Obesity and Lifespan Health—Importance of the Fetal Environment

Abstract
:1. Introduction
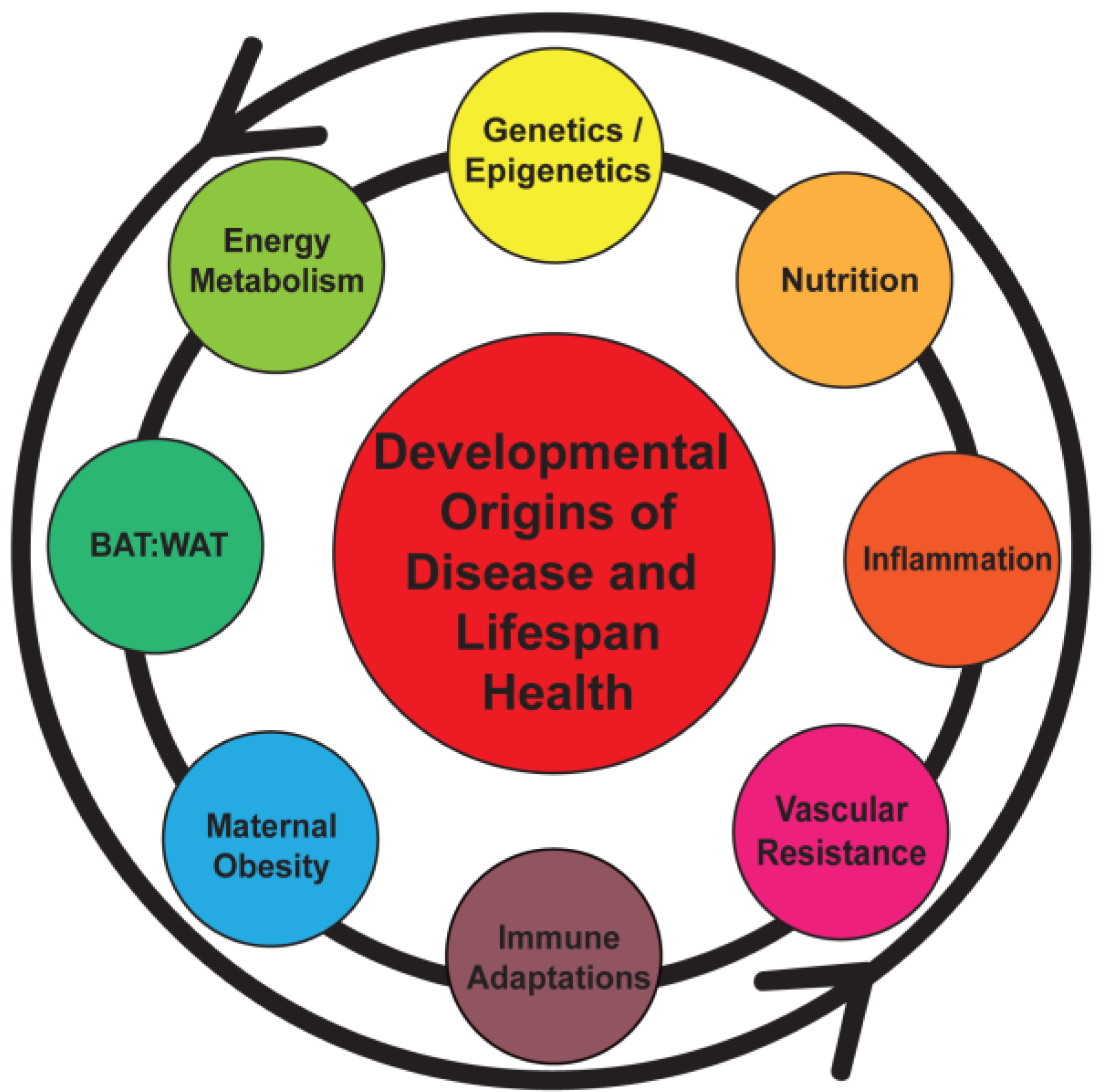
2. Pregnancy and Maternal Obesity

{kind=link}
{kind=link}
| Mechanisms | References |
|---|---|
| Endocrine changes | [20,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36] |
| Epigenetic modifications | [17,18,19,20,21] |
| Differential development of brown and white adipose tissue | [37,38,39,40,41,42] |
| Increased inflammatory burden | [22,23,29,31] |
| Immune system adaptations | [34,43] |
| Changes in vascular resistance and development | [22,29,32,44,45] |
| Ectopic fat accumulation | [28,46,47,48,49] |
| Nutritional modifications (e.g., fructose intake) | [30,50,51,52,53] |
| Energy metabolism | [54,55,56] |
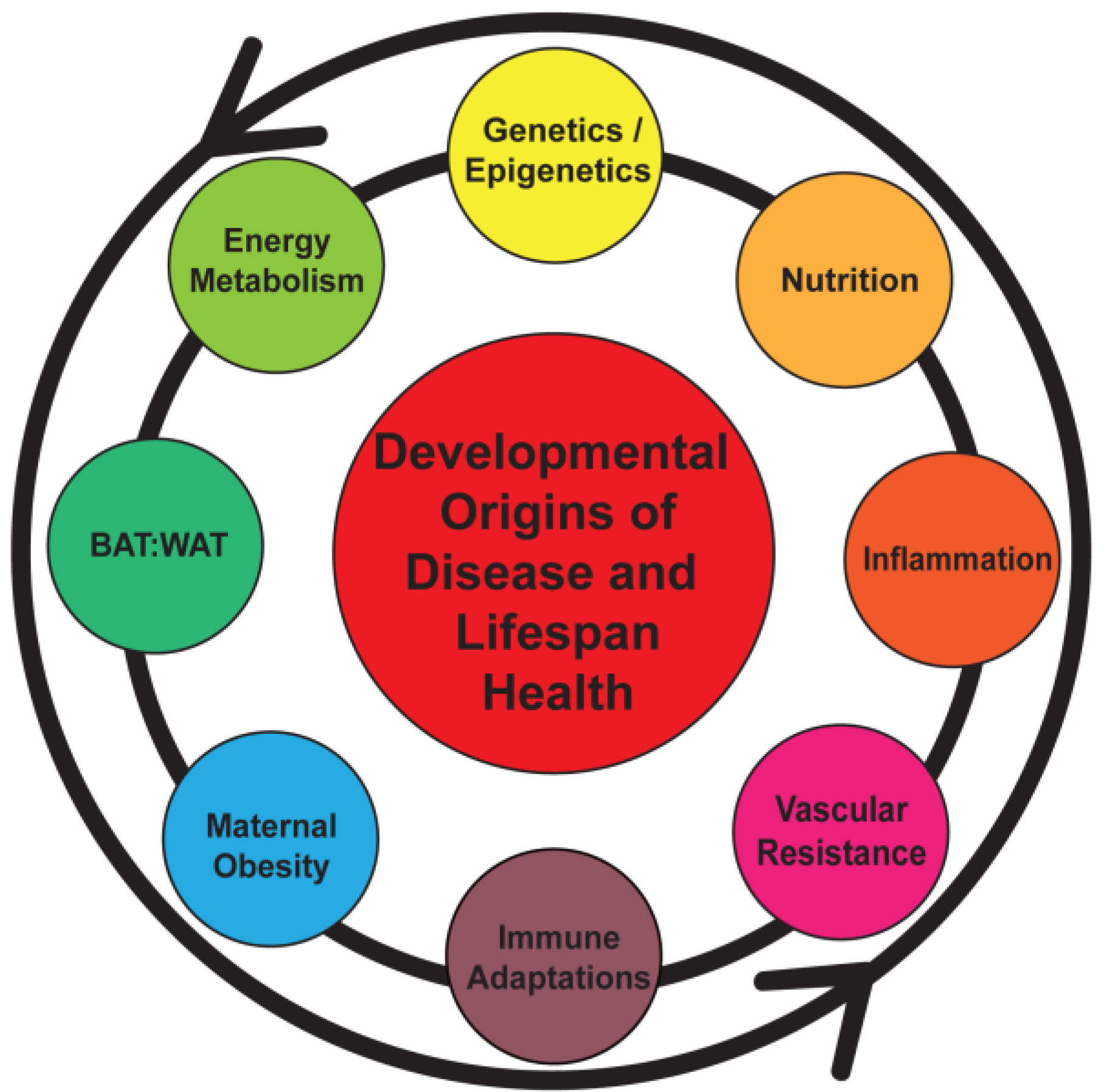
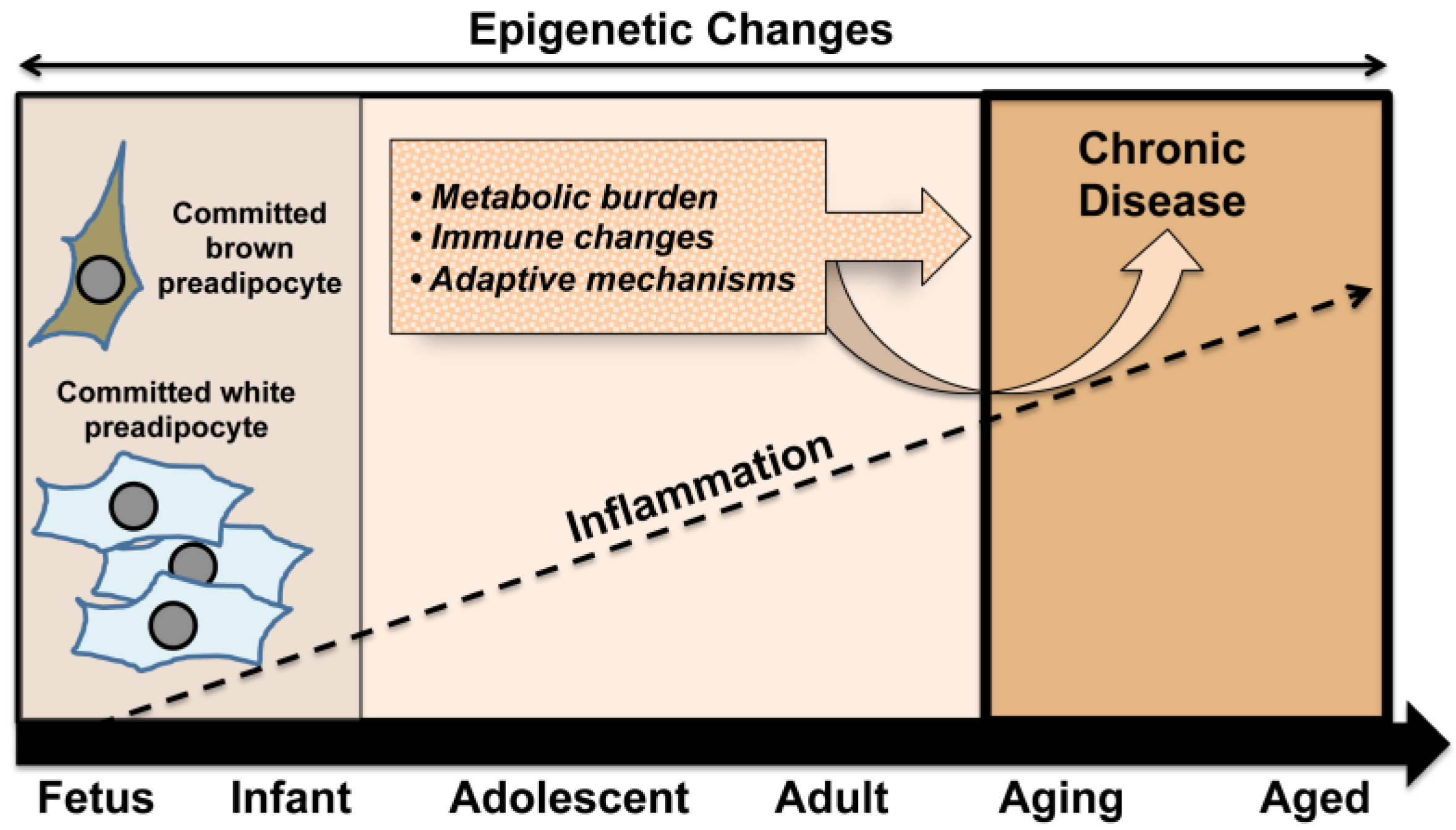
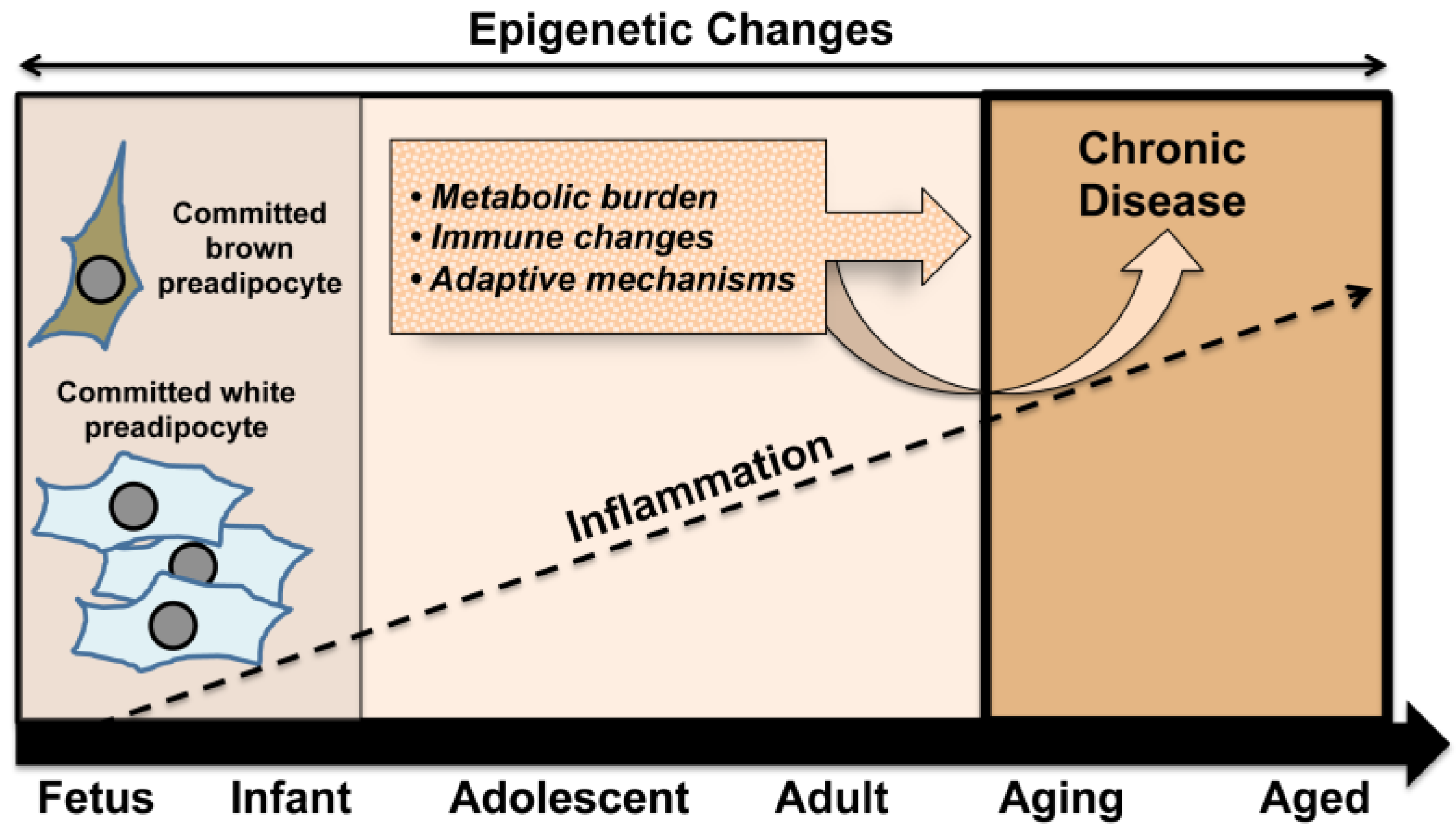
3. Obesity, Chronic Inflammation, and Pregnancy
4. Developmental Adipogenesis and Key Precursors
5. Relationship between BAT and WAT
6. Fetal Environment, Energy Metabolism, and Adiposity
7. Conclusions
Acknowledgements
Conflicts of Interest
References
- Hung, W.W.; Ross, J.S.; Boockvar, K.S.; Siu, A.L. Recent trends in chronic disease, impairment and disability among older adults in the United States. BMC Geriatr. 2011, 11, 47–59. [Google Scholar] [CrossRef]
- Sun, S.S.; Liang, R.; Huang, T.T.; Daniels, S.R.; Arsianian, S.; Liu, K.; Grave, G.D.; Siervogel, R.M. Childhood obesity predicts adult metabolic syndrome: The Fels Longitudinal Study. J. Pediatr. 2008, 152, 191–200. [Google Scholar] [CrossRef]
- Dhuper, S.; Buddhe, S.; Patel, S. Managing cardiovascular risk in overweight children and adolescents. Paediatr. Drugs 2013, 15, 181–190. [Google Scholar] [CrossRef]
- Barker, D.J. In utero programming of cardiovascular disease. Theriogenology 2000, 53, 555–574. [Google Scholar] [CrossRef]
