Melatonin Modulates the Microenvironment of Glioblastoma Multiforme by Targeting Sirtuin 1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. SRB Assay
2.4. MTT Assay
2.5. Cytosolic and Nuclear Extracts
2.6. Monocyte-Binding Assay
2.7. Western Blotting
2.8. Reverse Transcription and Real-Time PCR
2.9. Cell Transfection
2.10. Reporter Gene Assay
2.11. Enzyme-Linked Immunosorbent Assay (ELISA)
2.12. GEO Gene Expression Database
2.13. Statistics
3. Results
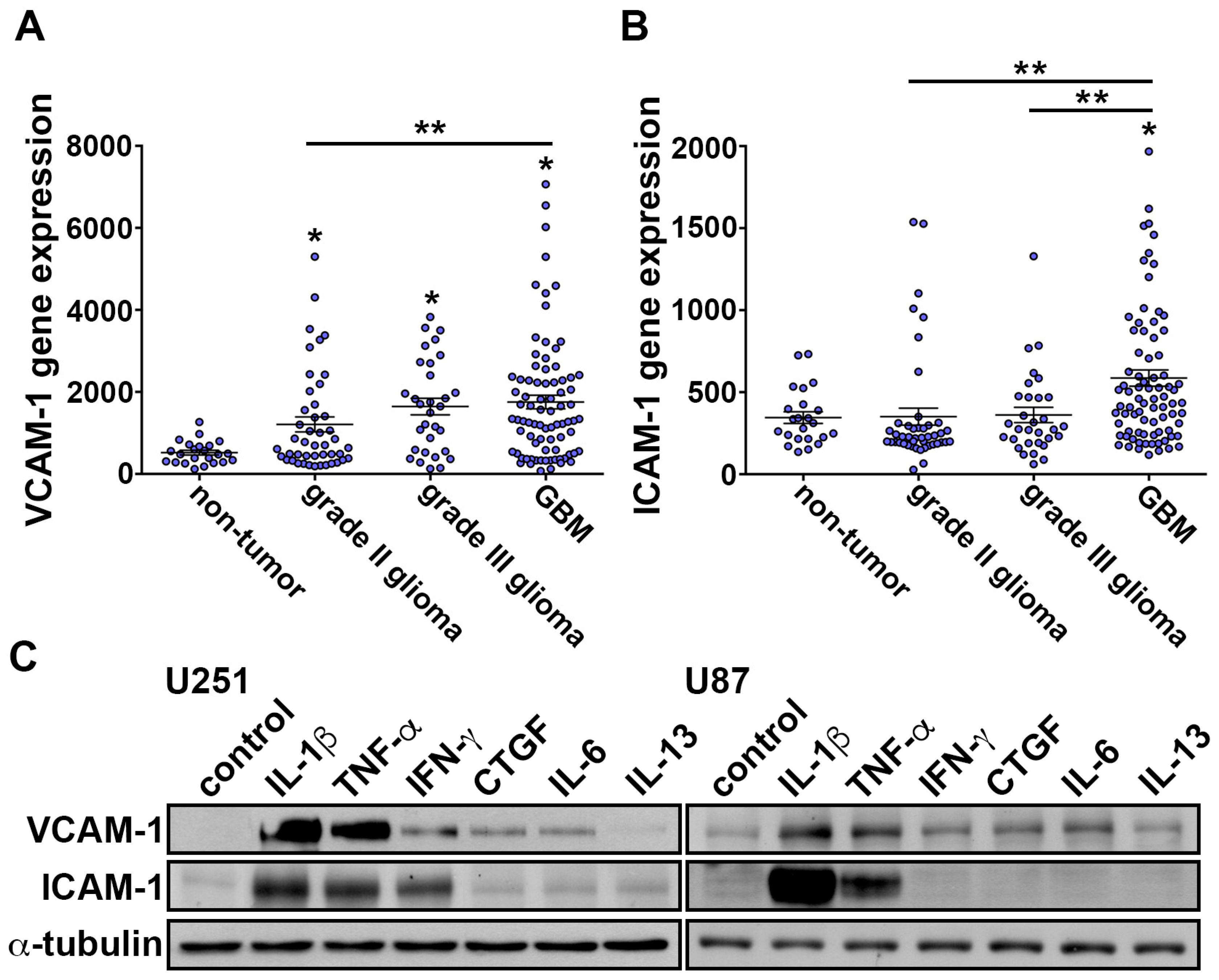
3.1. IL-1β Induces VCAM-1 and ICAM-1 Expression and Increases Monocyte Adhesion in GBM
3.2. Effects of Melatonin in IL-1β-Induced ICAM-1 and VCAM-1 Expression and Monocyte Adhesion
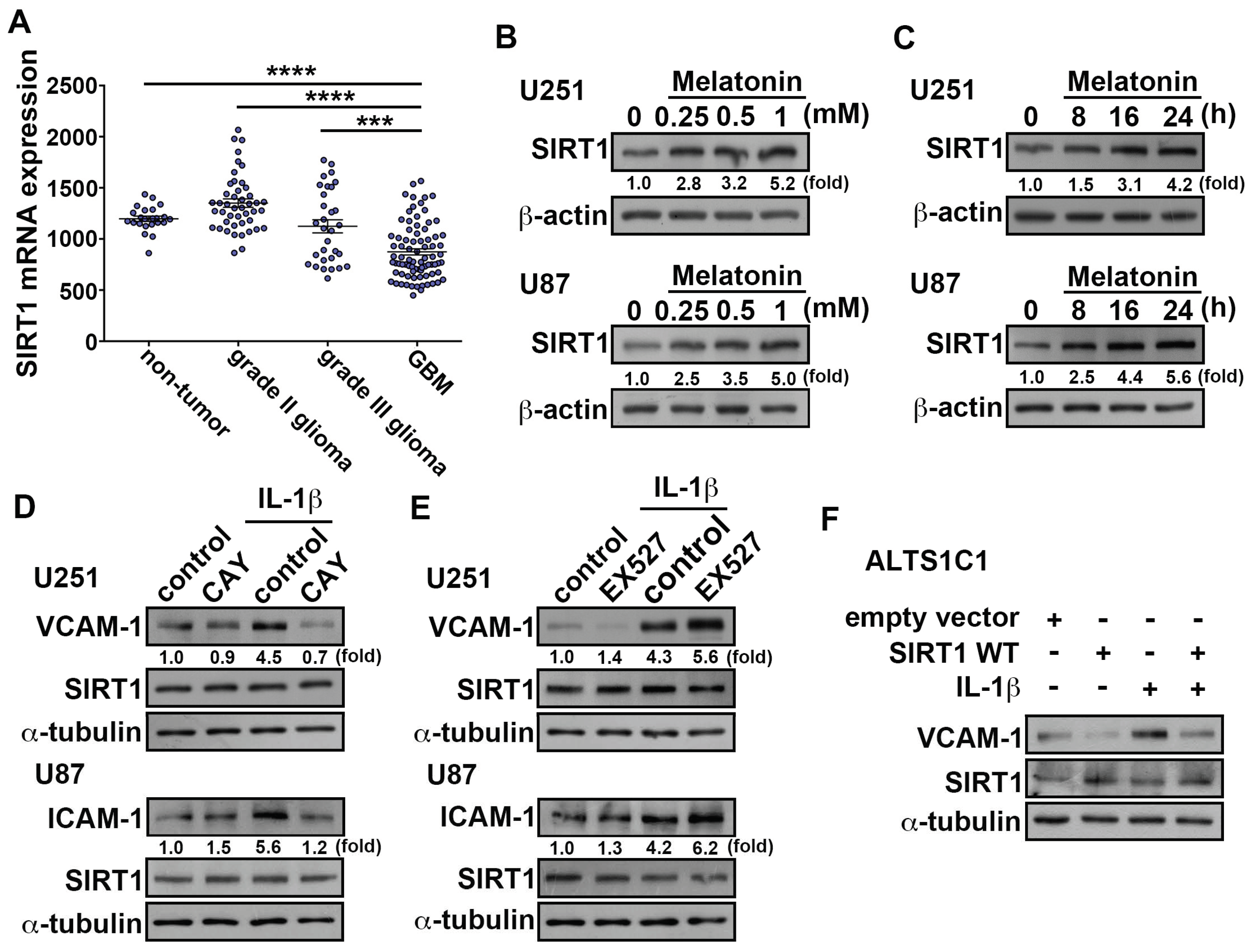
3.3. Upregulation of SIRT1 Inhibits ICAM-1 and VCAM-1 Expression in GBM
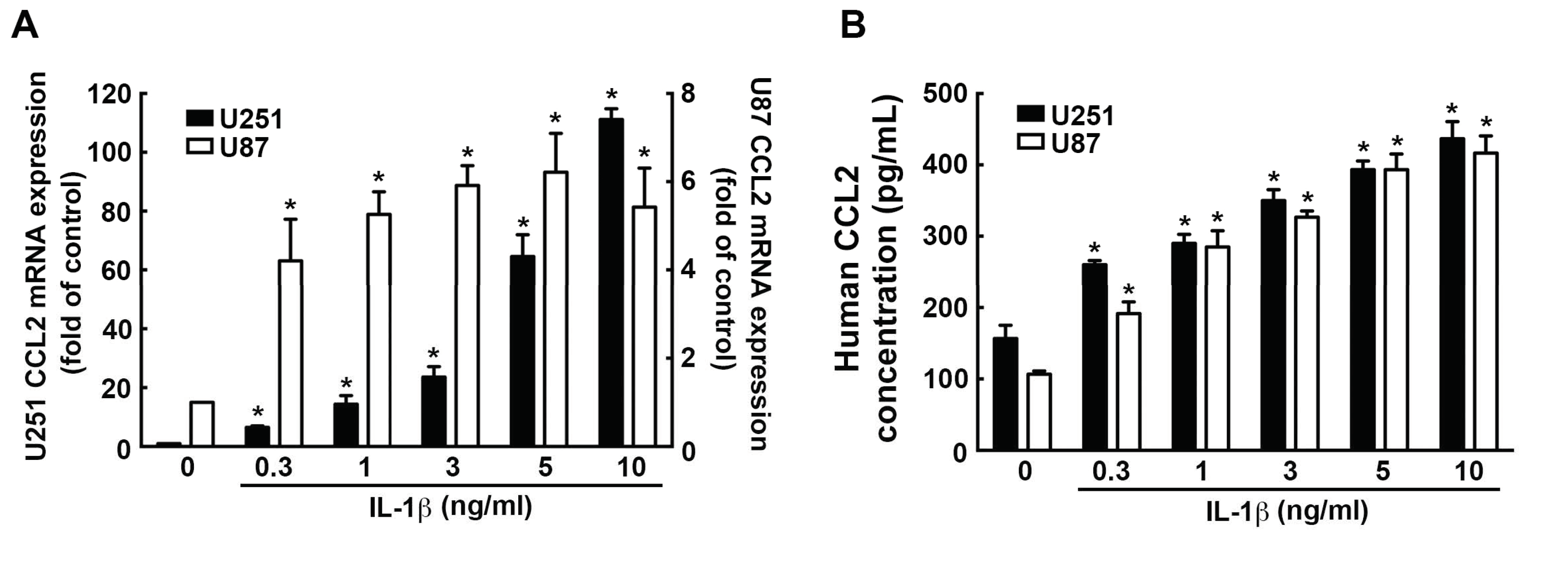
3.4. Involvement of CCL2 in the IL-1β-Induced ICAM-1 and VCAM-1 Expression and Monocyte Adhesion
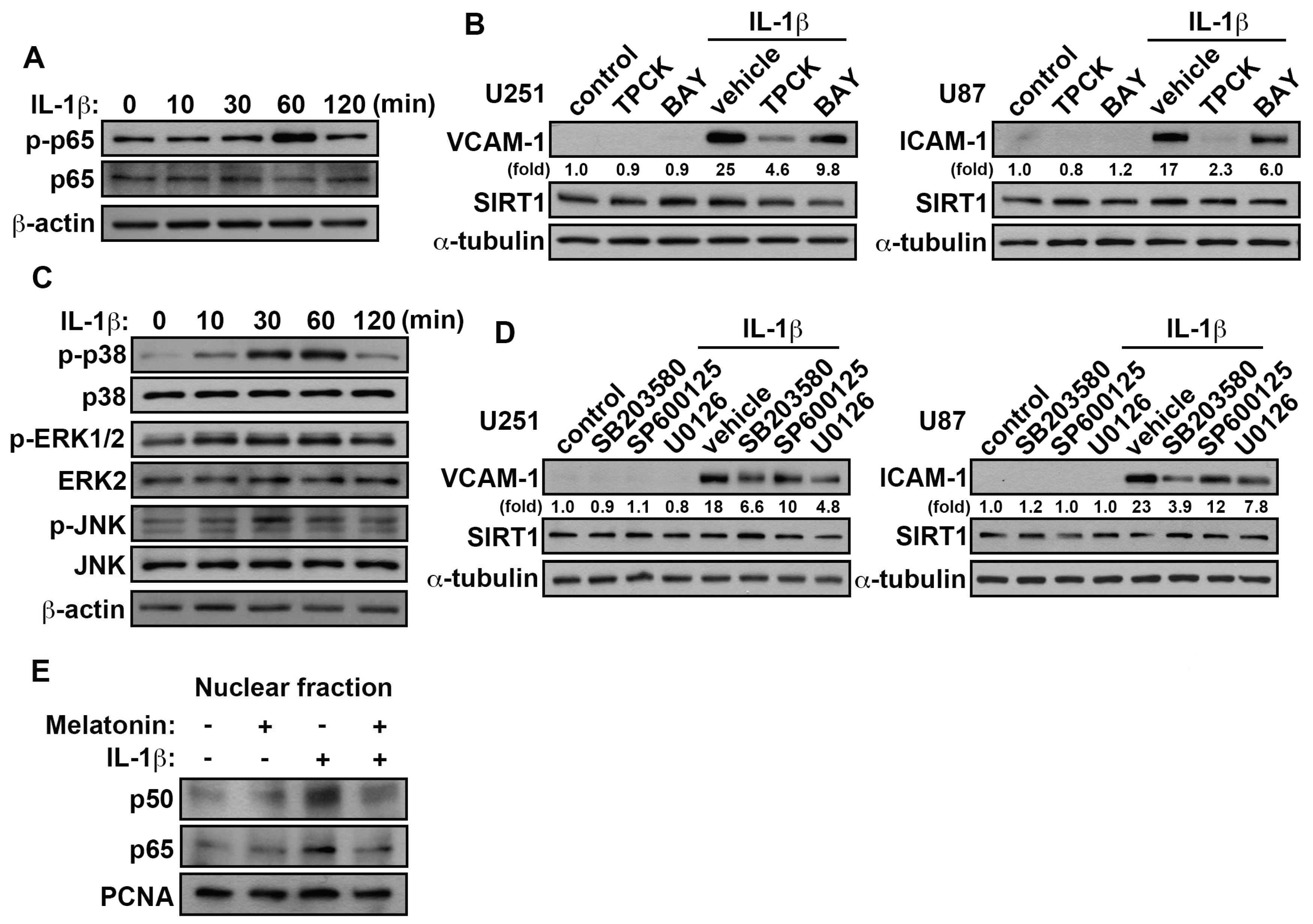
3.5. Involvement of p38/p65 in the IL-1β-Induced VCAM-1 and ICAM-1 in GBM
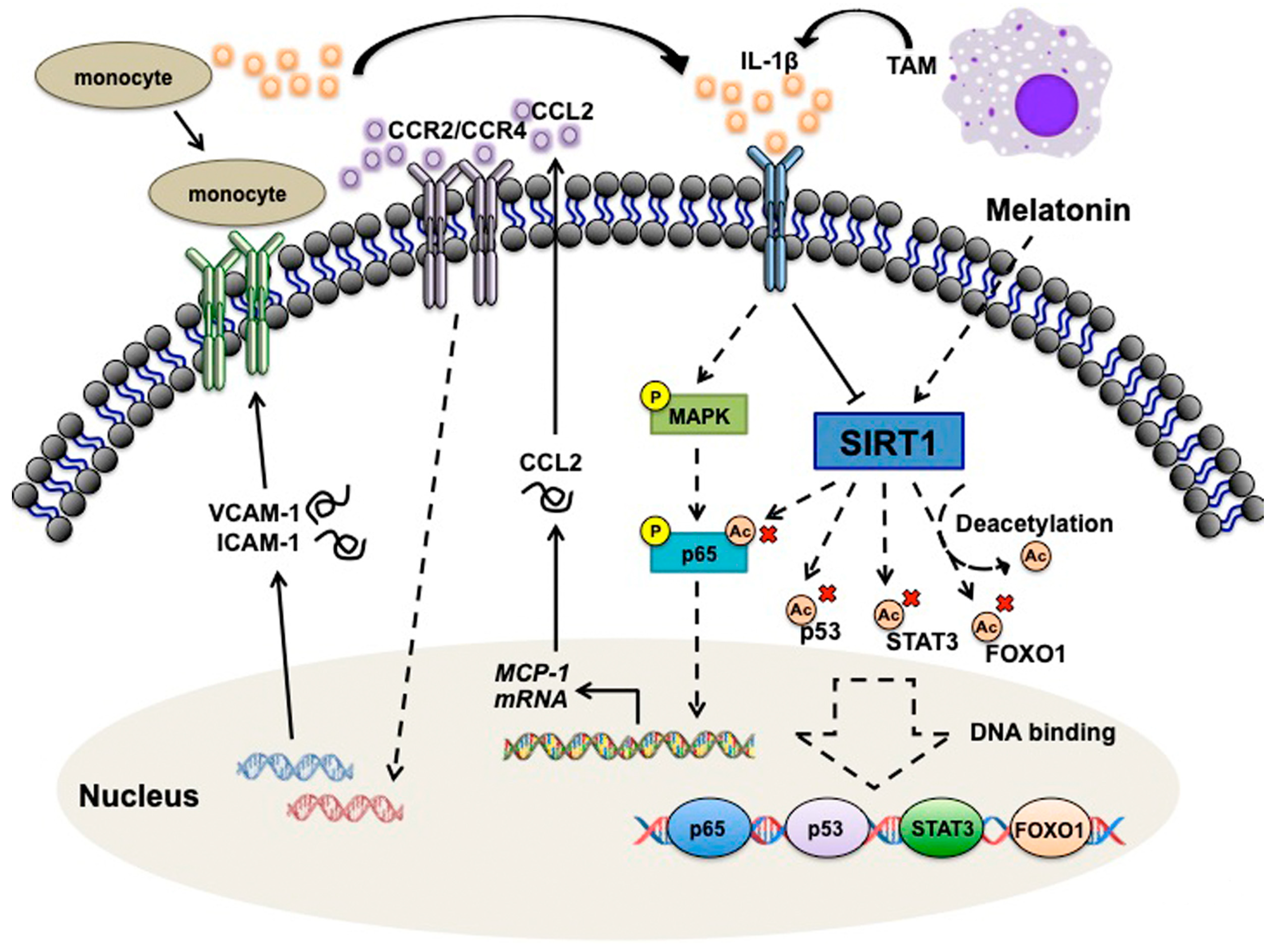
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, J.; Lee, S.M.; Shannon, S.; Gao, B.; Chen, W.; Chen, A.; Divekar, R.; McBurney, M.W.; Braley-Mullen, H.; Zaghouani, H.; et al. The type III histone deacetylase Sirt1 is essential for maintenance of T cell tolerance in mice. J. Clin. Investig. 2009, 119, 3048–3058. [Google Scholar] [CrossRef] [Green Version]
- Guarente, L.; Picard, F. Calorie restriction--the SIR2 connection. Cell 2005, 120, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Pillarisetti, S. A review of Sirt1 and Sirt1 modulators in cardiovascular and metabolic diseases. Recent Pat. Cardiovasc. Drug Discov. 2008, 3, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Klar, A.J.; Fogel, S.; Macleod, K. MAR1-a Regulator of the HMa and HMalpha Loci in SACCHAROMYCES CEREVISIAE. Genetics 1979, 93, 37–50. [Google Scholar] [PubMed]
- Demontis, F.; Perrimon, N. FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell 2010, 143, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Dryden, S.C.; Nahhas, F.A.; Nowak, J.E.; Goustin, A.S.; Tainsky, M.A. Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol. Cell. Biol. 2003, 23, 3173–3185. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, A.; Scher, M.B.; Lee, D.H.; Sutton, A.; Cheng, H.L.; Alt, F.W.; Serrano, L.; Sternglanz, R.; Reinberg, D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006, 20, 1256–1261. [Google Scholar] [CrossRef] [Green Version]
- Bishayee, A. Cancer prevention and treatment with resveratrol: From rodent studies to clinical trials. Cancer Prev. Res. 2009, 2, 409–418. [Google Scholar] [CrossRef]
