RhoA/ROCK Pathway Activation is Regulated by AT1 Receptor and Participates in Smooth Muscle Migration and Dedifferentiation via Promoting Actin Cytoskeleton Polymerization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
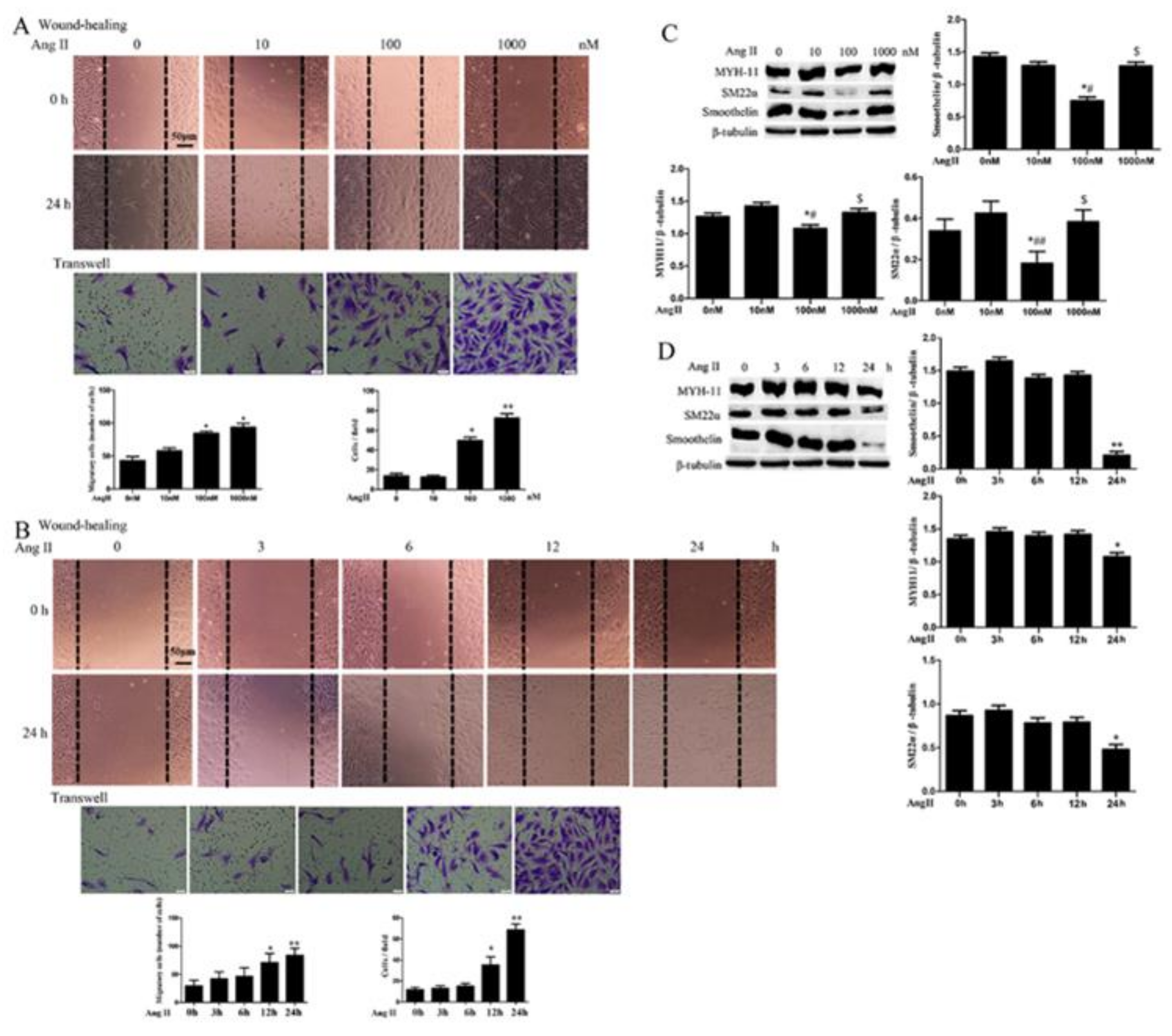
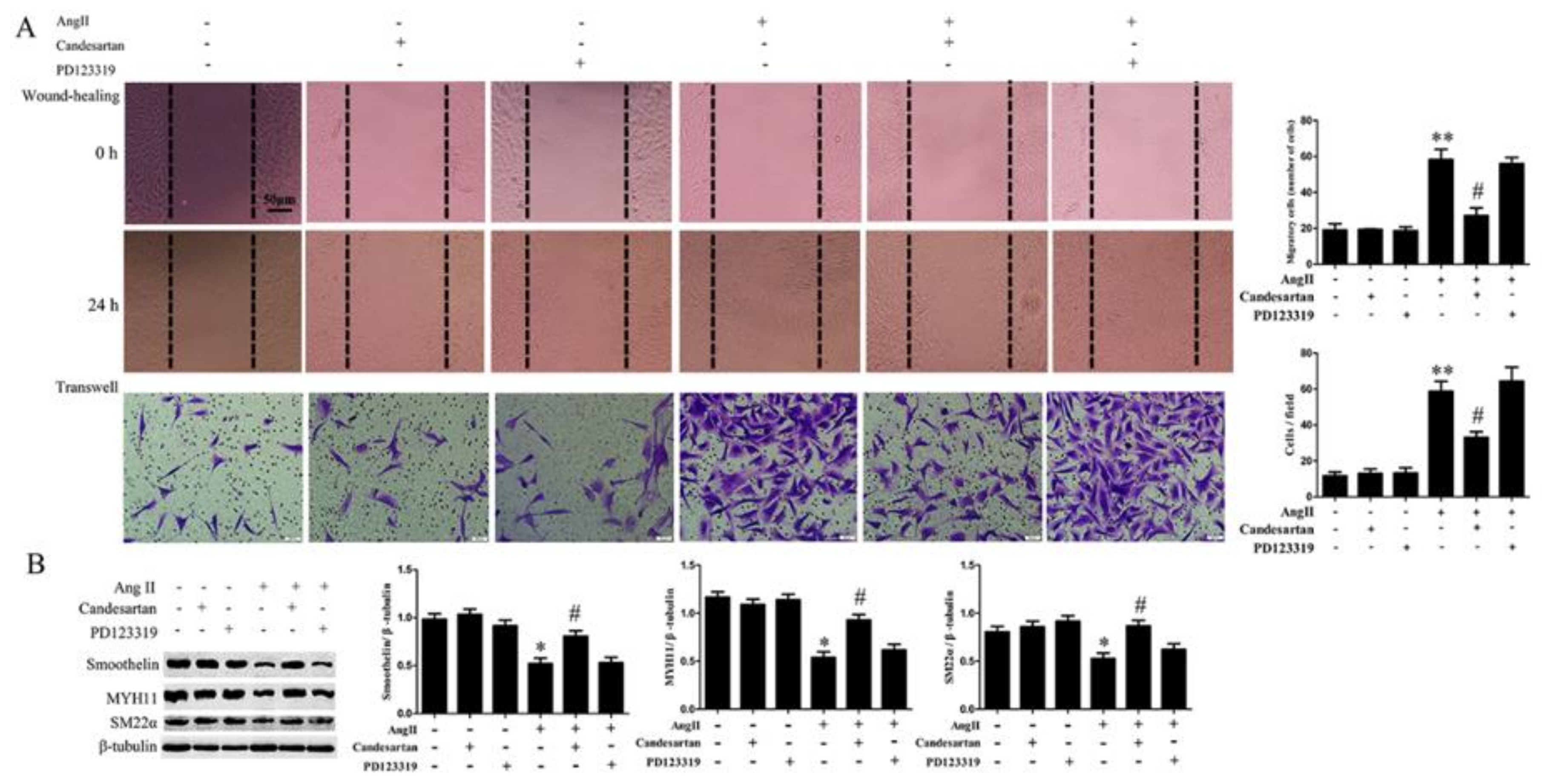
2.1. Ang II Promotes HA-VSMC Migration