- Calkins, K.; Devaskar, S.U. Fetal origins of adult disease. Curr. Probl. Pediatr. Adolesc. Health Care 2011, 41, 158–176. [Google Scholar] [CrossRef]
- Langley-Evans, S.C.; McMullen, S. Developmental origins of adult disease. Med. Princ. Pract. 2010, 19, 87–98. [Google Scholar] [CrossRef]
- Nicoletto, S.F.; Rinaldi, A. In the womb’s shadow. EMBO Rep. 2011, 12, 30–34. [Google Scholar] [CrossRef]
- Van Cleave, J.; Gortmaker, S.L.; Perrin, J.M. Dynamics of obesity and chronic health conditions among children and youth. JAMA 2010, 303, 623–630. [Google Scholar] [CrossRef]
- Desai, M.; Beall, M.; Ross, M.G. Developmental origins of obesity: Programmed adipogenesis. Curr. Diab. Rep. 2013, 13, 27–33. [Google Scholar] [CrossRef]
- Laitinen, J.; Jääskeläinen, A.; Hartikainen, A.L.; Sovio, U.; Vääräsmäki, M.; Pouta, A.; Kaakinen, M.; Järvelin, M.R. Maternal weight gain during the first half of pregnancy and offspring obesity at 16 years: A prospective cohort study. BJOG 2012, 119, 716–723. [Google Scholar] [CrossRef]
- Parlee, S.D.; Macdougald, O.A. Maternal nutrition and risk of obesity in offspring: The Trojan Horse of developmental plasticity. Biochim. Biophys. Acta 2014, 1842, 495–506. [Google Scholar] [CrossRef]
- Hanson, M.A.; Gluckman, P.D. Developmental origins of health and disease: Moving from biological concepts to interventions and policy. Int. J. Gynaecol. Obstet. 2011, 115, S3–S5. [Google Scholar] [CrossRef]
- Martin-Gronert, M.S.; Ozanne, S.E. Mechanisms underlying the developmental origins of disease. Rev. Endocr. Metab. Disord. 2012, 3, 85–92. [Google Scholar] [CrossRef]
- Schmatz, M.; Madan, J.; Marino, T.; Davis, J. Maternal obesity: The interplay between inflammation, mother and fetus. J. Perinatol. 2010, 30, 441–446. [Google Scholar] [CrossRef]