- Chauhan, D.; Bandi, M.; Singh, A.V.; Ray, A.; Raje, N.; Richardson, P.; Anderson, K.C. Preclinical evaluation of a novel SIRT1 modulator SRT1720 in multiple myeloma cells. Br. J. Haematol. 2011, 155, 588–598. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Lu, Y.; Zhang, Z.; Wang, J.; Yang, H.; Liu, G. Intercellular interplay between Sirt1 signalling and cell metabolism in immune cell biology. Immunology 2015, 145, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Yan, P.F.; Zhao, H.Y.; Zhang, F.C.; Zhao, W.H.; Feng, M. SIRT6 suppresses glioma cell growth via induction of apoptosis, inhibition of oxidative stress and suppression of JAK2/STAT3 signaling pathway activation. Oncol. Rep. 2016, 35, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, Z.; Shao, Y.; Pu, Y.; Miu, W.; Yao, J.; Wu, Y.; Yang, Z. MicroRNA-34a suppresses cell proliferation and induces apoptosis in U87 glioma stem cells. Technol. Cancer Res. Treat. 2012, 11, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H.; Sengupta, K.; Li, C.; Kim, H.S.; Cao, L.; Xiao, C.; Kim, S.; Xu, X.; Zheng, Y.; Chilton, B.; et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 2008, 14, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J. Pineal melatonin: Cell biology of its synthesis and of its physiological interactions. Endocr. Rev. 1991, 12, 151–180. [Google Scholar] [CrossRef] [PubMed]
- Stehle, J.H.; Saade, A.; Rawashdeh, O.; Ackermann, K.; Jilg, A.; Sebesteny, T.; Maronde, E. A survey of molecular details in the human pineal gland in the light of phylogeny, structure, function and chronobiological diseases. J. Pineal Res. 2011, 51, 17–43. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J. Melatonin: The chemical expression of darkness. Mol. Cell. Endocrinol. 1991, 79, C153–C158. [Google Scholar] [CrossRef]
- Carloni, S.; Favrais, G.; Saliba, E.; Albertini, M.C.; Chalon, S.; Longini, M.; Gressens, P.; Buonocore, G.; Balduini, W. Melatonin modulates neonatal brain inflammation through endoplasmic reticulum stress, autophagy, and miR-34a/silent information regulator 1 pathway. J. Pineal Res. 2016, 61, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, V.; Spence, D.W.; Pandi-Perumal, S.R.; Trakht, I.; Cardinali, D.P. Therapeutic actions of melatonin in cancer: Possible mechanisms. Integr. Cancer Ther. 2008, 7, 189–203. [Google Scholar] [CrossRef]
- Veneroso, C.; Tunon, M.J.; Gonzalez-Gallego, J.; Collado, P.S. Melatonin reduces cardiac inflammatory injury induced by acute exercise. J. Pineal Res. 2009, 47, 184–191. [Google Scholar] [CrossRef]
- Pozo, D.; Reiter, R.J.; Calvo, J.R.; Guerrero, J.M. Inhibition of cerebellar nitric oxide synthase and cyclic GMP production by melatonin via complex formation with calmodulin. J. Cell. Biochem. 1997, 65, 430–442. [Google Scholar] [CrossRef]
