Mitochondrial Structural Changes in the Pathogenesis of Diabetic Retinopathy

Abstract
:1. Introduction
2. Consequences of Mitochondrial Dysfunction in Microangiopathy
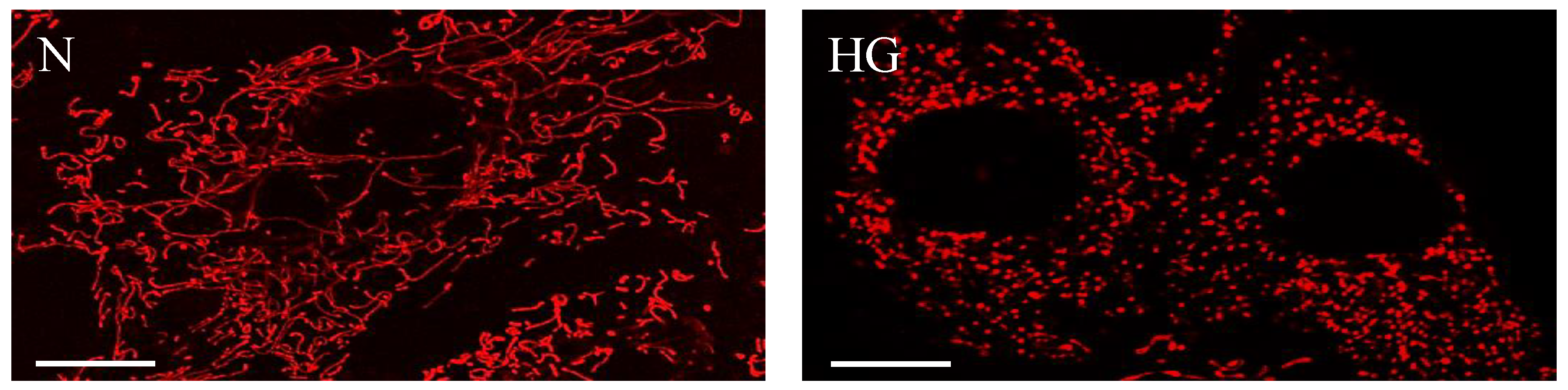
3. Changes in Mitochondrial Morphology in Diabetic Retinopathy
4. Alterations in Mitochondrial Structure: Fission, Fusion, and Fragmentation
5. HG-Induced Altered Mitochondrial Function Compromises Cellular Respiration
6. Mitochondrial Dysfunction-Mediated Apoptosis in Diabetic Retinopathy
7. Mitochondrial Cx43 (mtCx43) Abnormalities and Mitochondrial Fragmentation
8. Strategies Inhibiting Mitochondrial Fragmentation and Dysfunction
9. Conclusions
Funding
Conflicts of Interest
References
- Trudeau, K.; Molina, A.J.; Guo, W.; Roy, S. High glucose disrupts mitochondrial morphology in retinal endothelial cells: Implications for diabetic retinopathy. Am. J. Pathol. 2010, 177, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Trudeau, K.; Molina, A.J.; Roy, S. High glucose induces mitochondrial morphology and metabolic changes in retinal pericytes. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8657–8664. [Google Scholar] [CrossRef] [PubMed]
- Tien, T.; Zhang, J.; Muto, T.; Kim, D.; Sarthy, V.P.; Roy, S. High Glucose Induces Mitochondrial Dysfunction in Retinal Muller Cells: Implications for Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2915–2921. [Google Scholar] [CrossRef] [PubMed]
- Ayanga, B.A.; Badal, S.S.; Wang, Y.; Galvan, D.L.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Dynamin-Related Protein 1 Deficiency Improves Mitochondrial Fitness and Protects against Progression of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2733–2747. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef]
- Edwards, J.L.; Quattrini, A.; Lentz, S.I.; Figueroa-Romero, C.; Cerri, F.; Backus, C.; Hong, Y.; Feldman, E.L. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia 2010, 53, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xiang, H.; Chen, R.; Yang, J.; Yang, X.; Zhou, J.; Liu, H.; Zhao, S.; Xiao, J.; Chen, P.; et al. S1PR2 antagonist ameliorate high glucose-induced fission and dysfunction of mitochondria in HRGECs via regulating ROCK1. BMC Nephrol. 2019, 20, 135. [Google Scholar] [CrossRef]
- Choo, H.J.; Kim, J.H.; Kwon, O.B.; Lee, C.S.; Mun, J.Y.; Han, S.S.; Yoon, Y.S.; Yoon, G.; Choi, K.M.; Ko, Y.G. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 2006, 49, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Coughlan, M.T.; Nguyen, T.V.; Penfold, S.A.; Higgins, G.C.; Thallas-Bonke, V.; Tan, S.M.; Van Bergen, N.J.; Sourris, K.C.; Harcourt, B.E.; Thorburn, D.R.; et al. Mapping time-course mitochondrial adaptations in the kidney in experimental diabetes. Clin. Sci. (Lond.) 2016, 130, 711–720. [Google Scholar] [CrossRef]
- Dlaskova, A.; Spacek, T.; Santorova, J.; Plecita-Hlavata, L.; Berkova, Z.; Saudek, F.; Lessard, M.; Bewersdorf, J.; Jezek, P. 4Pi microscopy reveals an impaired three-dimensional mitochondrial network of pancreatic islet beta-cells, an experimental model of type-2 diabetes. Biochim. Biophys. Acta 2010, 1797, 1327–1341. [Google Scholar] [CrossRef]
- Harano, Y.; DePalma, R.G.; Miller, M. Fatty acid oxidation, citric acid cycle activity, and morphology of mitochondria in diabetic rat liver. Proc. Soc. Exp. Biol. Med. 1969, 131, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Jheng, H.F.; Tsai, P.J.; Guo, S.M.; Kuo, L.H.; Chang, C.S.; Su, I.J.; Chang, C.R.; Tsai, Y.S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [PubMed]