- Anuurad, E.; Enkhmaa, B.; Gungor, Z.; Zhang, W.; Tracy, R.P.; Pearson, T.A.; Kim, K.; Berglund, L. Age as a modulator of inflammatory cardiovascular risk factors. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2151–2156. [Google Scholar] [CrossRef]
- Kanterman, J.; Sade-Feldman, M.; Baniyash, M. New insights into chronic inflammation-induced immunosuppression. Semin. Cancer Biol. 2012, 22, 307–318. [Google Scholar] [CrossRef]
- Martínez, J.A.; Cordero, P.; Campión, J.; Milagro, F.I. Interplay of early-life nutritional programming on obesity, inflammation, and epigenetic outcomes. Proc. Nutr. Soc. 2012, 6, 1–8. [Google Scholar]
- Hochberg, Z.; Feil, R.; Constancia, M.; Fraga, M.; Junien, C.; Carel, J.C.; Boileau, P.; Le Bouc, Y.; Deal, C.L.; Lillycrop, K.; et al. Child health, developmental plasticity, and epigenetic programming. Endocr. Rev. 2011, 32, 159–224. [Google Scholar] [CrossRef]
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef]
- Khulan, B.; Drake, A.J. Glucocorticoids as mediators of developmental programming effects. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 689–700. [Google Scholar] [CrossRef]
- Mulligan, C.J.; D’Errico, N.C.; Stees, J.; Hughes, D.A. Methylation changes at NR3C1 in newborns associate with maternal prenatal stress exposure and newborn birth weight. Epigenetics 2012, 7, 853–857. [Google Scholar] [CrossRef]
- Denison, F.C.; Roberts, K.A.; Marr, S.M.; Norman, J.E. Obesity, pregnancy, inflammation, and vascular function. Reproduction 2010, 140, 373–385. [Google Scholar] [CrossRef]
- Basu, S.; Haghiac, M.; Surace, P.; Challier, J.-C.; Guerre-Millo, M.; Singh, K.; Waters, T.; Minium, J.; Presley, L.; Catalano, P.M.; Haughuel-de Mouzon, S. Pregravid obesity associated with increased maternal endotoxemia and metabolic inflammation. Obesity 2011, 19, 476–482. [Google Scholar] [CrossRef]