- Shah, S.A.; Khan, M.; Jo, M.H.; Jo, M.G.; Amin, F.U.; Kim, M.O. Melatonin Stimulates the SIRT1/Nrf2 Signaling Pathway Counteracting Lipopolysaccharide (LPS)-Induced Oxidative Stress to Rescue Postnatal Rat Brain. CNS Neurosci. Ther. 2017, 23, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.M.; Belancio, V.P.; Dauchy, R.T.; Xiang, S.; Brimer, S.; Mao, L.; Hauch, A.; Lundberg, P.W.; Summers, W.; Yuan, L.; et al. Melatonin: An inhibitor of breast cancer. Endocr. Relat. Cancer 2015, 22, R183–R204. [Google Scholar] [CrossRef] [PubMed]
- Motilva, V.; Garcia-Maurino, S.; Talero, E.; Illanes, M. New paradigms in chronic intestinal inflammation and colon cancer: Role of melatonin. J. Pineal Res. 2011, 51, 44–60. [Google Scholar] [CrossRef]
- Wang, J.; Hao, H.; Yao, L.; Zhang, X.; Zhao, S.; Ling, E.A.; Hao, A.; Li, G. Melatonin suppresses migration and invasion via inhibition of oxidative stress pathway in glioma cells. J. Pineal Res. 2012, 53, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Herrera, F.; Carrera-Gonzalez, P.; Garcia-Santos, G.; Antolin, I.; Rodriguez-Blanco, J.; Rodriguez, C. Intracellular signaling pathways involved in the cell growth inhibition of glioma cells by melatonin. Cancer Res. 2006, 66, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Lissoni, P.; Meregalli, S.; Nosetto, L.; Barni, S.; Tancini, G.; Fossati, V.; Maestroni, G. Increased survival time in brain glioblastomas by a radioneuroendocrine strategy with radiotherapy plus melatonin compared to radiotherapy alone. Oncology 1996, 53, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Cardinali, D.P.; Garcia, A.P.; Cano, P.; Esquifino, A.I. Melatonin role in experimental arthritis. Curr. Drug Targets Immune Endocr. Metab. Disord. 2004, 4, 1–10. [Google Scholar] [CrossRef]
- Maestroni, G.J. The immunotherapeutic potential of melatonin. Expert Opin. Investig. Drugs 2001, 10, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Inserra, P.F.; Liang, B.; Ardestani, S.K.; Elliott, K.K.; Molitor, M.; Watson, R.R. Melatonin, immune modulation and aging. Autoimmunity 1997, 26, 43–53. [Google Scholar] [CrossRef]
- Currier, N.L.; Sun, L.Z.; Miller, S.C. Exogenous melatonin: Quantitative enhancement in vivo of cells mediating non-specific immunity. J. Neuroimmunol. 2000, 104, 101–108. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Hilliard, B.; Ventura, E.; Rostami, A. Luzindole, a melatonin receptor antagonist, suppresses experimental autoimmune encephalomyelitis. Pathobiology 1997, 65, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Hayashi, Y.; Stephens, C.; Georgescu, M.M. Invasive glioblastoma cells acquire stemness and increased Akt activation. Neoplasia 2010, 12, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Cheng, L.; Guryanova, O.A.; Wu, Q.; Bao, S. Cancer stem cells in glioblastoma—Molecular signaling and therapeutic targeting. Protein Cell 2010, 1, 638–655. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Lorger, M. Tumor microenvironment in the brain. Cancers 2012, 4, 218–243. [Google Scholar] [CrossRef] [PubMed]