and Down-Regulates Differentiation Marker Proteins via AT1 Receptor
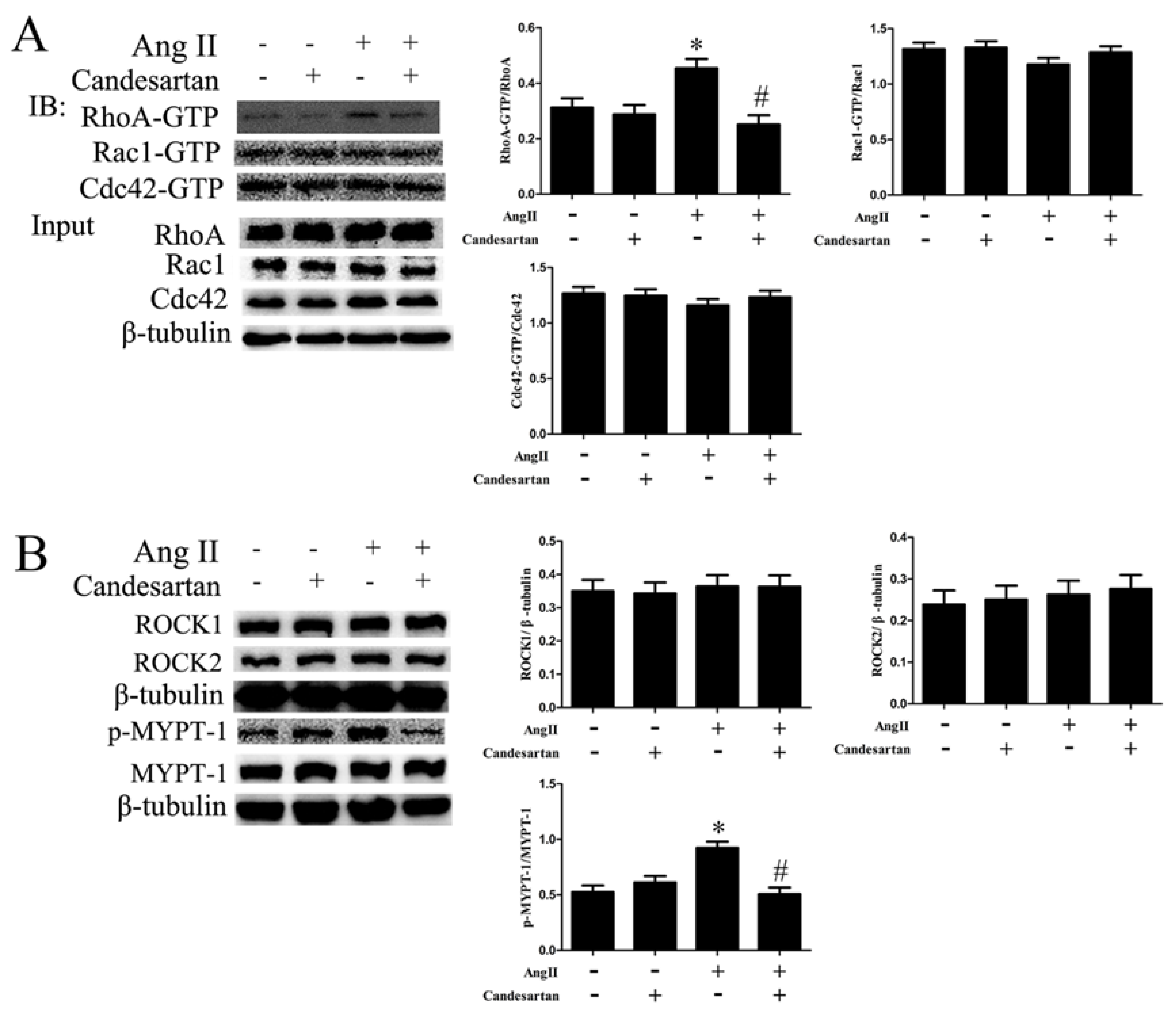