- Catalano, P.M. Obesity, insulin resistance, and pregnancy outcome. Reproduction 2010, 140, 365–371. [Google Scholar] [CrossRef]
- Catalano, P.M.; Hauguel-De Mouzon, S. Is it time to revisit the Pedersen hypothesis in the face of the obesity epidemic? Am. J. Obstet. Gynecol. 2011, 204, 479–487. [Google Scholar] [CrossRef]
- Djiane, J.; Attig, L. Role of leptin during perinatal metabolic programming and obesity. J. Physiol. Pharmacol. 2008, 59, 55–63. [Google Scholar]
- Drake, A.J.; Reynolds, R.M. Impact of maternal obesity on offspring obesity and cardiometabolic disease risk. Reproduction 2010, 140, 387–398. [Google Scholar] [CrossRef]
- Grant, W.F.; Gillingham, M.B.; Batra, A.K.; Fewkes, N.M.; Comstock, S.M.; Takahashi, D.; Braun, T.P.; Glrove, K.L.; Friedman, J.E.; Marks, D.L. Maternal high fat diet is associated with decreased plasma n-3 fatty acids and fetal hepatic apoptosis in nonhuman primates. PLoS One 2011, 6, e17261. [Google Scholar] [CrossRef]
- Jarvie, E.; Hauguel-De-Mouzon, S.; Nelson, S.M.; Sattar, N.; Catalano, P.M.; Freeman, D.J. Lipotoxicity in obese pregnancy and its potential role in adverse pregnancy outcome and obesity in the offspring. Clin. Sci. 2010, 119, 123–129. [Google Scholar] [CrossRef]
- McCurdy, C.E.; Bishop, J.M.; Williams, S.M.; Grayson, B.E.; Smith, M.S.; Friedman, J.E.; Grove, K.L. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J. Clin. Invest. 2009, 119, 323–335. [Google Scholar]
- Shankar, K.; Zhong, Y.; Kang, P.; Lau, F.; Blackburn, M.L.; Chen, J.R.; Borengasser, S.J.; Ronis, M.J.; Badger, T.M. Maternal obesity promotes a proinflammatory signature in rat uterus and blastocyst. Endocrinology 2011, 152, 4158–4170. [Google Scholar] [CrossRef]
- Thompson, J.A.; Regnault, T.R. In utero origins of adult insulin resistance and vascular dysfunction. Semin. Reprod. Med. 2011, 29, 211–224. [Google Scholar] [CrossRef]