- Shergalis, A.; Bankhead, A., 3rd; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2012, 60, 502–514. [Google Scholar] [CrossRef]
- Gil-Bernabe, A.M.; Ferjancic, S.; Tlalka, M.; Zhao, L.; Allen, P.D.; Im, J.H.; Watson, K.; Hill, S.A.; Amirkhosravi, A.; Francis, J.L.; et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood 2012, 119, 3164–3175. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Zhang, L.; Handel, M.V.; Schartner, J.M.; Hagar, A.; Allen, G.; Curet, M.; Badie, B. Regulation of IL-10 expression by upstream stimulating factor (USF-1) in glioma-associated microglia. J. Neuroimmunol. 2007, 184, 188–197. [Google Scholar] [CrossRef]
- Mestas, J.; Ley, K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 2008, 18, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Szulzewsky, F.; Yerevanian, A.; Chen, Z.; Heinzmann, D.; Rasmussen, R.D.; Alvarez-Garcia, V.; Kim, Y.; Wang, B.; Tamagno, I.; et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget 2015, 6, 15077–15094. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Yoshie, O. The chemokine superfamily revisited. Immunity 2012, 36, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Craig, M.J.; Loberg, R.D. CCL2 (Monocyte Chemoattractant Protein-1) in cancer bone metastases. Cancer Metastasis Rev. 2006, 25, 611–619. [Google Scholar] [CrossRef]
- Berman, J.W.; Guida, M.P.; Warren, J.; Amat, J.; Brosnan, C.F. Localization of monocyte chemoattractant peptide-1 expression in the central nervous system in experimental autoimmune encephalomyelitis and trauma in the rat. J. Immunol. 1996, 156, 3017–3023. [Google Scholar]
- Gourmala, N.G.; Buttini, M.; Limonta, S.; Sauter, A.; Boddeke, H.W. Differential and time-dependent expression of monocyte chemoattractant protein-1 mRNA by astrocytes and macrophages in rat brain: Effects of ischemia and peripheral lipopolysaccharide administration. J. Neuroimmunol. 1997, 74, 35–44. [Google Scholar] [CrossRef]
- Negus, R.P.; Stamp, G.W.; Hadley, J.; Balkwill, F.R. Quantitative assessment of the leukocyte infiltrate in ovarian cancer and its relationship to the expression of C-C chemokines. Am. J. Pathol. 1997, 150, 1723–1734. [Google Scholar]
- Fang, W.B.; Jokar, I.; Zou, A.; Lambert, D.; Dendukuri, P.; Cheng, N. CCL2/CCR2 chemokine signaling coordinates survival and motility of breast cancer cells through Smad3 protein- and p42/44 mitogen-activated protein kinase (MAPK)-dependent mechanisms. J. Biol. Chem. 2012, 287, 36593–36608. [Google Scholar] [CrossRef]