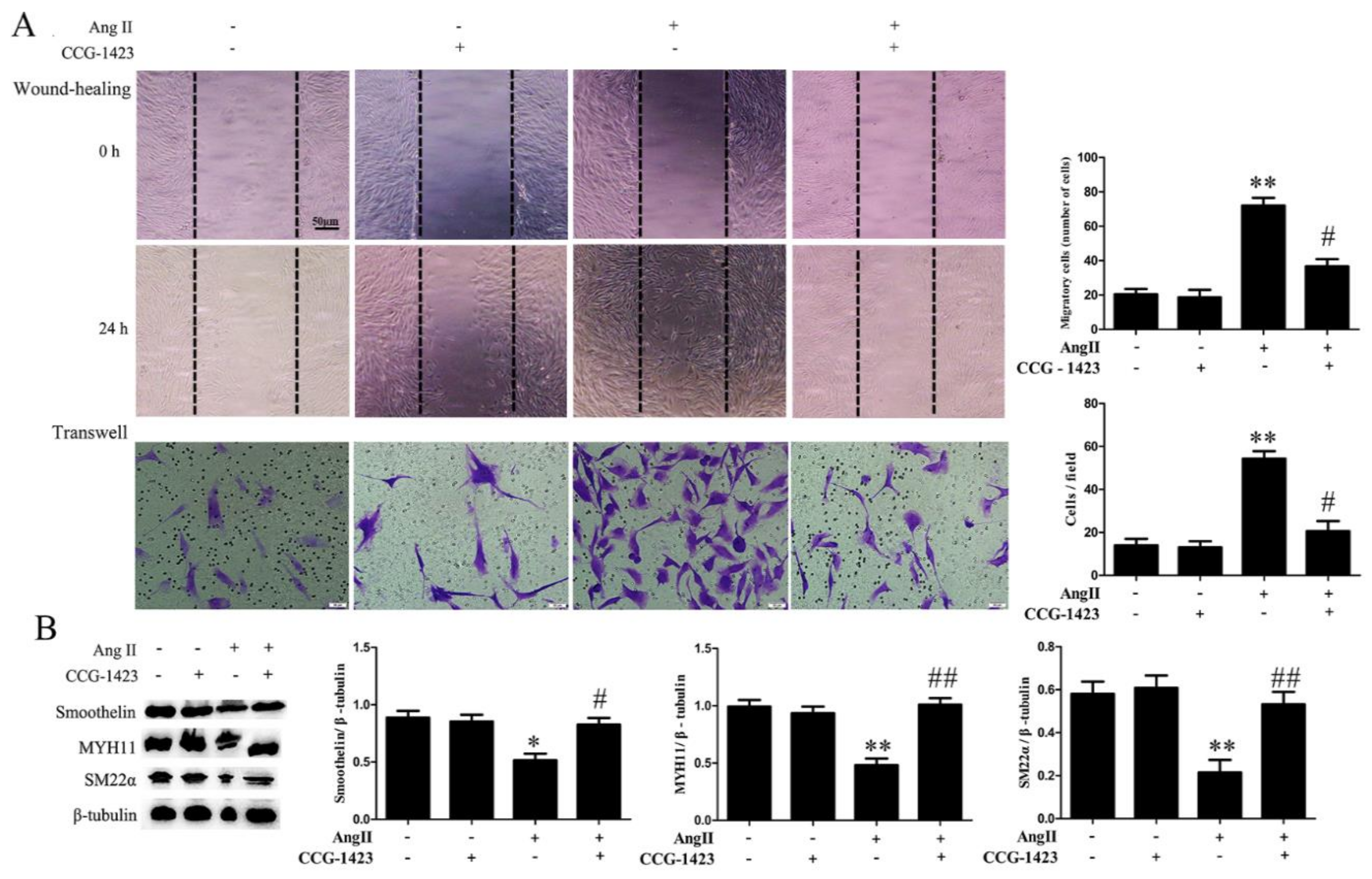
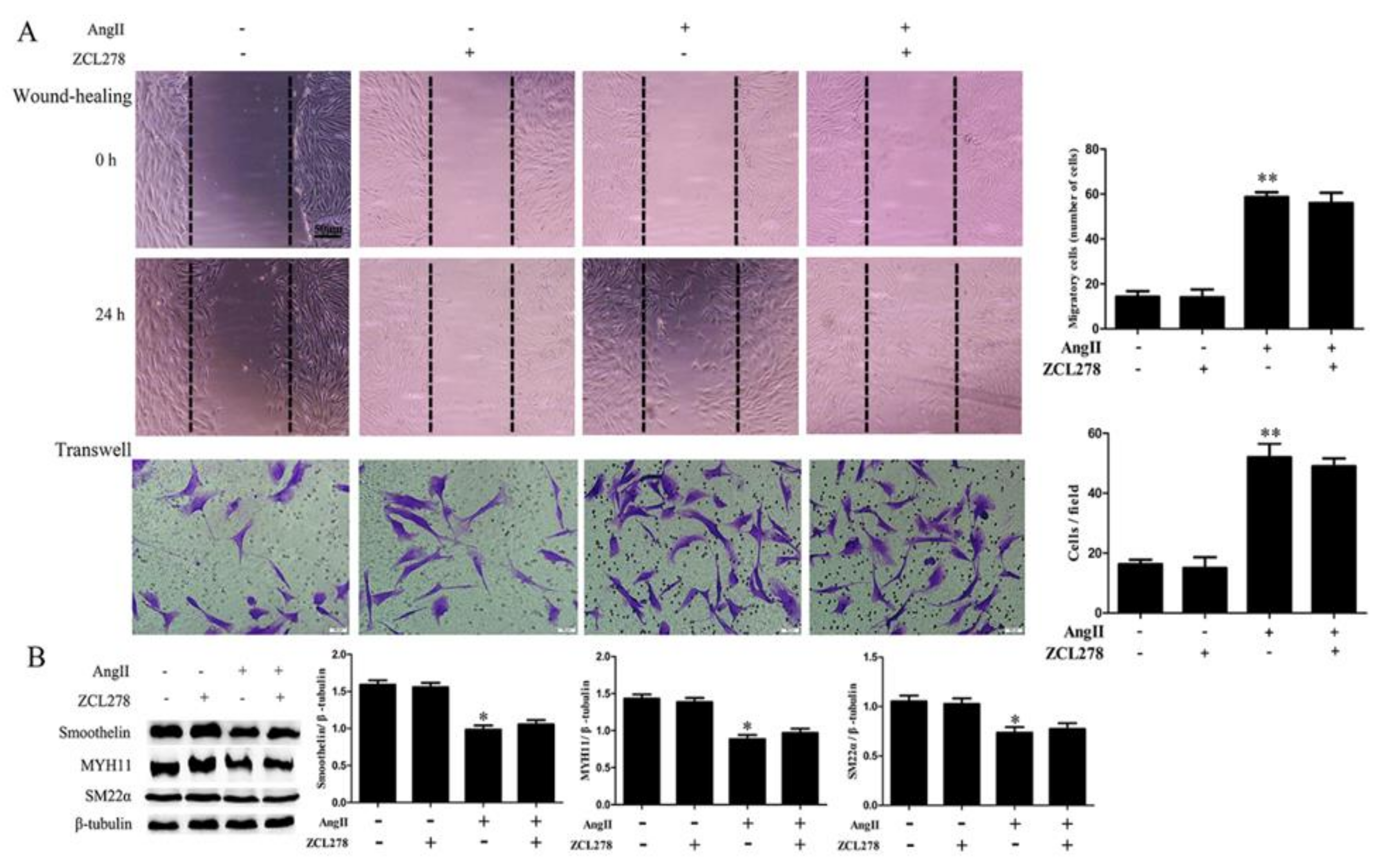
2.2. RhoA Activation Participates