CMH-Small Molecule Docks into SIRT1, Elicits Human IPF-Lung Fibroblast Cell Death, Inhibits Ku70-deacetylation, FLIP and Experimental Pulmonary Fibrosis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Lung Myofibroblasts
2.2. Animals
2.3. Oropharyngeal Aspiration (OA) and Induction of Lung Fibrosis in Mice
2.4. Isolation of Mouse Lung Myofibroblasts
2.5. Cell Death and Apoptosis
2.6. Immunohistochemistry (IHC) Staining of Lung Tissue Sections
2.7. FLIP Protein in Lung Myofibroblasts
2.8. Immunoprecipitation and Immunoblotting
2.9. Docking CMH into SIRT1
2.10. Data Analysis and Statistics
3. Results
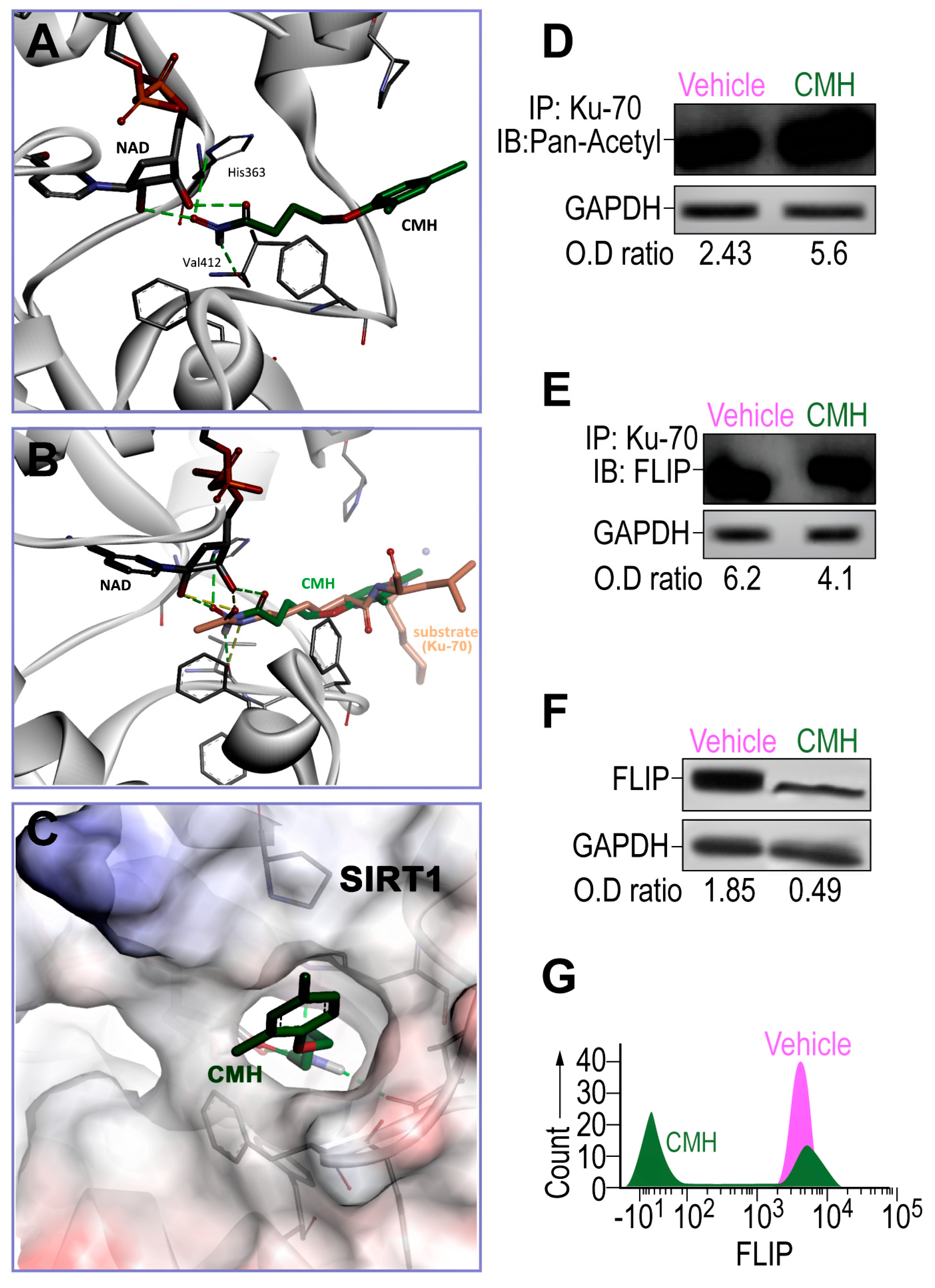
3.1. CMH Docks into SIRT1 Lysine Binding Site and Inhibits SIRT1 Activity of Ku70 Deacetylation, Destabilizes Ku70/FLIP Complex, and FLIP in IPF-Lung Myofibroblasts
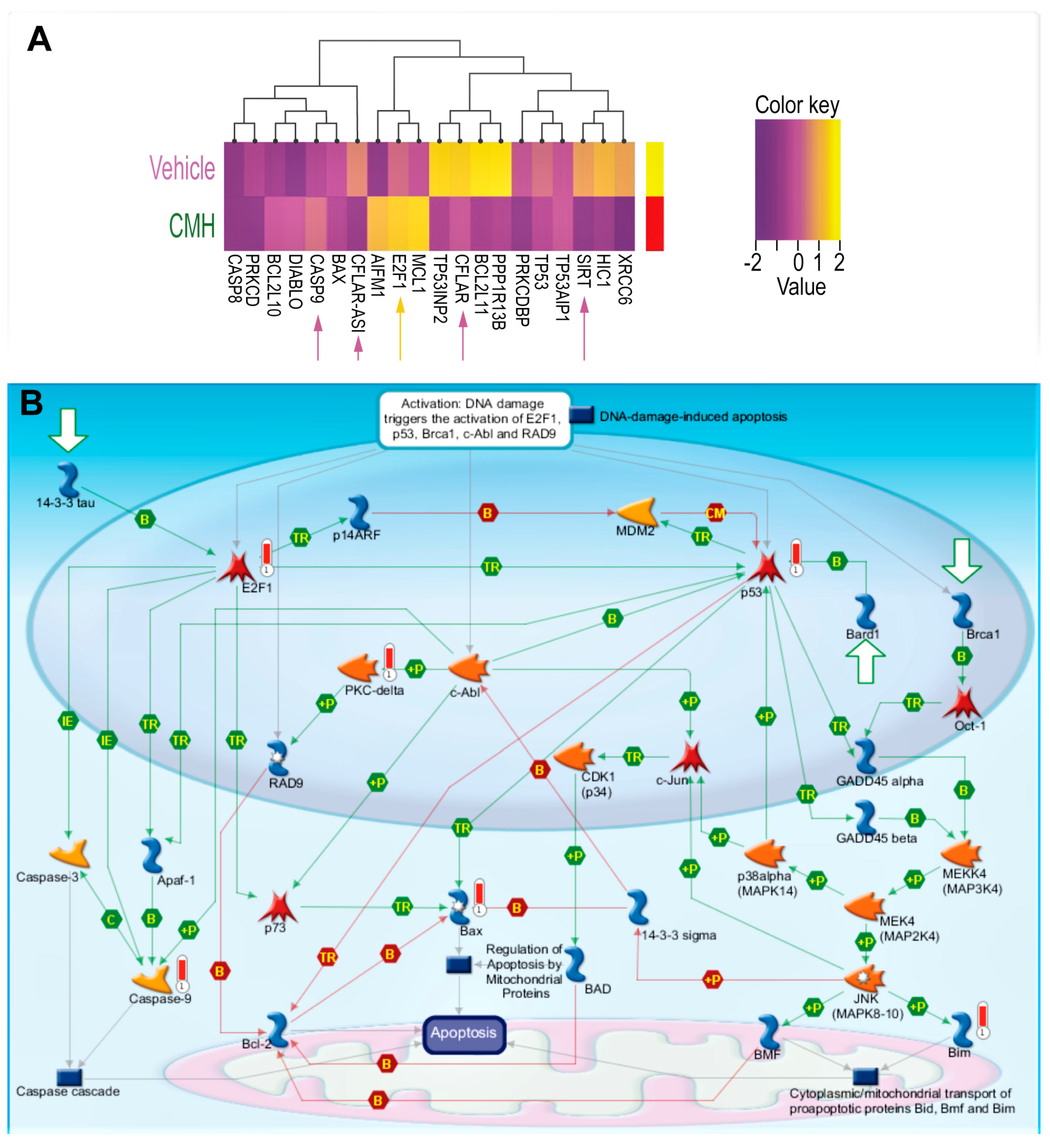
3.2. CMH Stimulates Apoptosis Pathways Regulated by SIRT1/Ku70 in Human IPF-Lung Myofibroblasts
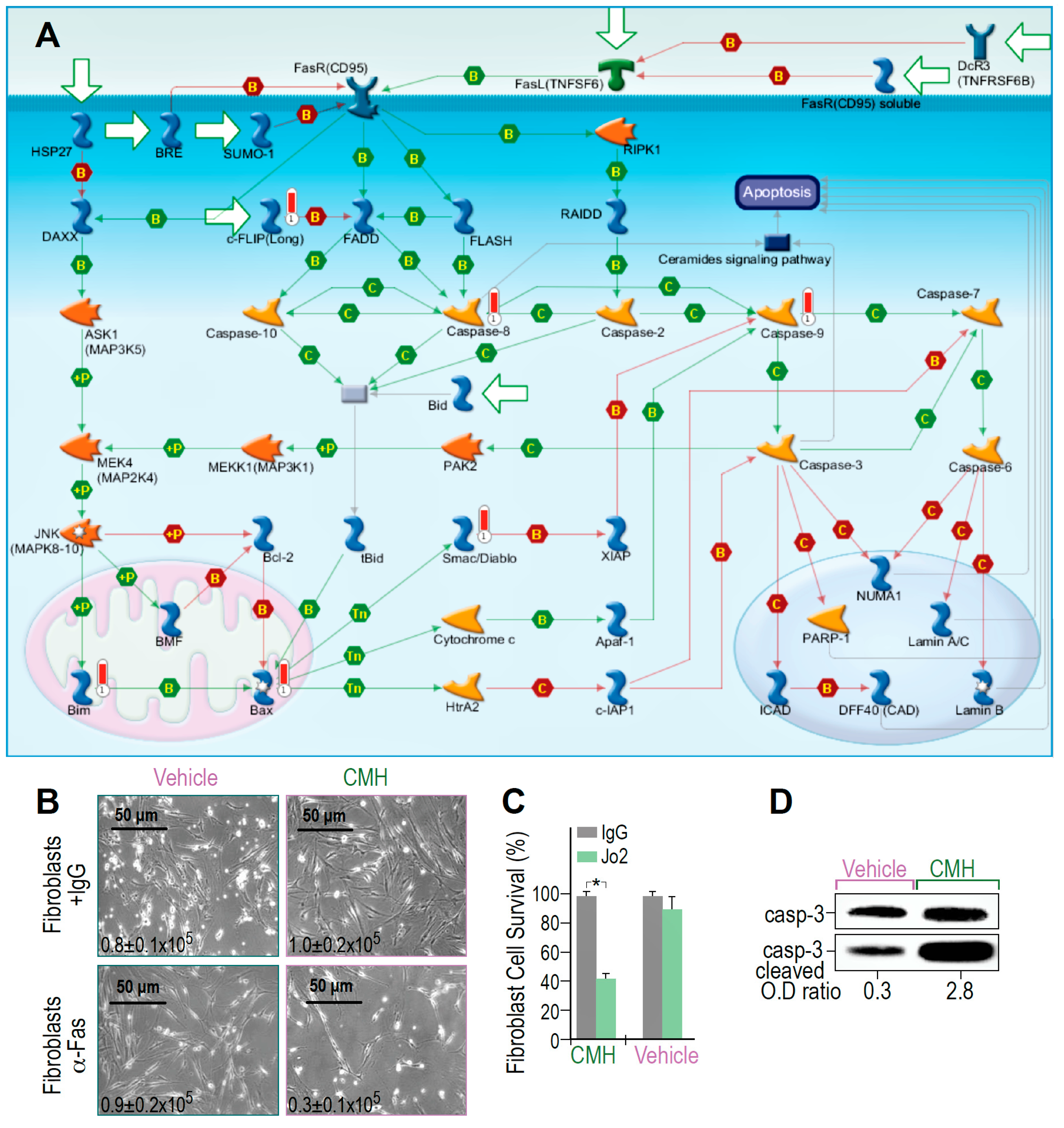
3.3. CMH Triggers and Boosts Fas-Death Signaling in Human IPF-Lung Myofibroblasts
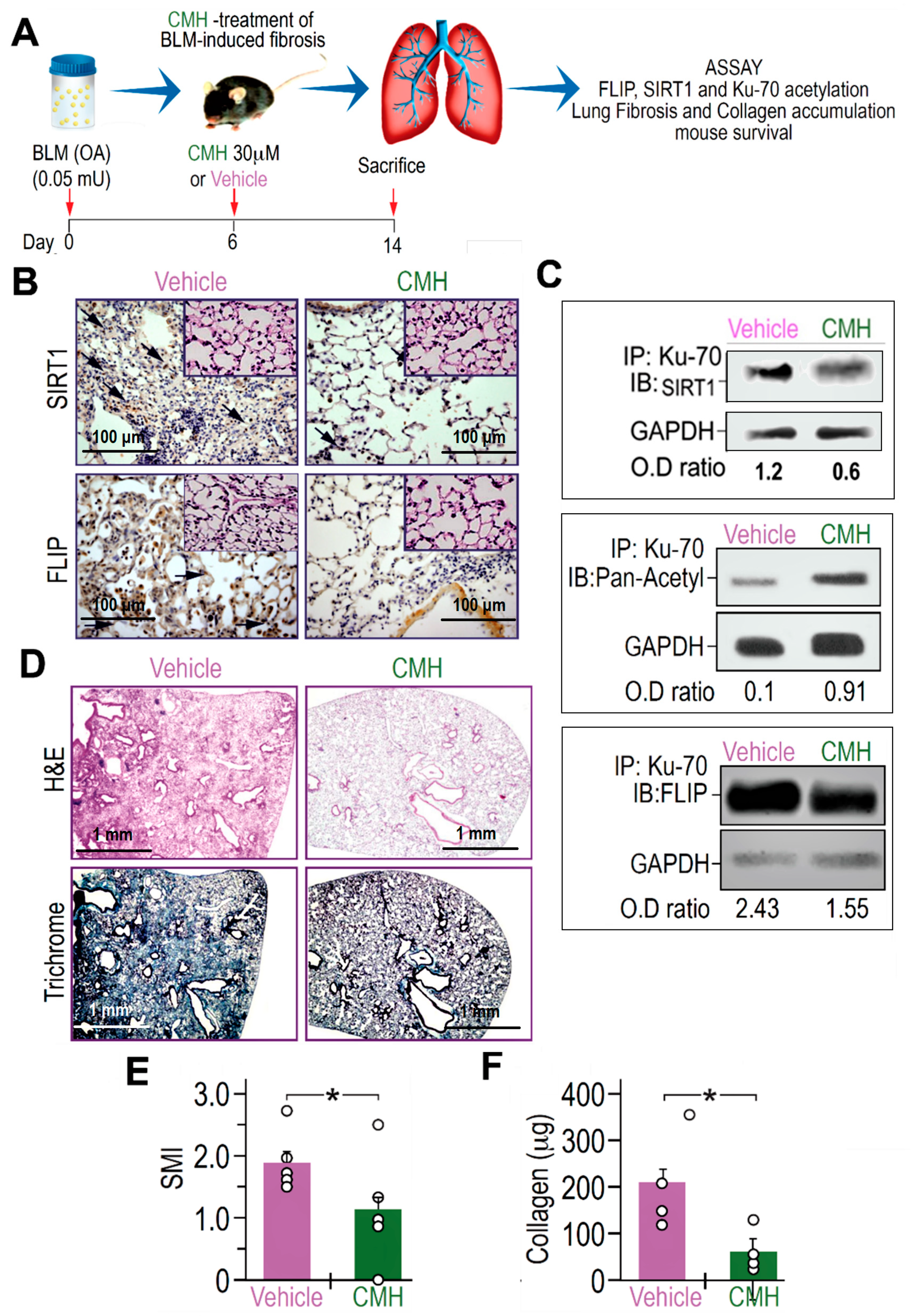
3.4. CMH Inhibits Fibrosis Evolution in BLM-Treated Mice and Ku70-Deacetylation, Ku70/FLIP Complex, FLIP Expression in Myofibroblasts Isolated From the Lungs at Day 14 Post BLM
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu, G.; Anstrom, K.J.; King, T.E.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, P.; Sverzellati, N.; Rossi, G.; Cavazza, A.; Tzouvelekis, A.; Crestani, B.; Vancheri, C. Idiopathic pulmonary fibrosis: An update. Ann. Med. 2015, 47, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H. Death receptors. Essays Biochem. 