- Boratynska, M. The role of monocyte chemotactic peptide (MCP-1) in chronic renal allograft rejection. Pol. Arch. Med. Wewn. 1998, 99, 272–280. [Google Scholar]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 2016, 7, 28697–28710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Somasundaram, R.; Berencsi, K.; Caputo, L.; Gimotty, P.; Rani, P.; Guerry, D.; Swoboda, R.; Herlyn, D. Migration of cytotoxic T lymphocytes toward melanoma cells in three-dimensional organotypic culture is dependent on CCL2 and CCR4. Eur. J. Immunol. 2006, 36, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Gavrilin, M.A.; Deucher, M.F.; Boeckman, F.; Kolattukudy, P.E. Monocyte chemotactic protein 1 upregulates IL-1beta expression in human monocytes. Biochem. Biophys. Res. Commun. 2000, 277, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Hanazawa, S.; Takeshita, A.; Chen, Y.; Watanabe, A.; Nishida, K.; Miyata, Y.; Kitano, S. Interleukin-1 beta and tumor necrosis factor-alpha stimulate synergistically the expression of monocyte chemoattractant protein-1 in fibroblastic cells derived from human periodontal ligament. Oral Microbiol. Immunol. 1996, 11, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.R.; Wang, J.; Kivisakk, P.; Rollins, B.J.; Ransohoff, R.M. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J. Exp. Med. 2001, 193, 713–726. [Google Scholar] [CrossRef]
- Fuentes, M.E.; Durham, S.K.; Swerdel, M.R.; Lewin, A.C.; Barton, D.S.; Megill, J.R.; Bravo, R.; Lira, S.A. Controlled recruitment of monocytes and macrophages to specific organs through transgenic expression of monocyte chemoattractant protein-1. J. Immunol. 1995, 155, 5769–5776. [Google Scholar]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef]
- Tsai, C.F.; Cheng, Y.K.; Lu, D.Y.; Wang, S.L.; Chang, C.N.; Chang, P.C.; Yeh, W.L. Inhibition of estrogen receptor reduces connexin 43 expression in breast cancers. Toxicol. Appl. Pharmacol. 2018, 338, 182–190. [Google Scholar] [CrossRef]
- Lin, H.Y.; Huang, B.R.; Yeh, W.L.; Lee, C.H.; Huang, S.S.; Lai, C.H.; Lin, H.; Lu, D.Y. Antineuroinflammatory effects of lycopene via activation of adenosine monophosphate-activated protein kinase-alpha1/heme oxygenase-1 pathways. Neurobiol. Aging 2014, 35, 191–202. [Google Scholar] [CrossRef]
- Yuan, P.; He, X.H.; Rong, Y.F.; Cao, J.; Li, Y.; Hu, Y.P.; Liu, Y.; Li, D.; Lou, W.; Liu, M.F. KRAS/NF-kappaB/YY1/miR-489 Signaling Axis Controls Pancreatic Cancer Metastasis. Cancer Res. 2017, 77, 100–111. [Google Scholar] [CrossRef]
- Bird, S.; Zou, J.; Wang, T.; Munday, B.; Cunningham, C.; Secombes, C.J. Evolution of interleukin-1beta. Cytokine Growth Factor Rev. 2002, 13, 483–502. [Google Scholar] [CrossRef]