- Makino, A.; Scott, B.T.; Dillmann, W.H. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia 2010, 53, 1783–1794. [Google Scholar] [CrossRef] [Green Version]
- Molina, A.J.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; et al. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes 2009, 58, 2303–2315. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Tao, L.; Xiaohui, X.; Zelin, Z.; Jiangang, L.; Zhao, S.; Weikang, H.; Hongchao, X.; Qiujing, W.; Xin, L. Polydatin impairs mitochondria fitness and ameliorates podocyte injury by suppressing Drp1 expression. J. Cell Physiol. 2017, 232, 2776–2787. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, K.; Matsuura, T.; Narama, I. Histochemical and morphometrical analysis of skeletal muscle in spontaneous diabetic WBN/Kob rat. Acta Neuropathol. 2001, 102, 264–270. [Google Scholar]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Sheu, S.S.; Robotham, J.L.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008, 79, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Devi, T.S.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP regulates mitophagy in retinal Muller cells under high-glucose conditions: Implications for diabetic retinopathy. Cell Death Dis. 2017, 8, e2777. [Google Scholar] [CrossRef] [PubMed]
- Devi, T.S.; Yumnamcha, T.; Yao, F.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP mediates high glucose-induced mitophagic flux and lysosome enlargement in human retinal pigment epithelial cells. Biol. Open 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, H.Y.; Kang, J.M.; Jun, H.H.; Kim, D.J.; Park, S.H.; Sung, M.J.; Heo, J.H.; Yang, D.H.; Lee, S.H.; Lee, S.Y. Chloroquine and amodiaquine enhance AMPK phosphorylation and improve mitochondrial fragmentation in diabetic tubulopathy. Sci. Rep. 2018, 8, 8774. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef]
- Parone, P.A.; Da Cruz, S.; Tondera, D.; Mattenberger, Y.; James, D.I.; Maechler, P.; Barja, F.; Martinou, J.C. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE 2008, 3, e3257. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.C.; Coughlan, M.T. Mitochondrial dysfunction and mitophagy: The beginning and end to diabetic nephropathy? Br. J. Pharmacol. 2014, 171, 1917–1942. [Google Scholar] [CrossRef] [PubMed]
- Soleimanpour, S.A.; Ferrari, A.M.; Raum, J.C.; Groff, D.N.; Yang, J.; Kaufman, B.A.; Stoffers, D.A. Diabetes Susceptibility Genes Pdx1 and Clec16a Function in a Pathway Regulating Mitophagy in beta-Cells. Diabetes 2015, 64, 3475–3484. [Google Scholar] [CrossRef] [PubMed]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef] [PubMed]
- Mitra, K.; Wunder, C.; Roysam, B.; Lin, G.; Lippincott-Schwartz, J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. USA 2009, 106, 11960–11965. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, T.; Yamasaki, Y. Ultra-high-resolution scanning electron microscopy of mitochondria and sarcoplasmic reticulum arrangement in human red, white, and intermediate muscle fibers. Anat. Rec. 1997, 248, 214–223. [Google Scholar] [CrossRef]
- Vanhorebeek, I.; De Vos, R.; Mesotten, D.; Wouters, P.J.; De Wolf-Peeters, C.; Van den Berghe, G. Protection of hepatocyte mitochondrial ultrastructure and function by strict blood glucose control with insulin in critically ill patients. Lancet 2005, 365, 53–59. [Google Scholar] [CrossRef]
- Van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Elgass, K.; Pakay, J.; Ryan, M.T.; Palmer, C.S. Recent advances into the understanding of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 150–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppins, S.; Lackner, L.; Nunnari, J. The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 2007, 76, 751–780. [Google Scholar] [CrossRef]
