. Role of Rho-specific guanine nucleotide dissociation inhibitor α regulation in cell migration. Acta Histochem. 2017, 119, 183–189. [Google Scholar] [CrossRef]
- Foster, R.; Hu, K.Q.; Lu, Y.; Nolan, K.M.; Thissen, J.; Settleman, J. Identification of a novel human Rho protein with unusual properties: GTPase deficiency and in-vivo farnesylation. Mol. Cell. Biol. 1996, 16, 2689–2699. [Google Scholar] [CrossRef] [PubMed]
- Fiegen, D.; Blumenstein, L.; Stege, P.; Vetter, I.R.; Ahmadian, M.R. Crystal structure of Rnd3/RhoE: Functional implications. FEBS Lett. 2002, 525, 100–104. [Google Scholar] [CrossRef]
- Goh, L.L.; Manser, E. The RhoA GEF Syx is a target of Rnd3 and regulated via a Raf1-like ubiquitin-related domain. PLoS ONE 2010, 5, e12409. [Google Scholar] [CrossRef]
- Wennerberg, K.; Forget, M.A.; Ellerbroek, S.M.; Arthur, W.T.; Burridge, K.; Settleman, J.; Der, C.J.; Hansen, S.H. Rnd proteins function as RhoA antagonists by activating p190 RhoGAP. Curr. Biol. 2003, 13, 1106–1115. [Google Scholar] [CrossRef]
- Nobes, C.D.; Lauritzen, I.; Mattei, M.G.; Paris, S.; Hall, A.; Chardin, P. A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J. Cell Biol. 1998, 141, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Brandwein, D.; Wang, Z. Interaction between Rho GTPases and 14-3-3 Proteins. Int. J. Mol. Sci. 2017, 18, 2148. [Google Scholar] [CrossRef] [PubMed]
- Riou, P.; Kjær, S.; Garg, R.; Purkiss, A.; George, R.; Cain, R.J.; Bineva, G.; Reymond, N.; McColl, B.; Thompson, A.J.; et al. 14-3-3 proteins interact with a hybrid prenyl-phosphorylation motif to inhibit G proteins. Cell 2013, 153, 640–653, Erratum: Cell 2013, 153, 1164. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Rivero, F. Atypical Rho GTPases of the RhoBTB subfamily: Roles in vesicle trafficking and tumorigenesis. Cells 2016, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Boureux, A.; Vignal, E.; Faure, S.; Fort, P. Evolution of the Rho family of Ras-like GTPases in eukaryotes. Mol. Biol. Evol. 2007, 24, 203–216. [Google Scholar] [CrossRef]
- Espinosa, E.J.; Calero, M.; Sridevi, K.; Pfeffer, S.R. RhoBTB3: A Rho GTPase-family ATPase required for endosome to Golgi transport. Cell 2009, 137, 938–948. [Google Scholar] [CrossRef]
- Berthold, J.; Schenková, K.; Ramos, S.; Miura, Y.; Furukawa, M.; Aspenström, P.; Rivero, F. Characterization of RhoBTB-dependent Cul3 ubiquitin ligase complexes—Evidence for an autoregulatory mechanism. Exp. Cell Res. 2008, 314, 3453–3465. [Google Scholar] [CrossRef] [PubMed]
- Aspenström, P.; Fransson, A.; Saras, J. Rho GTPases have diverse effects on the organization of the actin filament system. Biochem. J. 2004, 377, 327–337. [Google Scholar] [CrossRef]
- Wilkins, A.; Ping, Q.; Carpenter, C.L. RhoBTB2 is a substrate of the mammalian Cul3 ubiquitin ligase complex. Genes Dev. 2004, 18, 856–861. [Google Scholar] [CrossRef]
- Saras, J.; Wollberg, P.; Aspenström, P. Wrch1 is a GTPase-deficient Cdc42-like protein with unusual binding characteristics and cellular effects. Exp. Cell Res. 2004, 299, 356–369. [Google Scholar] [CrossRef]
- Shutes, A.; Berzat, A.C.; Cox, A.D.; Der, C.J. Atypical mechanism of regulation of the Wrch-1 Rho family small GTPase. Curr. Biol. 2004, 14, 2052–2056. [Google Scholar] [CrossRef] [PubMed]