2003, 39, 53–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djerbi, M.; Screpanti, V.; Catrina, A.I.; Bogen, B.; Biberfeld, P.; Grandien, A. The Inhibitor of Death Receptor Signaling, Flice-Inhibitory Protein Defines a New Class of Tumor Progression Factors. J. Exp. Med. 1999, 190, 1025–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, L.E.; Tschopp, J. Inhibition of Death Receptor Signaling by Flice-Inhibitory Protein as a Mechanism for Immune Escape of Tumors. J. Exp. Med. 1999, 190, 891–894. [Google Scholar] [CrossRef] [Green Version]
- Medema, J.P.; de Jong, J.; van Hall, T.; Melief, C.J.M.; Offringa, R. Immune Escape of Tumors in Vivo by Expression of Cellular Flice-Inhibitory Protein. J. Exp. Med. 1999, 190, 1033–1038. [Google Scholar] [CrossRef]
- Tepper, C.G.; Seldin, M.F. Modulation of caspase-8 and FLICE-inhibitory protein expression as a potential mechanism of Epstein-Barr virus tumorigenesis in Burkitt’s lymphoma. Blood 1999, 94, 1727–1737. [Google Scholar] [CrossRef]
- Tanaka, T.; Yoshimi, M.; Maeyama, T.; Hagimoto, N.; Kuwano, K.; Hara, N. Resistance to Fas-mediated apoptosis in human lung fibroblast. Eur. Respir. J. 2002, 20, 359–368. [Google Scholar] [CrossRef]
- Yeh, W.C.; Itie, A.; Elia, A.J.; Ng, M.; Shu, H.B.; Wakeham, A.; Mirtsos, C.; Suzuki, N.; Bonnard, M.; Goeddel, D.V.; et al. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity 2000, 12, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Kalous, K.S.; Wynia-Smith, S.L.; Olp, M.D.; Smith, B.C. Mechanism of Sirt1 NAD+-dependent Protein Deacetylase Inhibition by CysteineS-Nitrosation. J. Biol. Chem. 2016, 291, 25398–25410. [Google Scholar] [CrossRef] [Green Version]
- Ota, C.; Yamada, M.; Fujino, N.; Motohashi, H.; Tando, Y.; Takei, Y.; Suzuki, T.; Takahashi, T.; Kamata, S.; Makiguchi, T.; et al. Histone deacetylase inhibitor restores surfactant protein-C expression in alveolar-epithelial type II cells and attenuates bleomycin-induced pulmonary fibrosis in vivo. Exp. Lung Res. 2015, 41, 422–434. [Google Scholar] [CrossRef]