- Lee, S.E.; Ma, W.; Rattigan, E.M.; Aleshin, A.; Chen, L.; Johnson, L.L.; D’Agati, V.D.; Schmidt, A.M.; Barile, G.R. Ultrastructural features of retinal capillary basement membrane thickening in diabetic swine. Ultrastruct. Pathol. 2010, 34, 35–41. [Google Scholar] [CrossRef]
- Chang, J.Y.; Yu, F.; Shi, L.; Ko, M.L.; Ko, G.Y. Melatonin Affects Mitochondrial Fission/Fusion Dynamics in the Diabetic Retina. J. Diabetes Res. 2019, 2019, 8463125. [Google Scholar] [CrossRef]
- Civitarese, A.E.; Ravussin, E. Mitochondrial energetics and insulin resistance. Endocrinology 2008, 149, 950–954. [Google Scholar] [CrossRef]
- Duraisamy, A.J.; Mohammad, G.; Kowluru, R.A. Mitochondrial fusion and maintenance of mitochondrial homeostasis in diabetic retinopathy. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Rovira-Llopis, S.; Banuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Caino, M.C. Mitochondrial Dynamics in Type 2 Diabetes and Cancer. Front. Endocrinol. (Lausanne) 2018, 9, 211. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, L.; Li, X.; Jiang, Z.; Sun, L.; Zhao, G.; Zhou, G.; Zhang, H.; Shang, J.; Wang, T. Mitochondrial fusion/fission process involved in the improvement of catalpol on high glucose-induced hepatic mitochondrial dysfunction. Acta Biochim. Biophys. Sin. (Shanghai) 2015, 47, 730–740. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Q.; Kowluru, R.A. Diabetic retinopathy and damage to mitochondrial structure and transport machinery. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8739–8746. [Google Scholar] [CrossRef]
- Paltauf-Doburzynska, J.; Malli, R.; Graier, W.F. Hyperglycemic conditions affect shape and Ca2+ homeostasis of mitochondria in endothelial cells. J. Cardiovasc. Pharmacol. 2004, 44, 423–436. [Google Scholar] [CrossRef]
- TeSlaa, T.; Teitell, M.A. Techniques to monitor glycolysis. Methods Enzymol. 2014, 542, 91–114. [Google Scholar] [CrossRef]
- Osorio-Paz, I.; Uribe-Carvajal, S.; Salceda, R. In the Early Stages of Diabetes, Rat Retinal Mitochondria Undergo Mild Uncoupling due to UCP2 Activity. PLoS ONE 2015, 10, e0122727. [Google Scholar] [CrossRef]
- Mandarino, L.J.; Finlayson, J.; Hassell, J.R. High glucose downregulates glucose transport activity in retinal capillary pericytes but not endothelial cells. Investig. Ophthalmol. Vis. Sci. 1994, 35, 964–972. [Google Scholar]
- Jacobson, M.D.; Burne, J.F.; Raff, M.C. Programmed cell death and Bcl-2 protection in the absence of a nucleus. EMBO J. 1994, 13, 1899–1910. [Google Scholar] [CrossRef]
- Hockenbery, D.; Nunez, G.; Milliman, C.; Schreiber, R.D.; Korsmeyer, S.J. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 1990, 348, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Petit, P.; Zamzami, N.; Vayssiere, J.L.; Mignotte, B. The biochemistry of programmed cell death. FASEB J. 1995, 9, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef]
- Estaquier, J.; Arnoult, D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007, 14, 1086–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boengler, K.; Dodoni, G.; Rodriguez-Sinovas, A.; Cabestrero, A.; Ruiz-Meana, M.; Gres, P.; Konietzka, I.; Lopez-Iglesias, C.; Garcia-Dorado, D.; Di Lisa, F.; et al. Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc. Res. 2005, 67, 234–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boengler, K.; Schulz, R. Connexin 43 and Mitochondria in Cardiovascular Health and Disease. Adv. Exp. Med. Biol. 2017, 982, 227–246. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Stahlhofen, S.; van de Sand, A.; Gres, P.; Ruiz-Meana, M.; Garcia-Dorado, D.; Heusch, G.; Schulz, R. Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res. Cardiol. 2009, 104, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Gadicherla, A.K.; Wang, N.; Bulic, M.; Agullo-Pascual, E.; Lissoni, A.; De Smet, M.; Delmar, M.; Bultynck, G.; Krysko, D.V.; Camara, A.; et al. Mitochondrial Cx43 hemichannels contribute to mitochondrial calcium entry and cell death in the heart. Basic Res. Cardiol. 2017, 112, 27. [Google Scholar] [CrossRef]
- Garcia-Dorado, D.; Ruiz-Meana, M.; Rodriguez-Sinovas, A. Connexin 43 phosphorylation in subsarcolemmal mitochondria: A general cardioprotective signal targeted by fibroblast growth factor-2? Cardiovasc. Res. 2014, 103, 1–2. [Google Scholar] [CrossRef]
- Goubaeva, F.; Mikami, M.; Giardina, S.; Ding, B.; Abe, J.; Yang, J. Cardiac mitochondrial connexin 43 regulates apoptosis. Biochem. Biophys. Res. Commun. 2007, 352, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.N.; Kwon, H.J.; Im, S.W.; Son, Y.H.; Akindehin, S.; Jung, Y.S.; Lee, S.J.; Rhyu, I.J.; Kim, I.Y.; Seong, J.K.; et al. Connexin 43 is required for the maintenance of mitochondrial integrity in brown adipose tissue. Sci. Rep. 2017, 7, 7159. [Google Scholar] [CrossRef] [PubMed]
- Kirca, M.; Kleinbongard, P.; Soetkamp, D.; Heger, J.; Csonka, C.; Ferdinandy, P.; Schulz, R. Interaction between connexin 43 and nitric oxide synthase in mice heart mitochondria. J. Cell Mol. Med. 2015, 19, 815–825. [Google Scholar] [CrossRef]
- Lu, G.; Haider, H.; Porollo, A.; Ashraf, M. Mitochondria-specific transgenic overexpression of connexin-43 simulates preconditioning-induced cytoprotection of stem cells. Cardiovasc. Res. 2010, 88, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecoraro, M.; Pinto, A.; Popolo, A. Inhibition of Connexin 43 translocation on mitochondria accelerates CoCl2-induced apoptotic response in a chemical model of hypoxia. Toxicol. In Vitro 2018, 47, 120–128. [Google Scholar] [CrossRef]
- Pecoraro, M.; Sorrentino, R.; Franceschelli, S.; Del Pizzo, M.; Pinto, A.; Popolo, A. Doxorubicin-Mediated Cardiotoxicity: Role of Mitochondrial Connexin 43. Cardiovasc. Toxicol. 2015, 15, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Sinovas, A.; Boengler, K.; Cabestrero, A.; Gres, P.; Morente, M.; Ruiz-Meana, M.; Konietzka, I.; Miro, E.; Totzeck, A.; Heusch, G.; et al. Translocation of connexin 43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein 90-dependent TOM pathway and its importance for cardioprotection. Circ. Res. 2006, 99, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Sinovas, A.; Ruiz-Meana, M.; Denuc, A.; Garcia-Dorado, D. Mitochondrial Cx43, an important component of cardiac preconditioning. Biochim. Biophys. Acta Biomembr. 2018, 1860, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Meana, M.; Rodriguez-Sinovas, A.; Cabestrero, A.; Boengler, K.; Heusch, G.; Garcia-Dorado, D. Mitochondrial connexin43 as a new player in the pathophysiology of myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2008, 77, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Srisakuldee, W.; Makazan, Z.; Nickel, B.E.; Zhang, F.; Thliveris, J.A.; Pasumarthi, K.B.; Kardami, E. The FGF-2-triggered protection of cardiac subsarcolemmal mitochondria from calcium overload is mitochondrial connexin 43-dependent. Cardiovasc. Res. 2014, 103, 72–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trudeau, K.; Muto, T.; Roy, S. Downregulation of mitochondrial connexin 43 by high glucose triggers mitochondrial shape change and cytochrome C release in retinal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6675–6681. [Google Scholar] [CrossRef] [PubMed]