- Tounian, P. Programming towards childhood obesity. Ann. Nutr. Metab. 2011, 58, 30–41. [Google Scholar] [CrossRef]
- Williams, C.L.; Teeling, J.L.; Fleming, T.P. Mouse maternal systemic inflammation at the zygote stage causes blunted cytokine responsiveness in lipopolysaccharide-challenged adult offspring. BMC Biol. 2011, 9, 49. [Google Scholar] [CrossRef]
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of adipose tissue: An endocrine organ. Arch. Med. Sci. 2013, 9, 191–200. [Google Scholar]
- Balistreri, C.R.; Caruso, C.; Candore, G. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediators Inflamm. 2010, 2010, 802078. [Google Scholar] [CrossRef]
- Drubach, L.A.; Palmer, E.L., 3rd; Connolly, L.P.; Baker, A.; Zurakowski, D.; Cypess, A.M. Pediatric brown adipose tissue: Detection, epidemiology, and differences from adults. J. Pediatr. 2011, 159, 939–944. [Google Scholar]
- Billon, N.; Dani, C. Developmental origins of the adipocyte lineage: New insights from genetics and genomics studies. Stem Cell Rev. 2012, 8, 55–56. [Google Scholar] [CrossRef]
- Cawthorn, W.P.; Scheller, E.L.; MacDougald, O.A. Adipose tissue stem cells meet preadipocyte commitment: Going back to the future. J. Lipid Res. 2012, 53, 227–246. [Google Scholar] [CrossRef]
- Seale, P.; Kajimura, S.; Spiegelman, B.M. Transcriptional control of brown adipocyte development and physiological function—Of mice and men. Genes Dev. 2009, 23, 788–797. [Google Scholar] [CrossRef]
- Hu, H.H.; Tovar, J.P.; Pavlova, Z.; Smith, M.L.; Gilsanz, V. Unequivocal identification of brown adipose tissue in a human infant. J. Magn. Reson. Imaging 2012, 35, 938–942. [Google Scholar] [CrossRef]