- Pang, M.; Zhuang, S. Histone Deacetylase: A Potential Therapeutic Target for Fibrotic Disorders. J. Pharmacol. Exp. Ther. 2010, 335, 266–272. [Google Scholar] [CrossRef] [Green Version]
- Sanders, Y.Y.; Hagood, J.S.; Liu, H.; Zhang, W.; Ambalavanan, N.; Thannickal, V.J. Histone deacetylase inhibition promotes fibroblast apoptosis and ameliorates pulmonary fibrosis in mice. Eur. Respir. J. 2014, 43, 1448–1458. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, X.; Zhou, Q.; Huang, C.; Meng, X.; Xu, F.; Li, J. Silent information regulator 1 (SIRT1) ameliorates liver fibrosis via promoting activated stellate cell apoptosis and reversion. Toxicol. Appl. Pharmacol. 2015, 289, 163–176. [Google Scholar] [CrossRef]
- Zerr, P.; Palumbo-Zerr, K.; Huang, J.; Tomcik, M.; Sumova, B.; Distler, O.; Schett, G.; Distler, J.H.W. Sirt1 regulates canonical TGF-β signalling to control fibroblast activation and tissue fibrosis. Ann. Rheum. Dis. 2016, 75, 226–233. [Google Scholar] [CrossRef]
- Alves-Fernandes, D.K.; Jasiulionis, M.G. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. Int. J. Mol. Sci. 2019, 20, 3153. [Google Scholar] [CrossRef] [Green Version]
- Akgedik, R.; Akgedik, S.; Karamanlı, H.; Uysal, S.; Bozkurt, B.; Ozol, D.; Armutcu, F.; Yıldırım, Z. Effect of resveratrol on treatment of bleomycin-induced pulmonary fibrosis in rats. Inflammation 2012, 35, 1732–1741. [Google Scholar] [CrossRef]
- Sener, G.; Topaloğlu, N.; Sehirli, A.O.; Ercan, F.; Gedik, N. Resveratrol alleviates bleomycin-induced lung injury in rats. Pulm. Pharmacol. Ther. 2007, 20, 642–649. [Google Scholar] [CrossRef]
- Shetty, S.K.; Tiwari, N.; Marudamuthu, A.S.; Puthusseri, B.; Bhandary, Y.P.; Fu, J.; Levin, J.; Idell, S.; Shetty, S. p53 and miR-34a Feedback Promotes Lung Epithelial Injury and Pulmonary Fibrosis. Am. J. Pathol. 2017, 187, 1016–1034. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Cheng, S.; Chen, H.; Li, Q.; Hu, Y.; Wang, Q.; Zhu, X.; Wang, J. Activation and overexpression of Sirt1 attenuates lung fibrosis via P300. Biochem. Biophys. Res. Commun. 2017, 486, 1021–1026. [Google Scholar] [CrossRef]