- Wang, X.; Wang, B.; Xie, J.; Hou, D.; Zhang, H.; Huang, H. Melatonin inhibits epithelialtomesenchymal transition in gastric cancer cells via attenuation of IL1beta/NFkappaB/MMP2/MMP9 signaling. Int. J. Mol. Med. 2018, 42, 2221–2228. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Rizak, J.D.; Li, X.; Li, J.; Ma, Y. Melatonin treatment increases the transcription of cell proliferation-related genes prior to inducing cell death in C6 glioma cells in vitro. Oncol. Lett. 2013, 6, 347–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajes, M.; Gutierrez-Cuesta, J.; Ortuno-Sahagun, D.; Camins, A.; Pallas, M. Anti-aging properties of melatonin in an in vitro murine senescence model: Involvement of the sirtuin 1 pathway. J. Pineal Res. 2009, 47, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in tumor immunity. Int. J. Cancer 2010, 127, 759–767. [Google Scholar] [CrossRef]
- Ha, K.H.; Byun, M.S.; Choi, J.; Jeong, J.; Lee, K.J.; Jue, D.M. N-tosyl-L-phenylalanine chloromethyl ketone inhibits NF-kappaB activation by blocking specific cysteine residues of IkappaB kinase beta and p65/RelA. Biochemistry 2009, 48, 7271–7278. [Google Scholar] [CrossRef]
- Kim, S.H.; Oh, J.M.; No, J.H.; Bang, Y.J.; Juhnn, Y.S.; Song, Y.S. Involvement of NF-kappaB and AP-1 in COX-2 upregulation by human papillomavirus 16 E5 oncoprotein. Carcinogenesis 2009, 30, 753–757. [Google Scholar] [CrossRef]
- Jordan, J.T.; Sun, W.; Hussain, S.F.; DeAngulo, G.; Prabhu, S.S.; Heimberger, A.B. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol. Immunother. 2008, 57, 123–131. [Google Scholar] [CrossRef]
- Vakilian, A.; Khorramdelazad, H.; Heidari, P.; Sheikh Rezaei, Z.; Hassanshahi, G. CCL2/CCR2 signaling pathway in glioblastoma multiforme. Neurochem. Int. 2017, 103, 1–7. [Google Scholar] [CrossRef]
- Kawai, Y.; Kaidoh, M.; Yokoyama, Y.; Sano, K.; Ohhashi, T. Chemokine CCL2 facilitates ICAM-1-mediated interactions of cancer cells and lymphatic endothelial cells in sentinel lymph nodes. Cancer Sci. 2009, 100, 419–428. [Google Scholar] [CrossRef]
- Lin, Y.M.; Hsu, C.J.; Liao, Y.Y.; Chou, M.C.; Tang, C.H. The CCL2/CCR2 axis enhances vascular cell adhesion molecule-1 expression in human synovial fibroblasts. PLoS ONE 2012, 7, e49999. [Google Scholar] [CrossRef]
- Nduom, E.K.; Weller, M.; Heimberger, A.B. Immunosuppressive mechanisms in glioblastoma. Neuro-Oncology 2015, 17 (Suppl. 7), vii9–vii14. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Bollen, A.W.; Gupta, N. CC chemokine receptor-2A is frequently overexpressed in glioblastoma. J. Neuro-Oncol. 2008, 86, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Cutando, A.; Lopez-Valverde, A.; Arias-Santiago, S.; De Vicente, J.; De Diego, R.G. Role of melatonin in cancer treatment. Anticancer Res. 2012, 32, 2747–2753. [Google Scholar] [PubMed]