- Traut, T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell Biochem. 1994, 140, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Berzat, A.C.; Buss, J.E.; Chenette, E.J.; Weinbaum, C.A.; Shutes, A.; Der, C.J.; Minden, A.; Cox, A.D. Transforming activity of the Rho family GTPase, Wrch-1, a Wnt-regulated Cdc42 homolog, is dependent on a novel carboxyl-terminal palmitoylation motif. J. Biol. Chem. 2005, 280, 33055–33065. [Google Scholar] [CrossRef] [PubMed]
- Chenette, E.J.; Abo, A.; Der, C.J. Critical and distinct roles of amino- and carboxyl-terminal sequences in regulation of the biological activity of the Chp atypical Rho GTPase. J. Biol. Chem. 2005, 280, 13784–13792. [Google Scholar] [CrossRef] [PubMed]
- Chardin, P. Function and regulation of Rnd proteins. Nat. Rev. Mol. Cell Biol. 2006, 7, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Risse, S.L.; Vaz, B.; Burton, M.F.; Aspenström, P.; Piekorz, R.P.; Brunsveld, L.; Ahmadian, M.R. SH3-mediated targeting of Wrch1/RhoU by multiple adaptor proteins. Biol. Chem. 2013, 394, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Blom, M.; Reis, K.; Aspenström, P. RhoD localization and function is dependent on its GTP/GDP-bound state and unique N-terminal motif. Eur. J. Cell Biol. 2018, 97, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Qiu, R.G.; Chen, J.; McCormick, F.; Symons, M. A role for Rho in Ras transformation. Proc. Natl. Acad. Sci. USA 1995, 92, 11781–11785. [Google Scholar] [CrossRef] [PubMed]
- Khosravi-Far, R.; Solski, P.A.; Clark, G.J.; Kinch, M.S.; Der, C.J. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol. Cell. Biol. 1995, 15, 6443–6453. [Google Scholar] [CrossRef] [PubMed]
- Del Peso, L.; Hernández-Alcoceba, R.; Embade, N.; Carnero, A.; Esteve, P.; Paje, C.; Lacal, J.C. Rho proteins induce metastatic properties in vivo. Oncogene 1997, 15, 3047–3057. [Google Scholar] [CrossRef] [Green Version]
- Eva, A.; Aaronson, S.A. Isolation of a new human oncogene from a diffuse B-cell lymphoma. Nature 1985, 316, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Tronick, S.R.; Aaronson, S.A.; Eva, A. Molecular cloning and characterization of the human dbl proto-oncogene: Evidence that its overexpression is sufficient to transform NIH/3T3 cells. EMBO J. 1988, 7, 2465–2473. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, F.; Felline, A.; Fanelli, F. Catching functional modes and structural communication in Dbl family Rho guanine nucleotide exchange factors. J. Chem. Inf. Model. 2015, 55, 1878–1893. [Google Scholar] [CrossRef] [PubMed]
- Côté, J.F.; Vuori, K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J. Cell Sci. 2002, 115, 4901–4913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadea, G.; Blangy, A. Dock-family exchange factors in cell migration and disease. Eur. J. Cell Biol. 2014, 93, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Harding, M.A.; Theodorescu, D. RhoGDI signaling provides targets for cancer therapy. Eur. J. Cancer 2010, 46, 1252–1259. [Google Scholar] [CrossRef]
- Yuan, B.Z.; Miller, M.J.; Keck, C.L.; Zimonjic, D.B.; Thorgeirsson, S.S.; Popescu, N.C. Cloning, characterization, and chromosomal localization of a gene frequently deleted in human liver cancer (DLC-1) homologous to rat RhoGAP. Cancer Res. 1998, 58, 2196–2199. [Google Scholar]
