Effect of Quercetin on Dexamethasone-Induced C2C12 Skeletal Muscle Cell Injury

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Quercetin Reduces Growth Inhibition and Triggers Morphological Changes in Dexamethasone-Treated C2C12 Myotube Cells
2.2. Dexamethasone-Induced Loss of ΔΨm and the Elicited Caspase-Dependent Apoptosis in C2C12 Myotube Cells
2.3. Effects of Quercetin on ROS Production and the Apoptotic Situation in Dexamethasone-Treated C2C12 Myotube Cells
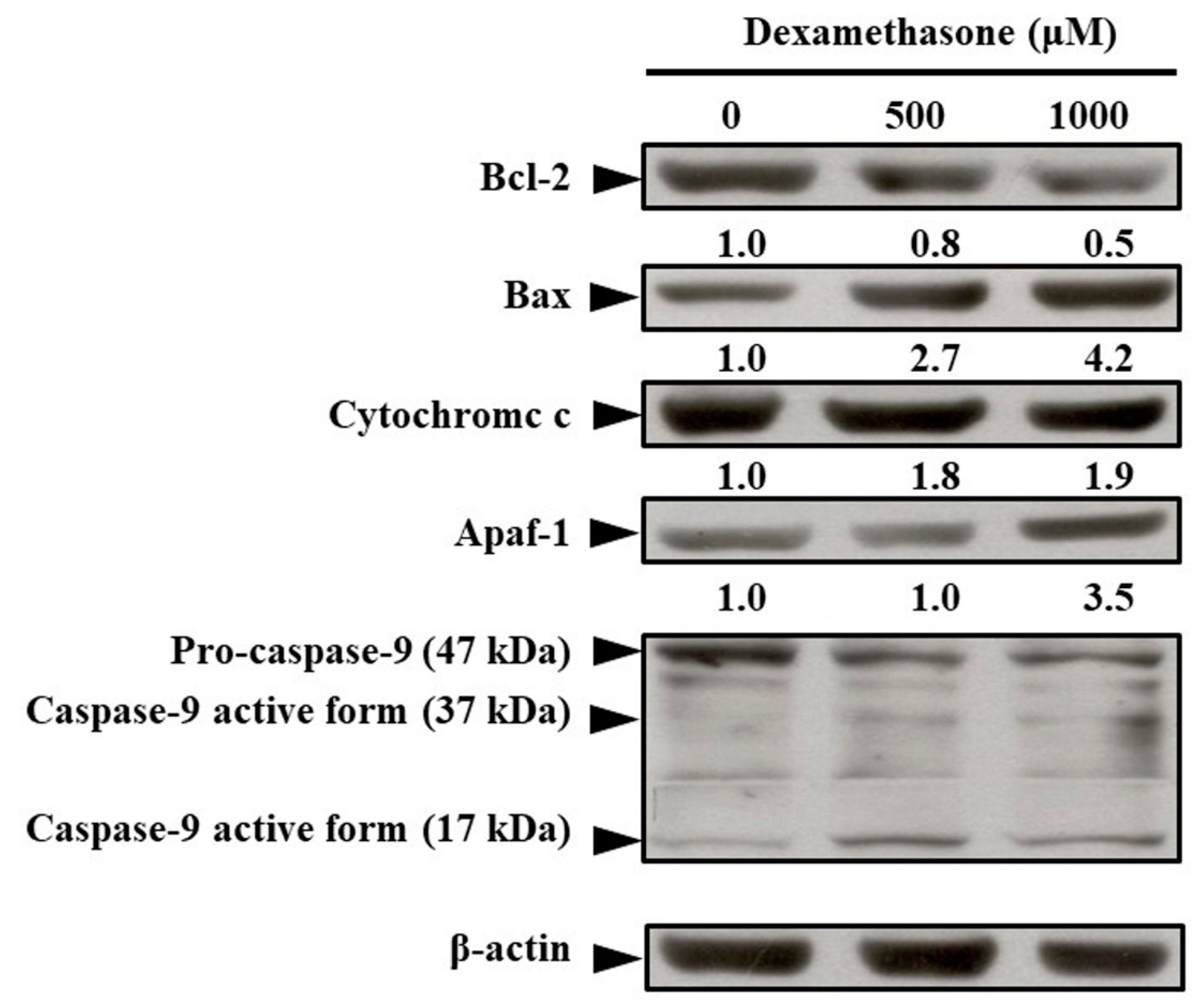
2.4. Effects of Dexamethasone on Changes in Apoptosis-Regulated Protein Levels in C2C12 Myotube Cells
2.5. Effects of Quercetin on Molecular Level Changes in Apoptosis-Regulated Protein and Caspase-3/9 Activities in Dexamethasone-Treated C2C12 Myotube Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cytotoxicity Assay
4.4. Morphological Assay
4.5. Apoptosis Analysis through TUNEL Assay
4.6. Caspase-3/9 Assay and Their Specific Inhibitor Activities
4.7. Western Blotting Analysis
4.8. Determination of ROS Levels through Flow Cytometry
4.9. Detection of Mitochondrial Electrical Potential (ΔΨm)
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Cyt C | Cytochrome C |
| GR | Glucocorticoid receptors |
| iNOS | Inducible nitric oxide synthase |
| MOMP | Mitochondrial outer membrane permeabilization |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| ROS | Reactive oxygen species |
| RNS | Reactive nitrogen species |
| ΔΨm | Mitochondrial Membrane Potential |
References
- Cruz-Topete, D.; Cidlowski, J.A. One Hormone, Two Actions: Anti- and Pro-Inflammatory Effects of Glucocorticoids. Neuroimmunomodulation 2015, 22, 20–32. [Google Scholar]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar]
- Nussinovitch, U.; de Carvalho, J.F.; Pereira, R.M.; Shoenfeld, Y. Glucocorticoids and the cardiovascular system: State of the art. Curr. Pharm. Des. 2010, 16, 3574–3585. [Google Scholar]
- Zheng, Y.; Xiong, S.; Jiang, P.; Liu, R.; Liu, X.; Qian, J.; Zheng, X.; Chu, Y. Glucocorticoids inhibit lipopolysaccharide-mediated inflammatory response by downregulating microRNA-155: A novel anti-inflammation mechanism. Free Radic. Biol. Med. 2012, 52, 1307–1317. [Google Scholar]
- Rauchhaus, U.; Schwaiger, F.W.; Panzner, S. Separating therapeutic efficacy from glucocorticoid side-effects in rodent arthritis using novel, liposomal delivery of dexamethasone phosphate: Long-term suppression of arthritis facilitates interval treatment. Arthritis Res. Ther. 2009, 11, R190. [Google Scholar]