- Drazic, A.; Myklebust, L.M.; Ree, R.; Arnesen, T. The world of protein acetylation. Biochim. Et Biophys. Acta 2016, 1864, 1372–1401. [Google Scholar] [CrossRef] [Green Version]
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015, 16, 258–264. [Google Scholar] [CrossRef]
- Kim, M.-J.; Hong, K.-S.; Kim, H.-B.; Lee, S.-H.; Bae, J.-H.; Kim, D.-W.; Dao, T.T.; Oh, W.K.; Kang, C.-D.; Kim, S.-H. Ku70 acetylation and modulation of c-Myc/ATF4/CHOP signaling axis by SIRT1 inhibition lead to sensitization of HepG2 cells to TRAIL through induction of DR5 and down-regulation of c-FLIP. Int. J. Biochem. Cell Biol. 2013, 45, 711–723. [Google Scholar] [CrossRef]
- Roth, M.; Wang, Z.; Chen, W.Y. SIRT1 and LSD1 competitively regulate KU70 functions in DNA repair and mutation acquisition in cancer cells. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Kerr, E.; Holohan, C.; McLaughlin, K.M.; Majkut, J.; Dolan, S.; Redmond, K.; Riley, J.; McLaughlin, K.; Stasik, I.; Crudden, M.; et al. Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell Death Differ. 2012, 19, 1317–1327. [Google Scholar] [CrossRef]
- Golan-Gerstl, R.; Wallach-Dayan, S.B.; Amir, G.; Breuer, R. Epithelial Cell Apoptosis by Fas Ligand–Positive Myofibroblasts in Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2007, 36, 270–275. [Google Scholar] [CrossRef] [Green Version]
- Wallach-Dayan, S.B.; Golan-Gerstl, R.; Breuer, R. Evasion of myofibroblasts from immune surveillance: A mechanism for tissue fibrosis. Proc. Natl. Acad. Sci. USA 2007, 104, 20460–20465. [Google Scholar] [CrossRef] [Green Version]