- Braun, A.C.; Olayioye, M.A. Rho regulation: DLC proteins in space and time. Cell Signal. 2015, 27, 1643–1651. [Google Scholar] [CrossRef]
- Lukasik, D.; Wilczek, E.; Wasiutynski, A.; Gornicka, B. Deleted in liver cancer protein family in human malignancies. Oncol. Lett. 2011, 2, 763–768. [Google Scholar] [CrossRef]
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [Google Scholar] [CrossRef]
- Jordan, P.; Brazåo, R.; Boavida, M.G.; Gespach, C.; Chastre, E. Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene 1999, 18, 6835–6839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnelzer, A.; Prechtel, D.; Knaus, U.; Dehne, K.; Gerhard, M.; Graeff, H.; Harbeck, N.; Schmitt, M.; Lengyel, E. Rac1 in human breast cancer: Overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 2000, 19, 3013–3020. [Google Scholar] [CrossRef] [PubMed]
- Fiegen, D.; Haeusler, L.C.; Blumenstein, L.; Herbrand, U.; Dvorsky, R.; Vetter, I.R.; Ahmadian, M.R. Alternative splicing of Rac1 generates Rac1b, a self-activating GTPase. J. Biol. Chem. 2004, 279, 4743–4749. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Karnoub, A.E.; Palmby, T.R.; Lengyel, E.; Sondek, J.; Der, C.J. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene 2004, 23, 9369–9380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlichenko, L.; Geyer, R.; Yanagisawa, M.; Khauv, D.; Radisky, E.S.; Anastasiadis, P.Z.; Radisky, D.C. The 19-amino acid insertion in the tumor-associated splice isoform Rac1b confers specific binding to p120 catenin. J. Biol. Chem. 2010, 285, 19153–19161. [Google Scholar] [CrossRef] [PubMed]
- Matos, P.; Collard, J.G.; Jordan, P. Tumor-related alternatively spliced Rac1b is not regulated by Rho-GDP dissociation inhibitors and exhibits selective downstream signaling. J. Biol. Chem. 2003, 278, 50442–50448. [Google Scholar] [CrossRef] [PubMed]
- Nimnual, A.S.; Taylor, L.J.; Nyako, M.; Jeng, H.H.; Bar-Sagi, D. Perturbation of cytoskeleton dynamics by the opposing effects of Rac1 and Rac1b. Small GTPases 2010, 1, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.T.; Park, J.T.; Choi, K.; Choi, H.J.C.; Jung, C.W.; Kim, G.R.; Lee, Y.S.; Park, S.C. Chemical screening identifies ROCK as a target for recovering mitochondrial function in Hutchinson-Gilford progeria syndrome. Aging Cell 2017, 16, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Chen, Q.K.; Lui, C.; Cichon, M.A.; Radisky, D.C.; Nelson, C.M. Matrix compliance regulates Rac1b localization, NADPH oxidase assembly, and epithelial-mesenchymal transition. Mol. Biol. Cell 2012, 23, 4097–4108. [Google Scholar] [CrossRef]
- Pethe, V.V.; Charames, G.S.; Bapat, B. Rac1b recruits Dishevelled and β-catenin to Wnt target gene promoters independent of Wnt3A stimulation. Int. J. Oncol. 2011, 39, 805–810. [Google Scholar] [CrossRef]
- Reinstein, J.; Schlichting, I.; Frech, M.; Goody, R.S.; Wittinghofer, A. P21 with a phenylalanine 28→leucine mutation reacts normally with the GTPase-activating protein GAP, but nevertheless has transforming properties. J. Biol. Chem. 1991, 266, 17700–17706. [Google Scholar] [PubMed]