- Lofberg, E.; Gutierrez, A.; Wernerman, J.; Anderstam, B.; Mitch, W.E.; Price, S.R.; Bergstrom, J.; Alvestrand, A. Effects of high doses of glucocorticoids on free amino acids, ribosomes and protein turnover in human muscle. Eur. J. Clin. Investig. 2002, 32, 345–353. [Google Scholar]
- Schakman, O.; Kalista, S.; Barbe, C.; Loumaye, A.; Thissen, J.P. Glucocorticoid-induced skeletal muscle atrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2163–2172. [Google Scholar]
- Mitsui, T.; Azuma, H.; Nagasawa, M.; Iuchi, T.; Akaike, M.; Odomi, M.; Matsumoto, T. Chronic corticosteroid administration causes mitochondrial dysfunction in skeletal muscle. J. Neurol. 2002, 249, 1004–1009. [Google Scholar]
- Rodriguez, J.; Vernus, B.; Chelh, I.; Cassar-Malek, I.; Gabillard, J.C.; Hadj Sassi, A.; Seiliez, I.; Picard, B.; Bonnieu, A. Myostatin and the skeletal muscle atrophy and hypertrophy signaling pathways. Cell. Mol. Life Sci. 2014, 71, 4361–4371. [Google Scholar]
- Artaza, J.N.; Bhasin, S.; Magee, T.R.; Reisz-Porszasz, S.; Shen, R.; Groome, N.P.; Meerasahib, M.F.; Gonzalez-Cadavid, N.F. Myostatin inhibits myogenesis and promotes adipogenesis in C3H 10T(1/2) mesenchymal multipotent cells. Endocrinology 2005, 146, 3547–3557. [Google Scholar]
- Ma, K.; Mallidis, C.; Bhasin, S.; Mahabadi, V.; Artaza, J.; Gonzalez-Cadavid, N.; Arias, J.; Salehian, B. Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E363–E371. [Google Scholar]
- Otsuka, Y.; Egawa, K.; Kanzaki, N.; Izumo, T.; Rogi, T.; Shibata, H. Quercetin glycosides prevent dexamethasone-induced muscle atrophy in mice. Biochem. Biophys. Rep. 2019, 18, 100618. [Google Scholar]
- Liu, Y.; Cheng, H.; Zhou, Y.; Zhu, Y.; Bian, R.; Chen, Y.; Li, C.; Ma, Q.; Zheng, Q.; Zhang, Y.; et al. Myostatin induces mitochondrial metabolic alteration and typical apoptosis in cancer cells. Cell Death Disease 2013, 4, e494. [Google Scholar]
- Han, Y.-Q.; Ming, S.-L.; Wu, H.-T.; Zeng, L.; Ba, G.; Li, J.; Lu, W.-F.; Han, J.; Du, Q.-J.; Sun, M.-M.; et al. Myostatin knockout induces apoptosis in human cervical cancer cells via elevated reactive oxygen species generation. Redox Biol. 2018, 19, 412–428. [Google Scholar]
- Calvani, R.; Joseph, A.-M.; Adhihetty, P.J.; Miccheli, A.; Bossola, M.; Leeuwenburgh, C.; Bernabei, R.; Marzetti, E. Mitochondrial pathways in sarcopenia of aging and disuse muscle atrophy. Biol. Chem. 2013, 394, 393–414. [Google Scholar]
- Hyatt, H.; Deminice, R.; Yoshihara, T.; Powers, S.K. Mitochondrial dysfunction induces muscle atrophy during prolonged inactivity: A review of the causes and effects. Arch. Biochem. Biophys. 2019, 662, 49–60. [Google Scholar]