- Golan-Gerstl, R.; Wallach-Dayan, S.B.; Zisman, P.; Cardoso, W.V.; Goldstein, R.H.; Breuer, R. Cellular FLICE-like inhibitory protein deviates myofibroblast fas-induced apoptosis toward proliferation during lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Bulvik, R.; Breuer, R.; Dvir-Ginzberg, M.; Reich, E.; Berkman, N.; Wallach-Dayan, S.B. SIRT1 Deficiency, Specifically in Fibroblasts, Decreases Apoptosis Resistance and Is Associated with Resolution of Lung-Fibrosis. Biomolecules 2020, in press. [Google Scholar]
- Haag, C.; Stadel, D.; Zhou, S.; Bachem, M.G.; Möller, P.; Debatin, K.-M.; Fulda, S. Identification of c-FLIP(L) and c-FLIP(S) as critical regulators of death receptor-induced apoptosis in pancreatic cancer cells. Gut 2011, 60, 225–237. [Google Scholar] [CrossRef]
- Bijangi-Vishehsaraei, K.; Huang, S.; Safa, A.R.; Saadatzadeh, M.R.; Murphy, M.P. 4-(4-Chloro-2-methylphenoxy)-N-hydroxybutanamide (CMH) targets mRNA of the c-FLIP variants and induces apoptosis in MCF-7 human breast cancer cells. Mol. Cell. Biochem. 2010, 342, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Wallach-Dayan, S.B.; Izbicki, G.; Cohen, P.Y.; Gerstl-Golan, R.; Fine, A.; Breuer, R. Bleomycin initiates apoptosis of lung epithelial cells by ROS but not by Fas/FasL pathway. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L790–L796. [Google Scholar] [CrossRef]
- Cohen, P.Y.; Breuer, R.; Wallach-Dayan, S.B. Thy1 up-regulates FasL expression in lung myofibroblasts via Src family kinases. Am. J. Respir. Cell Mol. Biol. 2009, 40, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Mawji, I.A.; Simpson, C.D.; Gronda, M.; Williams, M.A.; Hurren, R.; Henderson, C.J.; Datti, A.; Wrana, J.L.; Schimmer, A.D. A chemical screen identifies anisomycin as an anoikis sensitizer that functions by decreasing FLIP protein synthesis. Cancer Res. 2007, 67, 8307–8315. [Google Scholar] [CrossRef] [Green Version]