- Wasilewski, M.; Scorrano, L. The changing shape of mitochondrial apoptosis. Trends Endocrinol. Met. 2009, 20, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Sharma, S. Dynamin-related protein-1 as potential therapeutic target in various diseases. Inflammopharmacology 2017, 25, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Inhibitors of mitochondrial fission as a therapeutic strategy for diseases with oxidative stress and mitochondrial dysfunction. J. Alzheimers Dis. 2014, 40, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Senyilmaz-Tiebe, D.; Pfaff, D.H.; Virtue, S.; Schwarz, K.V.; Fleming, T.; Altamura, S.; Muckenthaler, M.U.; Okun, J.G.; Vidal-Puig, A.; Nawroth, P.; et al. Dietary stearic acid regulates mitochondria in vivo in humans. Nat. Commun. 2018, 9, 3129. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Tissue Type | Cell Type | Species | Changes in Mitochondrial Morphology | References |
|---|---|---|---|---|
| Retina | Retinal endothelial cells | Rat | Fragmentation | [1] |
| Retinal pericytes | Bovine | Fragmentation | [2] | |
| Retinal Müller cells | Rat | Fragmentation | [3] | |
| Retinal Müller cells | Human | Fragmentation | [21] | |
| Retinal pigmented epithelial cells | Human | Fragmentation | [22] | |
| Kidney | Renal glomerular cells | Human | Fragmentation | [7] |
| Proximal tubule epithelial cells | Rat | Fragmentation | [9] | |
| Proximal tubule epithelial cells | Human | Fragmentation | [23] | |
| Podocytes | Mouse | Fragmentation | [16] | |
| Liver | Hepatocytes | Rat | Increased mitochondria size and density | [11] |
| Epithelial cells | Rat | Fragmentation | [19,20] | |
| Heart | Coronary endothelial cells | Mouse | Fragmentation | [14] |
| Myoblasts | Rat | Fragmentation | [19] | |
| Ventricular myocytes | Rat | Fragmentation | [20] | |
| Aortic endothelial cells | Bovine | Fragmentation | [20] | |
| Aortic smooth muscle cells | Mouse | Fragmentation | [20] | |
| Aortic endothelial cells | Human | Fragmentation | [18] | |
| Venous endothelial cells | Human | Fragmentation | [18] | |
| Pancreas | β-islet cells | Rat | Fragmentation | [10] |
| β-islet cells | Mouse | Fragmentation | [15] | |
| Fat | Adipocytes | Mouse | Decreased mitochondria size | [8] |
| Skeletal Muscle | Myoblast | Mouse | Fragmentation | [12] |
| Skeletal muscle cells | Human | Decreased mitochondria size and larger vacuoles | [13] | |
| Skeletal muscle cells | Mouse | Swelling and lysis of mitochondrial cristae | [17] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, S.; Kim, D.; Sankaramoorthy, A. Mitochondrial Structural Changes in the Pathogenesis of Diabetic Retinopathy. J. Clin. Med. 2019, 8, 1363. https://doi.org/10.3390/jcm8091363
Roy S, Kim D, Sankaramoorthy A. Mitochondrial Structural Changes in the Pathogenesis of Diabetic Retinopathy. Journal of Clinical Medicine. 2019; 8(9):1363. https://doi.org/10.3390/jcm8091363
Chicago/Turabian StyleRoy, Sayon, Dongjoon Kim, and Aravind Sankaramoorthy. 2019. "Mitochondrial Structural Changes in the Pathogenesis of Diabetic Retinopathy" Journal of Clinical Medicine 8, no. 9: 1363. https://doi.org/10.3390/jcm8091363
APA StyleRoy, S., Kim, D., & Sankaramoorthy, A. (2019). Mitochondrial Structural Changes in the Pathogenesis of Diabetic Retinopathy. Journal of Clinical Medicine, 8(9), 1363. https://doi.org/10.3390/jcm8091363