- Murphy, K.T.; Koopman, R.; Naim, T.; Leger, B.; Trieu, J.; Ibebunjo, C.; Lynch, G.S. Antibody-directed myostatin inhibition in 21-mo-old mice reveals novel roles for myostatin signaling in skeletal muscle structure and function. FASEB J. 2010, 24, 4433–4442. [Google Scholar]
- Dirks, A.; Leeuwenburgh, C. Apoptosis in skeletal muscle with aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 282, R519–R527. [Google Scholar]
- Siu, P.M. Muscle apoptotic response to denervation, disuse, and aging. Med. Sci. Sports Exerc. 2009, 41, 1876–1886. [Google Scholar]
- Leeuwenburgh, C.; Gurley, C.M.; Strotman, B.A.; Dupont-Versteegden, E.E. Age-related differences in apoptosis with disuse atrophy in soleus muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1288–R1296. [Google Scholar]
- Marzetti, E.; Wohlgemuth, S.E.; Lees, H.A.; Chung, H.Y.; Giovannini, S.; Leeuwenburgh, C. Age-related activation of mitochondrial caspase-independent apoptotic signaling in rat gastrocnemius muscle. Mech. Ageing Dev. 2008, 129, 542–549. [Google Scholar]
- Bonnefoy-Berard, N.; Aouacheria, A.; Verschelde, C.; Quemeneur, L.; Marçais, A.; Marvel, J. Control of proliferation by Bcl-2 family members. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2004, 1644, 159–168. [Google Scholar]
- Amirouche, A.; Durieux, A.C.; Banzet, S.; Koulmann, N.; Bonnefoy, R.; Mouret, C.; Bigard, X.; Peinnequin, A.; Freyssenet, D. Down-regulation of Akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology 2009, 150, 286–294. [Google Scholar]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar]
- Powers, S.K. Can antioxidants protect against disuse muscle atrophy? Sports Med. 2014, 44 (Suppl. 2), S155–S165. [Google Scholar]
- Gwaltney-Brant, S.M. Chapter 8-Nutraceuticals in Renal Diseases. In Nutraceuticals; Gupta, R.C., Ed.; Academic Press: Boston, MA, USA, 2016; pp. 101–108. [Google Scholar]
- Nishimuro, H.; Ohnishi, H.; Sato, M.; Ohnishi-Kameyama, M.; Matsunaga, I.; Naito, S.; Ippoushi, K.; Oike, H.; Nagata, T.; Akasaka, H.; et al. Estimated daily intake and seasonal food sources of quercetin in Japan. Nutrients 2015, 7, 2345–2358. [Google Scholar]
- Kwak, J.-H.; Seo, J.M.; Kim, N.-H.; Arasu, M.V.; Kim, S.; Yoon, M.K.; Kim, S.-J. Variation of quercetin glycoside derivatives in three onion (Allium cepa L.) varieties. Saudi J. Biol. Sci. 2017, 24, 1387–1391. [Google Scholar]
- Xu, D.; Hu, M.-J.; Wang, Y.-Q.; Cui, Y.-L. Antioxidant Activities of Quercetin and Its Complexes for Medicinal Application. Molecules 2019, 24, 1123. [Google Scholar]
- Li, C.; Zhang, W.-J.; Choi, J.; Frei, B. Quercetin affects glutathione levels and redox ratio in human aortic endothelial cells not through oxidation but formation and cellular export of quercetin-glutathione conjugates and upregulation of glutamate-cysteine ligase. Redox Biol. 2016, 9, 220–228. [Google Scholar]