- Schimmer, A.D.; Thomas, M.P.; Hurren, R.; Gronda, M.; Pellecchia, M.; Pond, G.R.; Konopleva, M.; Gurfinkel, D.; Mawji, I.A.; Brown, E.; et al. Identification of small molecules that sensitize resistant tumor cells to tumor necrosis factor-family death receptors. Cancer Res. 2006, 66, 2367–2375. [Google Scholar] [CrossRef] [Green Version]
- Buler, M.; Andersson, U.; Hakkola, J. Who watches the watchmen? Regulation of the expression and activity of sirtuins. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 3942–3960. [Google Scholar] [CrossRef] [Green Version]
- Oppenheimer, H.; Gabay, O.; Meir, H.; Haze, A.; Kandel, L.; Liebergall, M.; Gagarina, V.; Lee, E.J.; Dvir-Ginzberg, M. 75kDa SirT1 Blocks TNFα-Mediated Apoptosis in Human Osteoarthritic Chondrocytes. Arthritis Rheum. 2012, 64, 718–728. [Google Scholar] [CrossRef]
- Hague, A.; Eveson, J.W.; Macfarlane, M.; Huntley, S.; Janghra, N.; Thavaraj, S. Caspase-3 expression is reduced, in the absence of cleavage, in terminally differentiated normal oral epithelium but is increased in oral squamous cell carcinomas and correlates with tumour stage. J. Pathol. 2004, 204, 175–182. [Google Scholar] [CrossRef]
- Wood, T.E.; Dalili, S.; Simpson, C.D.; Sukhai, M.A.; Hurren, R.; Anyiwe, K.; Mao, X.; Suarez Saiz, F.; Gronda, M.; Eberhard, Y.; et al. Selective inhibition of histone deacetylases sensitizes malignant cells to death receptor ligands. Mol. Cancer 2010, 9, 246–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Auwerx, J. Protein deacetylation by SIRT1: An emerging key post-translational modification in metabolic regulation. Pharmacol. Res. 2010, 62, 35–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Wu, H.; Yang, M.; Ye, S.; Li, L.; Zhang, H.; Hu, J.; Wang, X.; Xu, J.; Liang, A. SIRT1 inhibition impairs non-homologous end joining DNA damage repair by increasing Ku70 acetylation in chronic myeloid leukemia cells. Oncotarget 2016, 7, 13538–13550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Chen, L.; Hou, X.; Li, Z.; Kabra, N.; Ma, Y.; Nemoto, S.; Finkel, T.; Gu, W.; Cress, W.D.; et al. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat. Cell Biol. 2006, 8, 1025–1031. [Google Scholar] [CrossRef]