- Lin, R.; Bagrodia, S.; Cerione, R.; Manor, D. A novel Cdc42Hs mutant induces cellular transformation. Curr. Biol. 1997, 7, 794–797. [Google Scholar] [CrossRef]
- Xu, X.; Wang, Y.; Barry, D.C.; Chanock, S.J.; Bokoch, G.M. Guanine nucleotide binding properties of Rac2 mutant proteins and analysis of the responsiveness to guanine nucleotide dissociation stimulator. Biochemistry 1997, 36, 626–632. [Google Scholar] [CrossRef]
- Schlichting, I.; John, J.; Frech, M.; Chardin, P.; Wittinghofer, A.; Zimmermann, H.; Rösch, P. Proton NMR studies of transforming and nontransforming H-ras p21 mutants. Biochemistry 1990, 29, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Oswald, R.E. Solution structure of an oncogenic mutant of Cdc42Hs. Biochemistry 2006, 45, 2577–2583. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.J.; Ha, B.H.; Holman, E.C.; Halaban, R.; Schlessinger, J.; Boggon, T.J. RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc. Natl. Acad. Sci. USA 2013, 110, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.; Fansa, E.K.; Dvorsky, R.; Ahmadian, M.R. New insight into the molecular switch mechanism of human Rho family proteins: Shifting a paradigm. Biol. Chem. 2013, 394, 89–95. [Google Scholar] [CrossRef]
- Krengel, U.; Schlichting, I.; Scherer, A.; Schumann, R.; Frech, M.; John, J.; Kabsch, W.; Pai, E.F.; Wittinghofer, A. Three-dimensional structures of H-ras p21 mutants: Molecular basis for their inability to function as signal switch molecules. Cell 1990, 62, 539–548. [Google Scholar] [CrossRef]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Kawazu, M.; Ueno, T.; Kontani, K.; Ogita, Y.; Ando, M.; Fukumura, K.; Yamato, A.; Soda, M.; Takeuchi, K.; Miki, Y.; et al. Transforming mutations of RAC guanosine triphosphatases in human cancers. Proc. Natl. Acad. Sci. USA 2013, 110, 3029–3034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caye, A.; Strullu, M.; Guidez, F.; Cassinat, B.; Gazal, S.; Fenneteau, O.; Lainey, E.; Nouri, K.; Nakhaei-Rad, S.; Dvorsky, R.; et al. Juvenile myelomonocytic leukemia displays mutations in components of the Ras pathway and the PRC2 network. Nat. Genet. 2015, 47, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.Y.; Sung, M.K.; Lee, S.H.; Kim, S.; Lee, H.; Park, S.; Kim, S.C.; Lee, B.; Rho, K.; Lee, J.E.; et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet. 2014, 46, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Kontani, K.; Enami, T.; Kataoka, K.; Ishii, R.; Totoki, Y.; Kataoka, T.R.; Hirata, M.; Aoki, K.; Nakano, K.; et al. Variegated RHOA mutations in adult T-cell leukemia/lymphoma. Blood 2016, 127, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Stevers, M.; Rabban, J.T.; Garg, K.; Van Ziffle, J.; Onodera, C.; Grenert, J.P.; Yeh, I.; Bastian, B.C.; Zaloudek, C.; Solomon, D.A. Well-differentiated papillary mesothelioma of the peritoneum is genetically defined by mutually exclusive mutations in TRAF7 and CDC42. Mod. Pathol. 2018. [Google Scholar] [CrossRef]
- Mar, V.J.; Wong, S.Q.; Logan, A.; Nguyen, T.; Cebon, J.; Kelly, J.W.; Wolfe, R.; Dobrovic, A.; McLean, C.; McArthur, G.A. Clinical and pathological associations of the activating RAC1 P29S mutation in primary cutaneous melanoma. Pigment Cell Melanoma Res. 2014, 27, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Vu, H.L.; Rosenbaum, S.; Purwin, T.J.; Davies, M.A.; Aplin, A.E. Rac1 P29S regulates PD-L1 expression in melanoma. Pigment Cell Melanoma Res. 2015, 28, 590–598. [Google Scholar] [CrossRef]