in HA-VSMC Migration and Dedifferentiation Induced by Ang II-AT1R Signaling
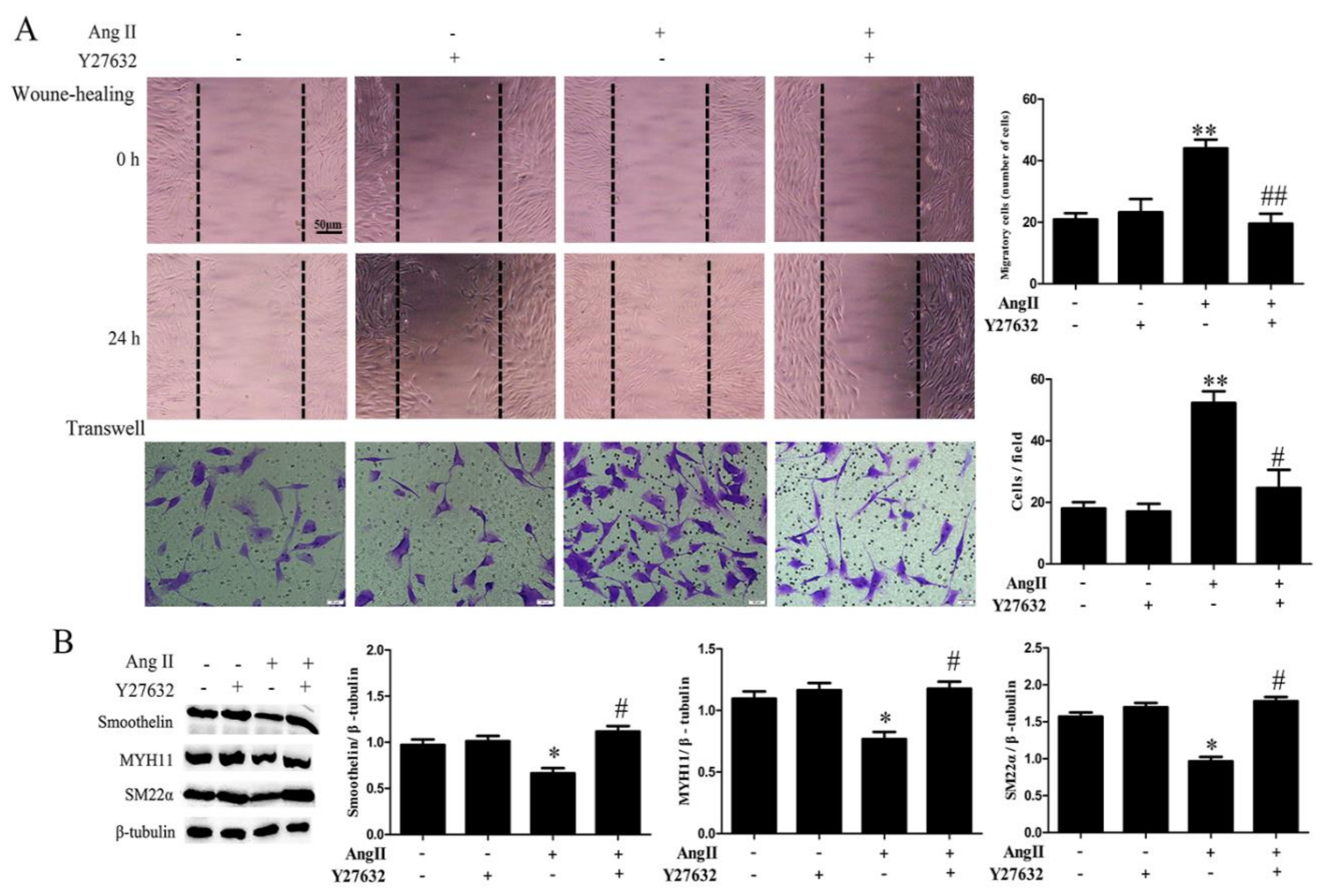