- Huang, S.K.; Scruggs, A.M.; Donaghy, J.; Horowitz, J.C.; Zaslona, Z.; Przybranowski, S.; White, E.S.; Peters-Golden, M. Histone modifications are responsible for decreased Fas expression and apoptosis resistance in fibrotic lung fibroblasts. Cell Death Dis. 2013, 4, e621. [Google Scholar] [CrossRef]
- Chen, H.P.; Zhao, Y.T.; Zhao, T.C. Histone deacetylases and mechanisms of regulation of gene expression. Crit. Rev. Oncog. 2015, 20, 35–47. [Google Scholar] [CrossRef]
- Yoon, S.; Kang, G.; Eom, G.H. HDAC Inhibitors: Therapeutic Potential in Fibrosis-Associated Human Diseases. Int. J. Mol. Sci. 2019, 20, 1329. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, J.L.; Stasik, I.; Kerr, E.M.; Holohan, C.; Redmond, K.M.; McLaughlin, K.M.; Busacca, S.; Barbone, D.; Broaddus, V.C.; Gray, S.G.; et al. Vorinostat/SAHA-induced apoptosis in malignant mesothelioma is FLIP/caspase 8-dependent and HR23B-independent. Eur. J. Cancer (Oxf. Engl. 1990) 2012, 48, 1096–1107. [Google Scholar] [CrossRef]
- Guo, W.; Shan, B.; Klingsberg, R.C.; Qin, X.; Lasky, J.A. Abrogation of TGF-β1-induced fibroblast-myofibroblast differentiation by histone deacetylase inhibition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L864–L870. [Google Scholar] [CrossRef] [Green Version]

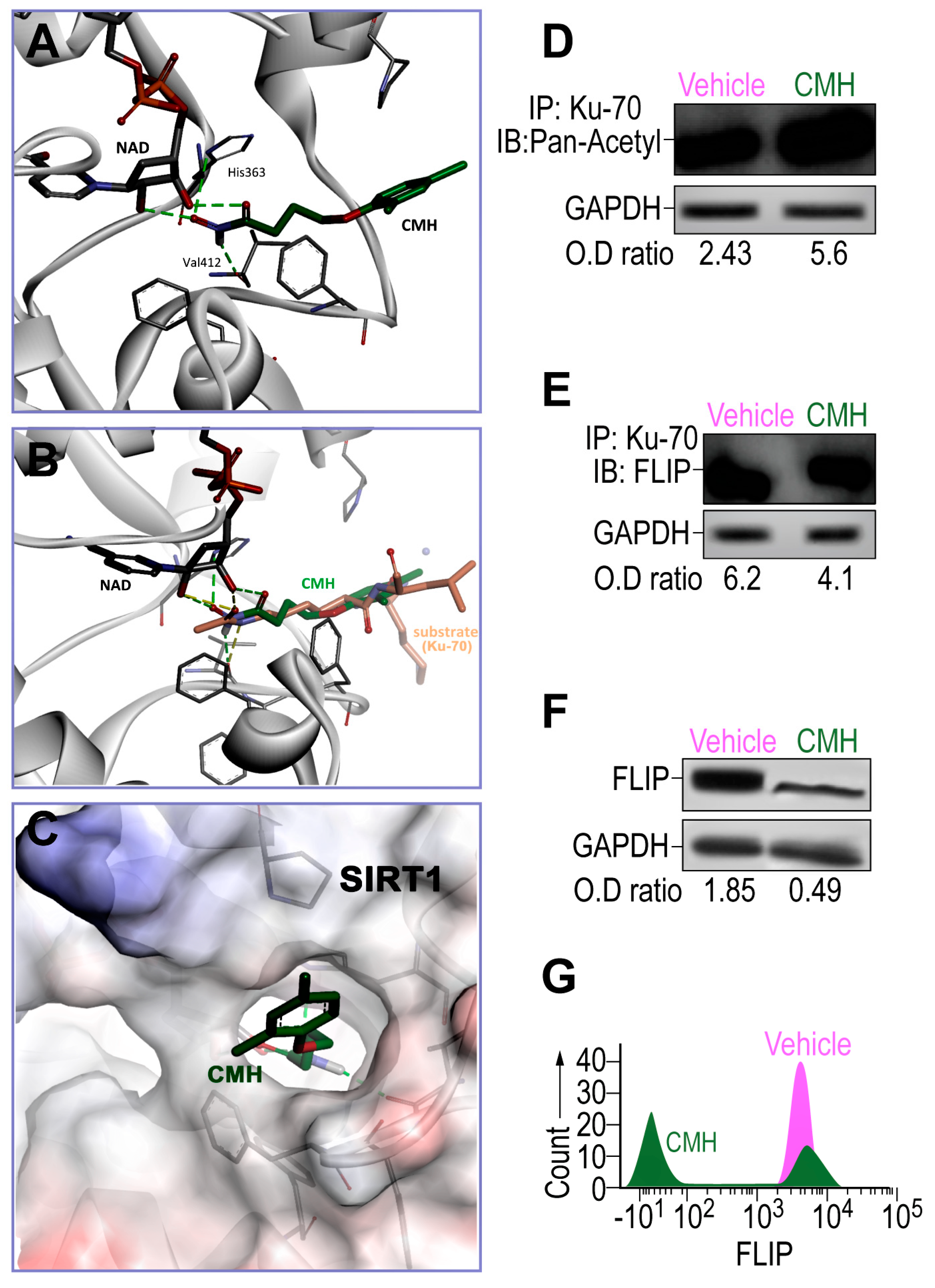{kind=link}
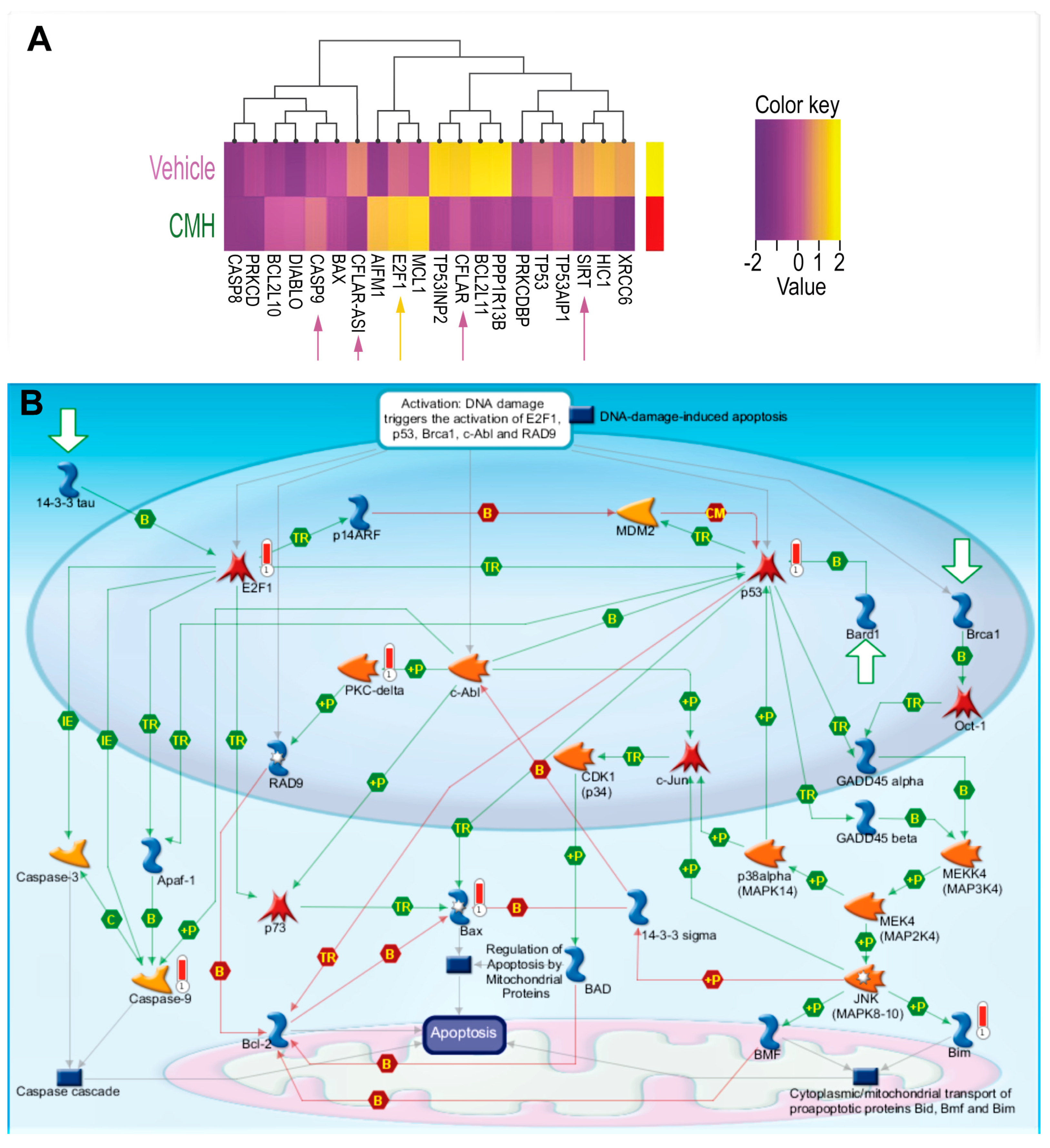{kind=link}
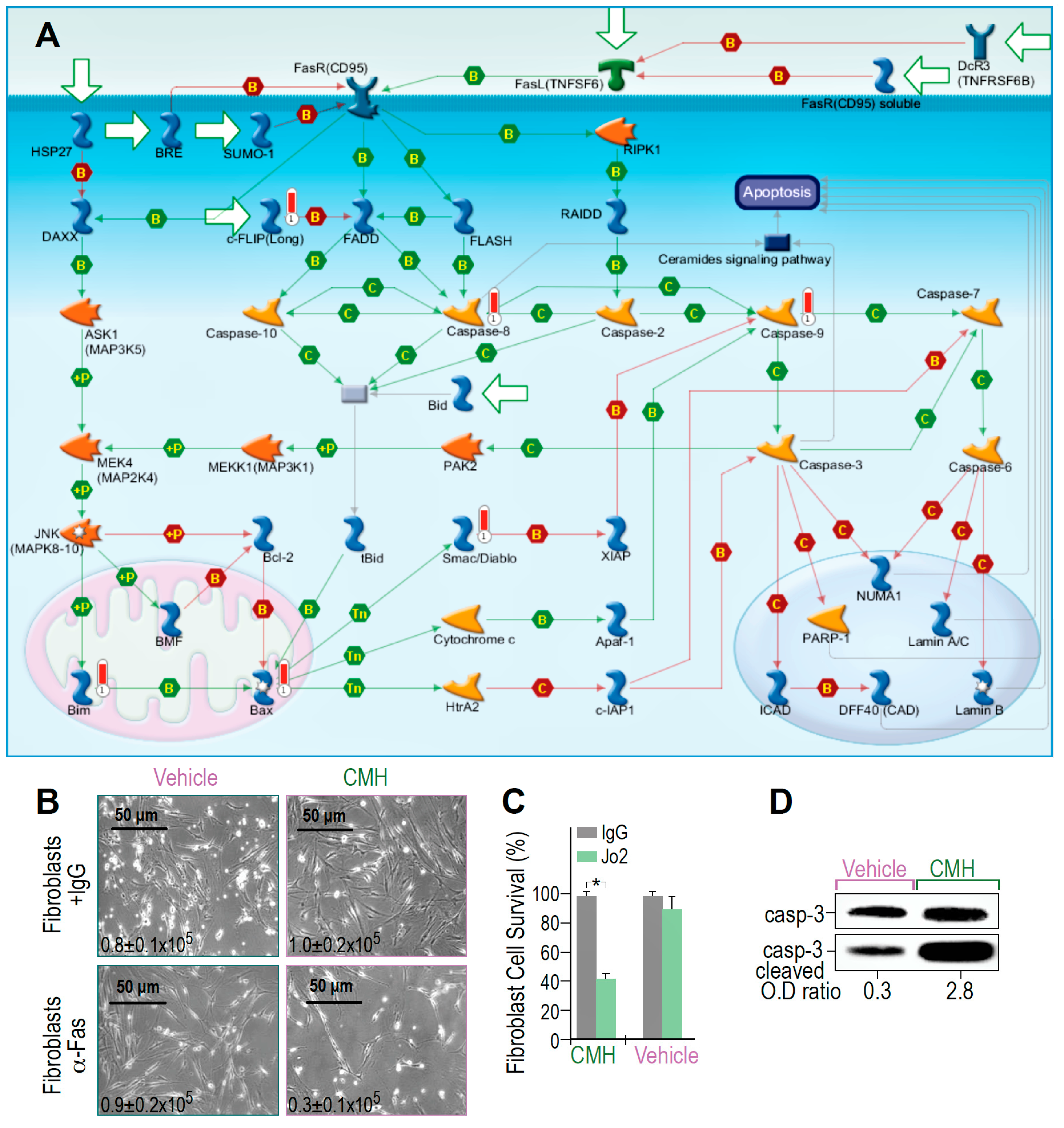{kind=link}
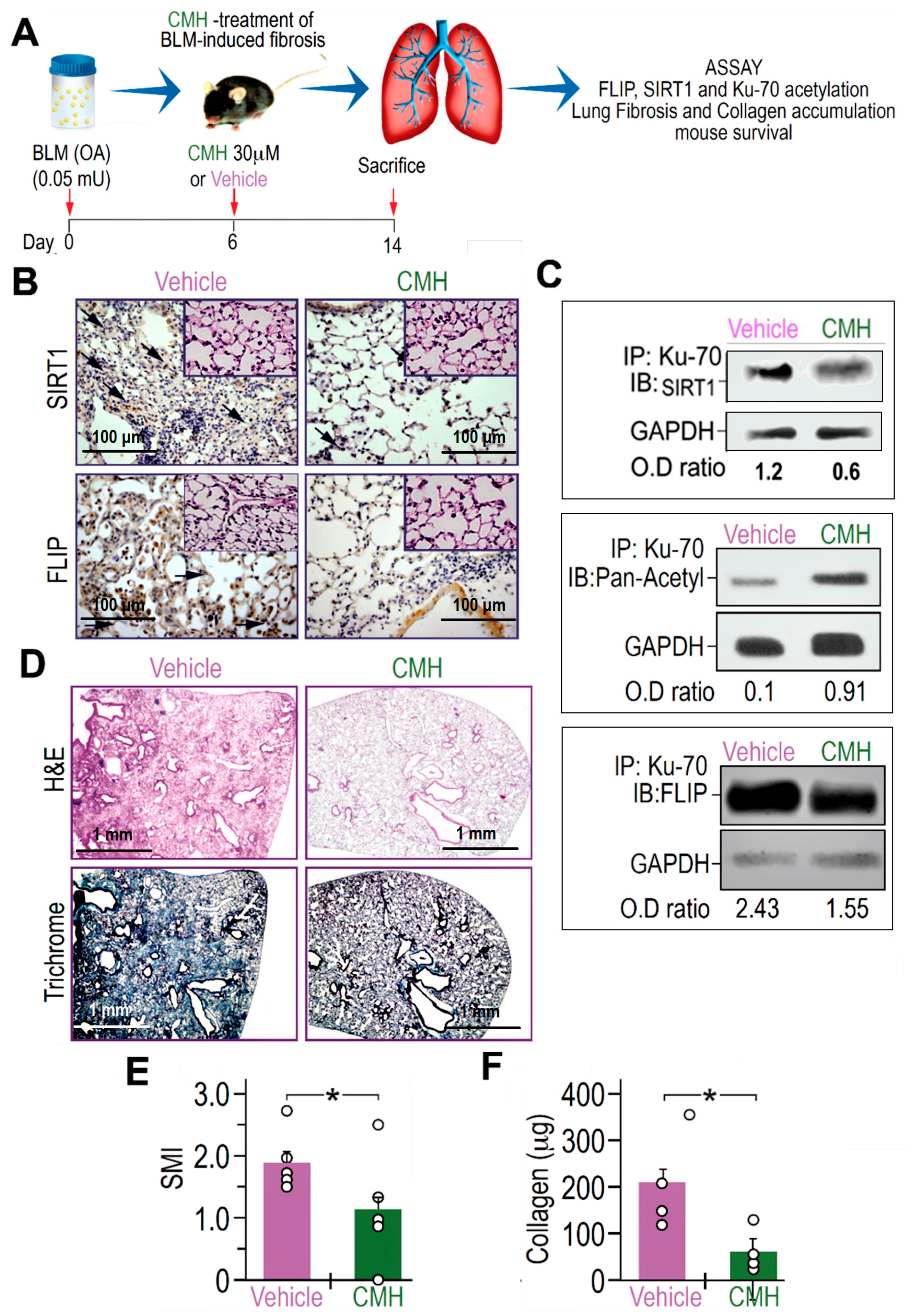{kind=link}
| Gene | Vehicle; M ± m | CMH; M ± m |
|---|---|---|
| XRCC6 | 192.46 ± 1.92 | 84.85 ± 22.88 |
| HIC1 | 5.55 ± 0.6 | 0.68 ± 0.44 |
| SIRT1 | 3.47 ± 0.1 | 0.48 ± 0.04 |
| TP53AIP1 | 1.67 ± 0.22 | 1.11 ± 0.2 |