- Yoneshiro, T.; Aita, S.; Matsushita, M.; Okamatsu-Ogura, Y.; Kameya, T.; Kawai, Y.; Miyagawa, M.; Tsujisaki, M.; Saito, M. Age-related decrease in cold-activated brown adipose tissue and accumulation of body fat in healthy humans. Obesity 2011, 19, 1755–1760. [Google Scholar] [CrossRef]
- Bories, G.; Caiazzo, R.; Derudas, B.; Copin, C.; Raverdy, V.; Pigeyre, M.; Pattou, F.; Staels, B.; Chinetti-Gbaguidi, G. Impaired alternative macrophage differentiation of peripheral blood mononuclear cells from obese subjects. Diab. Vasc. Dis. Res. 2012, 9, 189–195. [Google Scholar] [CrossRef]
- Acosta, J.C.; Hasas, D.M.; Saha, C.K.; Dimeglio, L.A.; Ingram, D.A.; Haneline, L.S. Gestational diabetes mellitus alters maternal and neonatal circulating endothelial progenitor cell subsets. Am. J. Obstet. Gynecol. 2011, 204, 254.e8–254.e15. [Google Scholar]
- Gu, P.; Xu, A. Interplay between adipose tissue and blood vessels in obesity and vascular dysfunction. Rev. Endocr. Metab. Disord. 2013, 14, 49–58. [Google Scholar] [CrossRef]
- Dey, D.; Wong, N.D.; Tamarappoo, B.; Nakazato, R.; Gransar, H.; Cheng, V.Y.; Ramesh, A.; Kakadiaris, I.; Germano, G.; Slomka, P.J.; Berman, D.A. Computer-aided non-contrast CT-based quantification of pericardial and thoracic fat and their associations with coronary calcium and metabolic syndrome. Atherosclerosis 2010, 209, 136–141. [Google Scholar] [CrossRef]
- Britton, K.A.; Fox, C.S. Ectopic fat depots and cardiovascular disease. Circulation 2011, 124, e837–e841. [Google Scholar] [CrossRef]
- Power, M.L.; Shulkin, J. Maternal obesity, metabolic disease, and allostatic load. Physiol. Behav. 2012, 106, 22–28. [Google Scholar] [CrossRef]
- Szendroedi, J.; Roden, M. Ectopic lipids and organ function. Curr. Opin. Lipidol. 2009, 20, 50–56. [Google Scholar] [CrossRef]