- Gao, W.; Pu, L.; Chen, M.; Wei, J.; Xin, Z.; Wang, Y.; Yao, Z.; Shi, T.; Guo, C. Glutathione homeostasis is significantly altered by quercetin via the Keap1/Nrf2 and MAPK signaling pathways in rats. J. Clin. Biochem. Nutr. 2018, 62, 56–62. [Google Scholar]
- Alamdari, N.; Aversa, Z.; Castillero, E.; Gurav, A.; Petkova, V.; Tizio, S.; Hasselgren, P.O. Resveratrol prevents dexamethasone-induced expression of the muscle atrophy-related ubiquitin ligases atrogin-1 and MuRF1 in cultured myotubes through a SIRT1-dependent mechanism. Biochem. Biophys. Res. Commun. 2012, 417, 528–533. [Google Scholar]
- Yoshioka, Y.; Kubota, Y.; Samukawa, Y.; Yamashita, Y.; Ashida, H. Glabridin inhibits dexamethasone-induced muscle atrophy. Arch. Biochem. Biophys. 2019, 664, 157–166. [Google Scholar]
- Luan, G.; Li, G.; Ma, X.; Jin, Y.; Hu, N.; Li, J.; Wang, Z.; Wang, H. Dexamethasone-Induced Mitochondrial Dysfunction and Insulin Resistance-Study in 3T3-L1 Adipocytes and Mitochondria Isolated from Mouse Liver. Molecules 2019, 24, 1982. [Google Scholar]
- Liu, J.; Peng, Y.; Wang, X.; Fan, Y.; Qin, C.; Shi, L.; Tang, Y.; Cao, K.; Li, H.; Long, J.; et al. Mitochondrial Dysfunction Launches Dexamethasone-Induced Skeletal Muscle Atrophy via AMPK/FOXO3 Signaling. Mol. Pharm. 2016, 13, 73–84. [Google Scholar]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 2013, 6, 25–39. [Google Scholar]
- Sartorelli, V.; Fulco, M. Molecular and cellular determinants of skeletal muscle atrophy and hypertrophy. Sci. STKE 2004, 2004, re11. [Google Scholar]
- Amthor, H.; Otto, A.; Vulin, A.; Rochat, A.; Dumonceaux, J.; Garcia, L.; Mouisel, E.; Hourde, C.; Macharia, R.; Friedrichs, M.; et al. Muscle hypertrophy driven by myostatin blockade does not require stem/precursor-cell activity. Proc. Natl. Acad. Sci. USA 2009, 106, 7479–7484. [Google Scholar]
- Blaauw, B.; Canato, M.; Agatea, L.; Toniolo, L.; Mammucari, C.; Masiero, E.; Abraham, R.; Sandri, M.; Schiaffino, S.; Reggiani, C. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J. 2009, 23, 3896–3905. [Google Scholar]
- McCarthy, J.J.; Mula, J.; Miyazaki, M.; Erfani, R.; Garrison, K.; Farooqui, A.B.; Srikuea, R.; Lawson, B.A.; Grimes, B.; Keller, C.; et al. Effective fiber hypertrophy in satellite cell-depleted skeletal muscle. Development 2011, 138, 3657–3666. [Google Scholar]
- Raffaello, A.; Milan, G.; Masiero, E.; Carnio, S.; Lee, D.; Lanfranchi, G.; Goldberg, A.L.; Sandri, M. JunB transcription factor maintains skeletal muscle mass and promotes hypertrophy. J. Cell Biol. 2010, 191, 101–113. [Google Scholar]