- Cuzzocrea, S.; Costantino, G.; Mazzon, E.; Micali, A.; De Sarro, A.; Caputi, A.P. Beneficial effects of melatonin in a rat model of splanchnic artery occlusion and reperfusion. J. Pineal Res. 2000, 28, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Baydas, G.; Nedzvetsky, V.S.; Nerush, P.A.; Kirichenko, S.V.; Demchenko, H.M.; Reiter, R.J. A novel role for melatonin: Regulation of the expression of cell adhesion molecules in the rat hippocampus and cortex. Neurosci. Lett. 2002, 326, 109–112. [Google Scholar] [CrossRef]
- Park, H.J.; Kim, H.J.; Ra, J.; Hong, S.J.; Baik, H.H.; Park, H.K.; Yim, S.V.; Nah, S.S.; Cho, J.J.; Chung, J.H. Melatonin inhibits lipopolysaccharide-induced CC chemokine subfamily gene expression in human peripheral blood mononuclear cells in a microarray analysis. J. Pineal Res. 2007, 43, 121–129. [Google Scholar] [CrossRef]
- Puliyappadamba, V.T.; Hatanpaa, K.J.; Chakraborty, S.; Habib, A.A. The role of NF-kappaB in the pathogenesis of glioma. Mol. Cell. Oncol. 2014, 1, e963478. [Google Scholar] [CrossRef]
- McNeill, R.S.; Canoutas, D.A.; Stuhlmiller, T.J.; Dhruv, H.D.; Irvin, D.M.; Bash, R.E.; Angus, S.P.; Herring, L.E.; Simon, J.M.; Skinner, K.R.; et al. Combination therapy with potent PI3K and MAPK inhibitors overcomes adaptive kinome resistance to single agents in preclinical models of glioblastoma. Neuro-Oncology 2017, 19, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Mawrin, C.; Diete, S.; Treuheit, T.; Kropf, S.; Vorwerk, C.K.; Boltze, C.; Kirches, E.; Firsching, R.; Dietzmann, K. Prognostic relevance of MAPK expression in glioblastoma multiforme. Int. J. Oncol. 2003, 23, 641–648. [Google Scholar] [CrossRef]
- Chen, J.H.; Huang, S.M.; Chen, C.C.; Tsai, C.F.; Yeh, W.L.; Chou, S.J.; Hsieh, W.T.; Lu, D.Y. Ghrelin induces cell migration through GHS-R, CaMKII, AMPK, and NF-kappaB signaling pathway in glioma cells. J. Cell. Biochem. 2011, 112, 2931–2941. [Google Scholar] [CrossRef] [PubMed]
- Tsou, H.K.; Su, C.M.; Chen, H.T.; Hsieh, M.H.; Lin, C.J.; Lu, D.Y.; Tang, C.H.; Chen, Y.H. Integrin-linked kinase is involved in TNF-alpha-induced inducible nitric-oxide synthase expression in myoblasts. J. Cell. Biochem. 2010, 109, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Liu, Y.; Liu, Z.; Liu, J.; Liu, X.; Chen, X.; Li, C.; Zeng, Y. p38gamma overexpression in gliomas and its role in proliferation and apoptosis. Sci. Rep. 2013, 3, 2089. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.S.; Lin, H.Y.; Lai, S.W.; Huang, C.Y.; Huang, B.R.; Chen, P.Y.; Wei, K.C.; Lu, D.Y. MiR-181b modulates EGFR-dependent VCAM-1 expression and monocyte adhesion in glioblastoma. Oncogene 2017, 36, 5006–5022. [Google Scholar] [CrossRef] [PubMed]