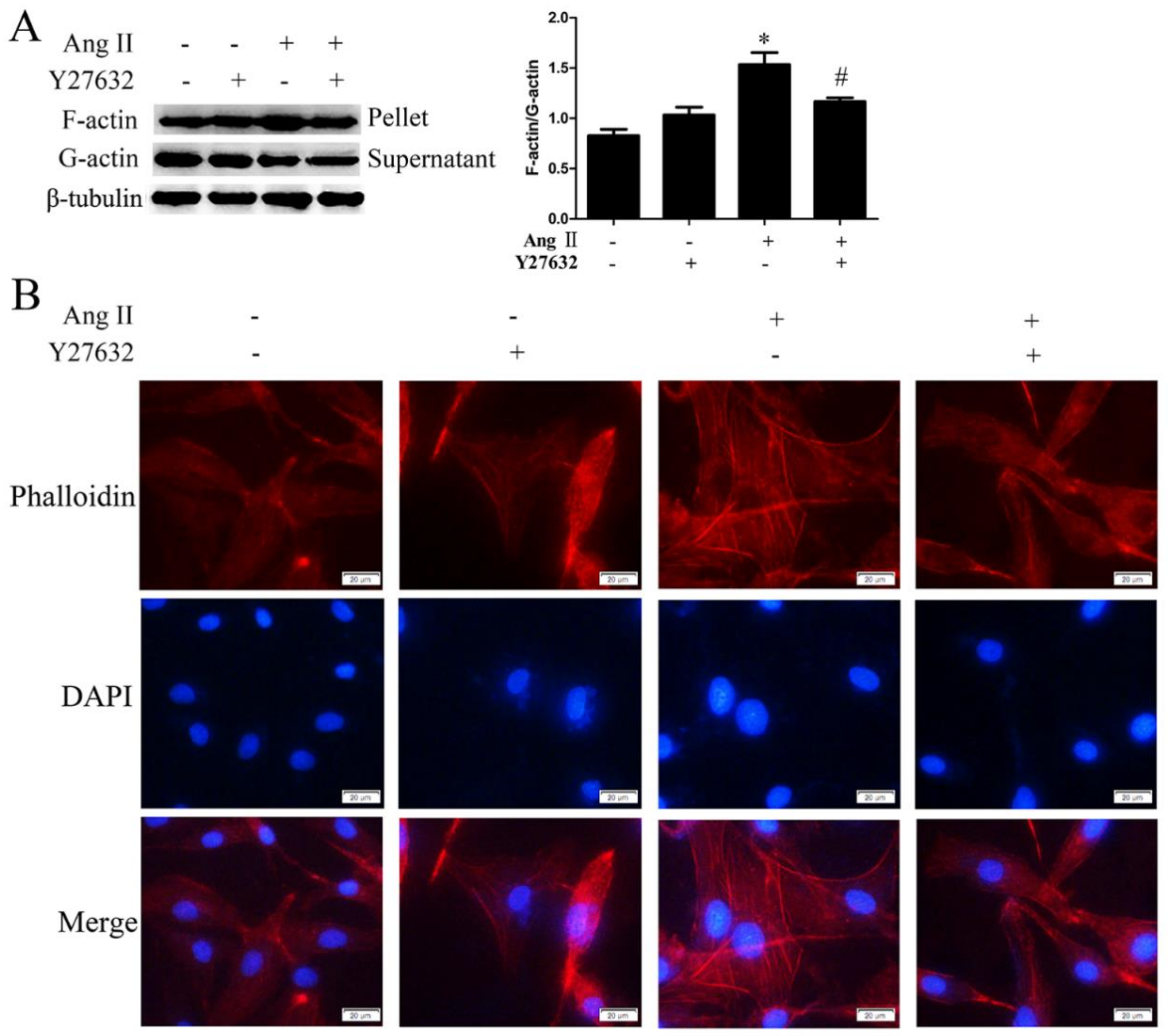
2.3. ROCK Activation Induced by Ang II-AT1R Signaling Participates in