- Larqué, E.; Ruiz-Palacios, M.; Koletzko, B. Placental regulation of fetal nutrient supply. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 292–297. [Google Scholar] [CrossRef]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.; McGahan, J.P.; Seibert, A.; et al. Effects of consuming fructose- or glucose-sweetened beverages for 10 weeks on lipids, insulin sensitivity and adiposity. J. Clin. Invest. 2009, 119, 1322–1334. [Google Scholar] [CrossRef]
- Dekker, M.J.; Su, Q.; Baker, C.; Rutledge, A.C.; Adeli, K. Fructose, a highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E685–E694. [Google Scholar] [CrossRef]
- Stanhope, K.L.; Schwarz, J.M.; Havel, P.J. Adverse metabolic effects of dietary fructose: Results from the recent epidemiological, clinical, and mechanistic studies. Curr. Opin. Lipidol. 2013, 24, 198–206. [Google Scholar] [CrossRef]
- Coletta, D.K.; Mandarino, L.J. Mitochondrial dysfunction and insulin resistance from the outside in: extracellular matrix, the cytoskeleton, and mitochondria. Am. J. Phys. Endocrinol. Metab. 2011, 301, E749–E755. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Invest. 2013, 123, 951–957. [Google Scholar] [CrossRef]
- Herrera, E.; Amusquivar, E. Lipid metabolism in the fetus and newborn. Diab. Metab. Res. Rev. 2000, 16, 202–210. [Google Scholar] [CrossRef]
- Dattilo, A.M.; Birch, L.; Krebs, N.F.; Lake, A.; Taveras, E.M.; Saavedra, J.M. Need for early interventions in the prevention of pediatric overweight: A review and upcoming directions. J. Obes. 2012, 2012, 123023. [Google Scholar] [CrossRef]
- Hult, M.; Tornhammar, P.; Ueda, P.; Chima, C.; Bonamy, A.K.; Ozumba, B.; Norman, M. Hypertension, diabetes and overweight: Looming legacies of the Biafran famine. PLoS One 2010, 5, e13582. [Google Scholar]
- Van Abeelen, A.F.; Elias, S.G.; Bossuyt, P.M.; Grobbee, D.E.; van der Schouw, Y.T.; Roseboom, T.J.; Uiterwaal, C.S. Cardiovascular consequences of famine in the young. Eur. Heart J. 2012, 33, 538–545. [Google Scholar] [CrossRef]