- Sartori, R.; Milan, G.; Patron, M.; Mammucari, C.; Blaauw, B.; Abraham, R.; Sandri, M. Smad2 and 3 transcription factors control muscle mass in adulthood. Am. J. Physiol. Cell Physiol. 2009, 296, C1248–C1257. [Google Scholar]
- Flint, M.S.; Baum, A.; Chambers, W.H.; Jenkins, F.J. Induction of DNA damage, alteration of DNA repair and transcriptional activation by stress hormones. Psychoneuroendocrinology 2007, 32, 470–479. [Google Scholar]
- Hara, M.R.; Kovacs, J.J.; Whalen, E.J.; Rajagopal, S.; Strachan, R.T.; Grant, W.; Towers, A.J.; Williams, B.; Lam, C.M.; Xiao, K.; et al. A stress response pathway regulates DNA damage through beta2-adrenoreceptors and beta-arrestin-1. Nature 2011, 477, 349–353. [Google Scholar]
- Gidron, Y.; Russ, K.; Tissarchondou, H.; Warner, J. The relation between psychological factors and DNA-damage: A critical review. Biol. Psychol. 2006, 72, 291–304. [Google Scholar]
- Bagchi, D.; Carryl, O.R.; Tran, M.X.; Bagchi, M.; Garg, A.; Milnes, M.M.; Williams, C.B.; Balmoori, J.; Bagchi, D.J.; Mitra, S.; et al. Acute and chronic stress-induced oxidative gastrointestinal mucosal injury in rats and protection by bismuth subsalicylate. Mol. Cell. Biochem. 1999, 196, 109–116. [Google Scholar]
- Pomies, P.; Blaquiere, M.; Maury, J.; Mercier, J.; Gouzi, F.; Hayot, M. Involvement of the FoxO1/MuRF1/Atrogin-1 Signaling Pathway in the Oxidative Stress-Induced Atrophy of Cultured Chronic Obstructive Pulmonary Disease Myotubes. PLoS ONE 2016, 11, e0160092. [Google Scholar]
- Damiano, S.; Muscariello, E.; La Rosa, G.; Di Maro, M.; Mondola, P.; Santillo, M. Dual Role of Reactive Oxygen Species in Muscle Function: Can Antioxidant Dietary Supplements Counteract Age-Related Sarcopenia? Int. J. Mol. Sci. 2019, 20, 3815. [Google Scholar]
- Dahlgren, C.; Karlsson, A.; Bylund, J. Intracellular Neutrophil Oxidants: From Laboratory Curiosity to Clinical Reality. J. Immunol. 2019, 202, 3127–3134. [Google Scholar]
- Boots, A.W.; Haenen, G.R.; Bast, A. Health effects of quercetin: From antioxidant to nutraceutical. Eur. J. Pharmacol. 2008, 585, 325–337. [Google Scholar]
- Niu, X.; Brahmbhatt, H.; Mergenthaler, P.; Zhang, Z.; Sang, J.; Daude, M.; Ehlert, F.G.R.; Diederich, W.E.; Wong, E.; Zhu, W.; et al. A Small-Molecule Inhibitor of Bax and Bak Oligomerization Prevents Genotoxic Cell Death and Promotes Neuroprotection. Cell Chem. Biol. 2017, 24, 493–506.e495. [Google Scholar]
- Dewson, G.; Kluck, R.M. Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J. Cell Sci. 2009, 122, 2801. [Google Scholar]