| TP53 | 8.14 ± 1.32 | 1.8 ± 1.19 |
| PRKCDBP | 107.67 ± 31.83 | 21.87 ± 9.21 |
| PPP1R13B | 1.72 ± 1.19 | 0.1 ± 0.05 |
| BCL2L11 | 3.28 ± 1.36 | 0.58 ± 0.09 |
| CFLAR | 7.36 ± 2.29 | 3.5 ± 1.42 |
| TP53INP2 | 12.53 ± 1.61 | 8.16 ± 0.54 |
| MCL1 | 52.13 ± 4.05 | 67.57 ± 34.04 |
| E2F1 | 0.21 ± 0.12 | 0.39 ± 0.21 |
| AIFM1 | 10.92 ± 2.11 | 12.3 ± 2.89 |
| CFLAR-AS1 | 0.09 ± 0.03 | 0.11 ± 0.04 |
| BAX | 196.43 ± 24.62 | 319.29 ± 78.06 |
| CASP9 | 0.38 ± 0.24 | 9.57 ± 6.69 |
| DIABLO | 3.32 ± 1.79 | 13.48 ± 3.27 |
| BCL2L10 | 0 ± 0 | 0.13 ± 0.05 |
| PRKCD | 6.51 ± 0.7 | 9 ± 4 |
| CASP8 | 5.4 ± 3.16 | 7.13 ± 4.18 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konikov-Rozenman, J.; Breuer, R.; Kaminski, N.; Wallach-Dayan, S.B. CMH-Small Molecule Docks into SIRT1, Elicits Human IPF-Lung Fibroblast Cell Death, Inhibits Ku70-deacetylation, FLIP and Experimental Pulmonary Fibrosis. Biomolecules 2020, 10, 997. https://doi.org/10.3390/biom10070997
Konikov-Rozenman J, Breuer R, Kaminski N, Wallach-Dayan SB. CMH-Small Molecule Docks into SIRT1, Elicits Human IPF-Lung Fibroblast Cell Death, Inhibits Ku70-deacetylation, FLIP and Experimental Pulmonary Fibrosis. Biomolecules. 2020; 10(7):997. https://doi.org/10.3390/biom10070997
Chicago/Turabian StyleKonikov-Rozenman, Jenya, Raphael Breuer, Naftali Kaminski, and Shulamit B. Wallach-Dayan. 2020. "CMH-Small Molecule Docks into SIRT1, Elicits Human IPF-Lung Fibroblast Cell Death, Inhibits Ku70-deacetylation, FLIP and Experimental Pulmonary Fibrosis" Biomolecules 10, no. 7: 997. https://doi.org/10.3390/biom10070997
APA StyleKonikov-Rozenman, J., Breuer, R., Kaminski, N., & Wallach-Dayan, S. B. (2020). CMH-Small Molecule Docks into SIRT1, Elicits Human IPF-Lung Fibroblast Cell Death, Inhibits Ku70-deacetylation, FLIP and Experimental Pulmonary Fibrosis. Biomolecules, 10(7), 997. https://doi.org/10.3390/biom10070997