- Anuurad, E.; Ozturk, Z.; Enkhmaa, B.; Pearson, T.A.; Berglund, L. Association of lipoprotein-associated phospholipase A2 with coronary artery disease in African-Americans and Caucasians. J. Clin. Endocrinol. Metab. 2010, 95, 2376–2383. [Google Scholar] [CrossRef]
- Anuurad, E.; Chiem, A.; Pearson, T.A.; Berglund, L. Metabolic syndrome components in African-American and European-American patients and its relation to coronary artery disease. Am. J. Cardiol. 2007, 100, 830–834. [Google Scholar] [CrossRef]
- Poissonnet, C.M.; Burdi, A.R.; Garn, S.M. The chronology of adipose tissue appearance and distribution in the human fetus. Early Hum. Dev. 1984, 10, 1–11. [Google Scholar] [CrossRef]
- Poissonnet, C.M.; LaVelle, M.; Burdi, A.R. Growth and development of adipose. J. Pediatr. 1988, 113, 1–9. [Google Scholar] [CrossRef]
- Symonds, M.E.; Budge, H.; Perkins, A.C.; Lomax, M.A. Adipose tissue development-impact of the early life environment. Prog. Biophys. Mol. Biol. 2011, 106, 300–306. [Google Scholar] [CrossRef]
- Grün, F.; Blumberg, B. Minireview: The case for obesogens. Mol. Endocrinol. 2009, 23, 1127–1134. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tarantal, A.F.; Berglund, L. Obesity and Lifespan Health—Importance of the Fetal Environment. Nutrients 2014, 6, 1725-1736. https://doi.org/10.3390/nu6041725
Tarantal AF, Berglund L. Obesity and Lifespan Health—Importance of the Fetal Environment. Nutrients. 2014; 6(4):1725-1736. https://doi.org/10.3390/nu6041725
Chicago/Turabian StyleTarantal, Alice F., and Lars Berglund. 2014. "Obesity and Lifespan Health—Importance of the Fetal Environment" Nutrients 6, no. 4: 1725-1736. https://doi.org/10.3390/nu6041725
APA StyleTarantal, A. F., & Berglund, L. (2014). Obesity and Lifespan Health—Importance of the Fetal Environment. Nutrients, 6(4), 1725-1736. https://doi.org/10.3390/nu6041725