- Flaherty, R.L.; Owen, M.; Fagan-Murphy, A.; Intabli, H.; Healy, D.; Patel, A.; Allen, M.C.; Patel, B.A.; Flint, M.S. Glucocorticoids induce production of reactive oxygen species/reactive nitrogen species and DNA damage through an iNOS mediated pathway in breast cancer. Breast Cancer Res. 2017, 19, 35. [Google Scholar]
- Song, I.-H.; Buttgereit, F. Non-genomic glucocorticoid effects to provide the basis for new drug developments. Mol. Cell. Endocrinol. 2006, 246, 142–146. [Google Scholar]
- Stahn, C.; Buttgereit, F. Genomic and nongenomic effects of glucocorticoids. Nat. Clin. Pract. Rheumatol. 2008, 4, 525–533. [Google Scholar]
- Tait, S.W.; Green, D.R. Mitochondrial regulation of cell death. Cold Spring Harb Perspect Biol 2013, 5, a008706. [Google Scholar]
- Ly, J.D.; Grubb, D.R.; Lawen, A. The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis 2003, 8, 115–128. [Google Scholar]
- Romanello, V.; Guadagnin, E.; Gomes, L.; Roder, I.; Sandri, C.; Petersen, Y.; Milan, G.; Masiero, E.; Del Piccolo, P.; Foretz, M.; et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010, 29, 1774–1785. [Google Scholar]
- Powers, S.K.; Wiggs, M.P.; Duarte, J.A.; Zergeroglu, A.M.; Demirel, H.A. Mitochondrial signaling contributes to disuse muscle atrophy. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E31–E39. [Google Scholar]
- Min, K.; Smuder, A.J.; Kwon, O.-S.; Kavazis, A.N.; Szeto, H.H.; Powers, S.K. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J. Appl. Physiol. 2011, 111, 1459–1466. [Google Scholar]
- Halliwell, B.; Rafter, J.; Jenner, A. Health promotion by flavonoids, tocopherols, tocotrienols, and other phenols: Direct or indirect effects? Antioxidant or not? Am. J. Clin. Nutr. 2005, 81, 268S–276S. [Google Scholar]
- Fraga, C.G.; Galleano, M.; Verstraeten, S.V.; Oteiza, P.I. Basic biochemical mechanisms behind the health benefits of polyphenols. Mol. Asp. Med. 2010, 31, 435–445. [Google Scholar]
- Halliwell, B. Are polyphenols antioxidants or pro-oxidants? What do we learn from cell culture and in vivo studies? Arch. Biochem. Biophys. 2008, 476, 107–112. [Google Scholar]
- Mukai, R.; Matsui, N.; Fujikura, Y.; Matsumoto, N.; Hou, D.X.; Kanzaki, N.; Shibata, H.; Horikawa, M.; Iwasa, K.; Hirasaka, K.; et al. Preventive effect of dietary quercetin on disuse muscle atrophy by targeting mitochondria in denervated mice. J. Nutr. Biochem. 2016, 31, 67–76. [Google Scholar]
- Costa, L.G.; Garrick, J.M.; Roque, P.J.; Pellacani, C. Mechanisms of Neuroprotection by Quercetin: Counteracting Oxidative Stress and More. Oxid. Med. Cell Longev. 2016, 2016, 2986796. [Google Scholar]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001, 3, 1014–1019. [Google Scholar]