- King, S.J.; Asokan, S.B.; Haynes, E.M.; Zimmerman, S.P.; Rotty, J.D.; Alb, J.G., Jr.; Tagliatela, A.; Blake, D.R.; Lebedeva, I.P.; Marston, D.; et al. Lamellipodia are crucial for haptotactic sensing and response. J. Cell Sci. 2016, 129, 2329–2342. [Google Scholar] [CrossRef] [Green Version]
- Revach, O.Y.; Winograd-Katz, S.E.; Samuels, Y.; Geiger, B. The involvement of mutant Rac1 in the formation of invadopodia in cultured melanoma cells. Exp. Cell Res. 2016, 343, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Moreno, V.; Gadea, G.; Ahn, J.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac activation and inactivation control plasticity of tumor cell movement. Cell 2008, 135, 510–523. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Proteins Known to Bind to Rac1b | Rac1-Binding Proteins that Do Not Bind Rac1b | ||
|---|---|---|---|
| Protein | Reference | Protein | Reference |
| smgGDS | [65] | Pak1 | [63,64] |
| RACK1 | RhoGDI | [63,64,65,66] | |
| p120 ctn | IQGAP1 | [65] | |
| ROCK1 * | [68] | GIT-1 | |
| Cytochrome C * | |||
| NADPH oxidase * | [69] | ||
| Dishevelled-3 * | [70] | ||
| β-Catenin * | |||
| TCF-4 * | |||
| Small GTPase | Mutant | Cell Type | Reference |
|---|---|---|---|
| Rac1 | P29S | Sun-exposed melanoma | [79] |
| Melanoma | [80] | ||
| Mouth, squamous cell carcinoma | [81] | ||
| MDA-MB-157 cell line | |||
| N92I | HT1080 fibrosarcoma cell line | [81] | |
| C157Y | Lung, adenocarcinoma | ||
| P179L | Skin, squamous cell carcinoma | ||
| Rac2 | P29L | Melanoma | [80] |
| I21M | Larynx, squamous cell carcinoma | [81] | |
| P29L | HCC1143 cell line | ||
| P29Q | KCL-22 cell line | ||
| D47Y | Glioma | ||
| P106H | Malignant melanoma | ||
| D63V | Juvenile myelomonocytic leukemia | [82] | |
| RhoA | G17V | Angioimmunoblastic T cell lymphoma | [83] |
| A161E | |||
| R5Q | Angioimmunoblastic T cell lymphoma | [84] | |
| G17V | Adult T-cell leukemia/lymphoma | [85] | |
| G14V | |||
| C16R | |||
| C16F | |||
| C16G | |||
| C16L | |||
| T19I | |||
| A56V | |||
| R68L | |||
| C83Y | |||
| N117I | |||
| K118E | |||
| K118Q | |||
| D120N | |||
| A161P | |||
| A161V | |||
| K162E | |||
| Cdc42 | Cdc42/Q61R | Papillary mesothelioma | [86] |
| Cdc42/P34Q | |||
| Cdc42/G12D | Melanoma | [80] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aspenström, P. Activated Rho GTPases in Cancer—The Beginning of a New Paradigm. Int. J. Mol. Sci. 2018, 19, 3949. https://doi.org/10.3390/ijms19123949
Aspenström P. Activated Rho GTPases in Cancer—The Beginning of a New Paradigm. International Journal of Molecular Sciences. 2018; 19(12):3949. https://doi.org/10.3390/ijms19123949
Chicago/Turabian StyleAspenström, Pontus. 2018. "Activated Rho GTPases in Cancer—The Beginning of a New Paradigm" International Journal of Molecular Sciences 19, no. 12: 3949. https://doi.org/10.3390/ijms19123949
APA StyleAspenström, P. (2018). Activated Rho GTPases in Cancer—The Beginning of a New Paradigm. International Journal of Molecular Sciences, 19(12), 3949. https://doi.org/10.3390/ijms19123949