- Krakstad, C.; Chekenya, M. Survival signalling and apoptosis resistance in glioblastomas: Opportunities for targeted therapeutics. Mol. Cancer 2010, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Hiratsuka, M.; Osaki, M.; Yamada, H.; Kishimoto, I.; Yamaguchi, S.; Nakano, S.; Katoh, M.; Ito, H.; Oshimura, M. SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene 2007, 26, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, C.; Zwaans, B.M.; Silberman, D.M.; Gymrek, M.; Goren, A.; Zhong, L.; Ram, O.; Truelove, J.; Guimaraes, A.R.; Toiber, D.; et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 2012, 151, 1185–1199. [Google Scholar] [CrossRef] [PubMed]
- Villalba, J.M.; Alcain, F.J. Sirtuin activators and inhibitors. Biofactors 2012, 38, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Adeberg, S.; Bernhardt, D.; Harrabi, S.B.; Nicolay, N.H.; Horner-Rieber, J.; Konig, L.; Repka, M.; Mohr, A.; Abdollahi, A.; Weber, K.J.; et al. Metformin Enhanced in Vitro Radiosensitivity Associates with G2/M Cell Cycle Arrest and Elevated Adenosine-5′-monophosphate-activated Protein Kinase Levels in Glioblastoma. Radiol. Oncol. 2017, 51, 431–437. [Google Scholar] [CrossRef]
- Yang, S.R.; Wright, J.; Bauter, M.; Seweryniak, K.; Kode, A.; Rahman, I. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: Implications for chronic inflammation and aging. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2007, 292, L567–L576. [Google Scholar] [CrossRef]
- Stein, S.; Schafer, N.; Breitenstein, A.; Besler, C.; Winnik, S.; Lohmann, C.; Heinrich, K.; Brokopp, C.E.; Handschin, C.; Landmesser, U.; et al. SIRT1 reduces endothelial activation without affecting vascular function in ApoE-/- mice. Aging 2010, 2, 353–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, S.-W.; Liu, Y.-S.; Lu, D.-Y.; Tsai, C.-F. Melatonin Modulates the Microenvironment of Glioblastoma Multiforme by Targeting Sirtuin 1. Nutrients 2019, 11, 1343. https://doi.org/10.3390/nu11061343
Lai S-W, Liu Y-S, Lu D-Y, Tsai C-F. Melatonin Modulates the Microenvironment of Glioblastoma Multiforme by Targeting Sirtuin 1. Nutrients. 2019; 11(6):1343. https://doi.org/10.3390/nu11061343
Chicago/Turabian StyleLai, Sheng-Wei, Yu-Shu Liu, Dah-Yuu Lu, and Cheng-Fang Tsai. 2019. "Melatonin Modulates the Microenvironment of Glioblastoma Multiforme by Targeting Sirtuin 1" Nutrients 11, no. 6: 1343. https://doi.org/10.3390/nu11061343
APA StyleLai, S.-W., Liu, Y.-S., Lu, D.-Y., & Tsai, C.-F. (2019). Melatonin Modulates the Microenvironment of Glioblastoma Multiforme by Targeting Sirtuin 1. Nutrients, 11(6), 1343. https://doi.org/10.3390/nu11061343