- Clarke, B.A.; Drujan, D.; Willis, M.S.; Murphy, L.O.; Corpina, R.A.; Burova, E.; Rakhilin, S.V.; Stitt, T.N.; Patterson, C.; Latres, E.; et al. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007, 6, 376–385. [Google Scholar]
- Sacheck, J.M.; Ohtsuka, A.; McLary, S.C.; Goldberg, A.L. IGF-I stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, atrogin-1 and MuRF1. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E591–E601. [Google Scholar]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar]
- Schakman, O.; Gilson, H.; Thissen, J.P. Mechanisms of glucocorticoid-induced myopathy. J. Endocrinol. 2008, 197, 1–10. [Google Scholar]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar]
- O’Neal, P.; Alamdari, N.; Smith, I.; Poylin, V.; Menconi, M.; Hasselgren, P.O. Experimental hyperthyroidism in rats increases the expression of the ubiquitin ligases atrogin-1 and MuRF1 and stimulates multiple proteolytic pathways in skeletal muscle. J. Cell. Biochem. 2009, 108, 963–973. [Google Scholar]
- Allen, D.L.; Unterman, T.G. Regulation of myostatin expression and myoblast differentiation by FoxO and SMAD transcription factors. Am. J. Physiol. Cell Physiol. 2007, 292, C188–C199. [Google Scholar]
- Liu, B.; Chen, Y.; St Clair, D.K. ROS and p53: A versatile partnership. Free Radic. Biol. Med. 2008, 44, 1529–1535. [Google Scholar]
- Zhang, Z.P.; Shen, C.C.; Gao, F.L.; Wei, H.; Ren, D.F.; Lu, J. Isolation, Purification and Structural Characterization of Two Novel Water-Soluble Polysaccharides from Anredera cordifolia. Molecules 2017, 22, 1276. [Google Scholar]
- Song, J.; Wang, Y.; Yuan, X.; Ji, Q.; Fan, C.; Zhao, H.; Hao, W.; Ren, D. Stretching magnitude-dependent inactivation of AKT by ROS led to enhanced p53 mitochondrial translocation and myoblast apoptosis. Mol. Biol. Cell 2019, 30, 1182–1197. [Google Scholar]
- Jäger, S.; Handschin, C.; Pierre, J.S.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar]
- Dudylina, A.L.; Ivanova, M.V.; Shumaev, K.B.; Ruuge, E.K. Superoxide Formation in Cardiac Mitochondria and Effect of Phenolic Antioxidants. Cell Biochem. Biophys. 2019, 77, 99–107. [Google Scholar]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar]
- Son, Y.H.; Jang, E.J.; Kim, Y.W.; Lee, J.H. Sulforaphane prevents dexamethasone-induced muscle atrophy via regulation of the Akt/Foxo1 axis in C2C12 myotubes. Biomed. Pharm. 2017, 95, 1486–1492. [Google Scholar]
- Grela, E.; Kozlowska, J.; Grabowiecka, A. Current methodology of MTT assay in bacteria-A review. Acta Histochem. 2018, 120, 303–311. [Google Scholar]
- Pascua-Maestro, R.; Corraliza-Gomez, M.; Diez-Hermano, S.; Perez-Segurado, C.; Ganfornina, M.D.; Sanchez, D. The MTT-formazan assay: Complementary technical approaches and in vivo validation in Drosophila larvae. Acta Histochem. 2018, 120, 179–186. [Google Scholar]
- Lee, C.F.; Yang, J.S.; Tsai, F.J.; Chiang, N.N.; Lu, C.C.; Huang, Y.S.; Chen, C.; Chen, F.A. Kaempferol induces ATM/p53-mediated death receptor and mitochondrial apoptosis in human umbilical vein endothelial cells. Int. J. Oncol. 2016, 48, 2007–2014. [Google Scholar]
- Lu, C.C.; Yang, J.S.; Chiang, J.H.; Hour, M.J.; Lin, K.L.; Lin, J.J.; Huang, W.W.; Tsuzuki, M.; Lee, T.H.; Chung, J.G. Novel quinazolinone MJ-29 triggers endoplasmic reticulum stress and intrinsic apoptosis in murine leukemia WEHI-3 cells and inhibits leukemic mice. PLoS ONE 2012, 7, e36831. [Google Scholar]
- Ma, Y.S.; Weng, S.W.; Lin, M.W.; Lu, C.C.; Chiang, J.H.; Yang, J.S.; Lai, K.C.; Lin, J.P.; Tang, N.Y.; Lin, J.G.; et al. Antitumor effects of emodin on LS1034 human colon cancer cells in vitro and in vivo: Roles of apoptotic cell death and LS1034 tumor xenografts model. Food Chem. Toxicol. 2012, 50, 1271–1278. [Google Scholar]
- Lu, C.C.; Yang, J.S.; Chiang, J.H.; Hour, M.J.; Lin, K.L.; Lee, T.H.; Chung, J.G. Cell death caused by quinazolinone HMJ-38 challenge in oral carcinoma CAL 27 cells: Dissections of endoplasmic reticulum stress, mitochondrial dysfunction and tumor xenografts. Biochim. Biophys. Acta 2014, 1840, 2310–2320. [Google Scholar]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Yang, J.-S.; Lu, C.-C.; Chiu, Y.-J.; Chen, H.-C.; Chung, M.-I.; Wu, Y.-T.; Chen, F.-A. Effect of Quercetin on Dexamethasone-Induced C2C12 Skeletal Muscle Cell Injury. Molecules 2020, 25, 3267. https://doi.org/10.3390/molecules25143267
Chen C, Yang J-S, Lu C-C, Chiu Y-J, Chen H-C, Chung M-I, Wu Y-T, Chen F-A. Effect of Quercetin on Dexamethasone-Induced C2C12 Skeletal Muscle Cell Injury. Molecules. 2020; 25(14):3267. https://doi.org/10.3390/molecules25143267
Chicago/Turabian StyleChen, Chun, Jai-Sing Yang, Chi-Cheng Lu, Yu-Jen Chiu, Hung-Che Chen, Mei-Ing Chung, Yu-Tse Wu, and Fu-An Chen. 2020. "Effect of Quercetin on Dexamethasone-Induced C2C12 Skeletal Muscle Cell Injury" Molecules 25, no. 14: 3267. https://doi.org/10.3390/molecules25143267
APA StyleChen, C., Yang, J.-S., Lu, C.-C., Chiu, Y.-J., Chen, H.-C., Chung, M.-I., Wu, Y.-T., & Chen, F.-A. (2020). Effect of Quercetin on Dexamethasone-Induced C2C12 Skeletal Muscle Cell Injury. Molecules, 25(14), 3267. https://doi.org/10.3390/molecules25143267