HA-VSMC Migration and Dedifferentiation by Promoting Actin Polymerization
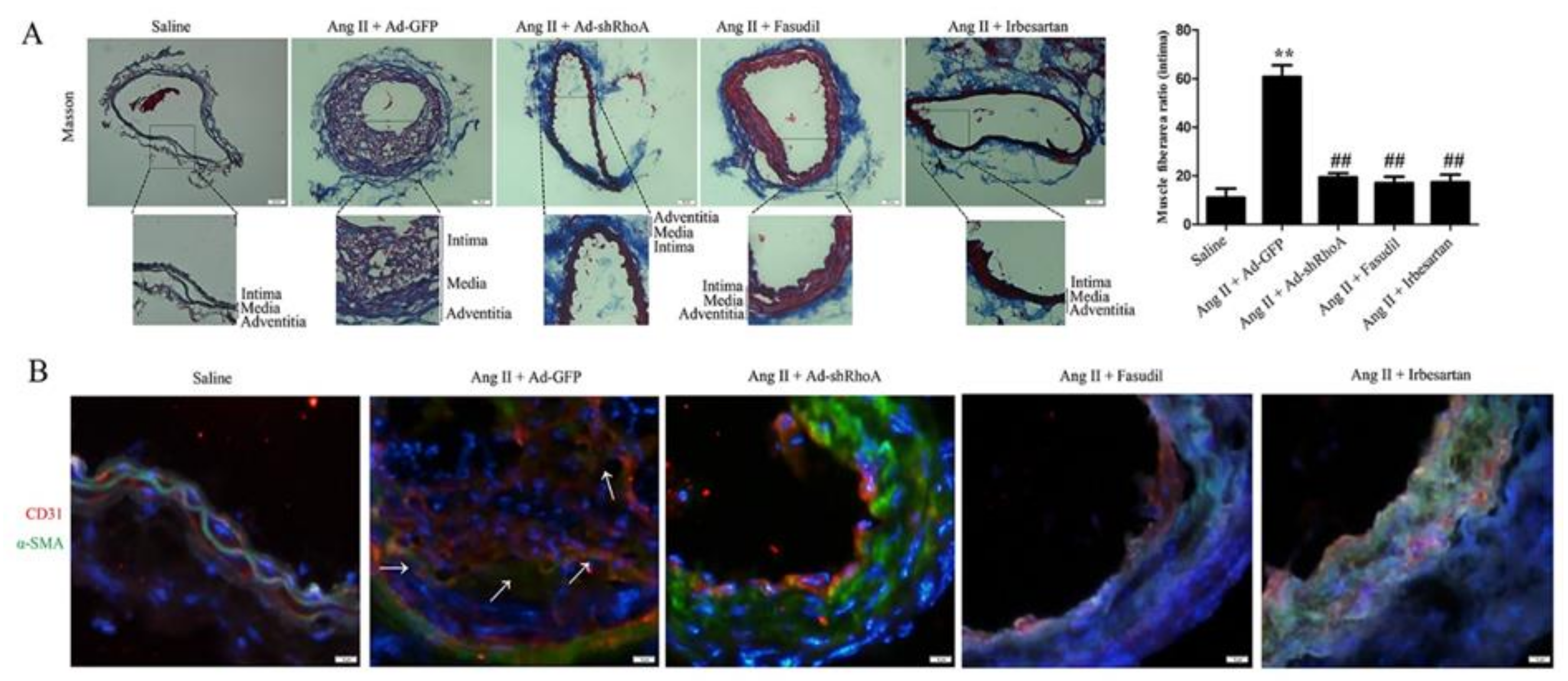
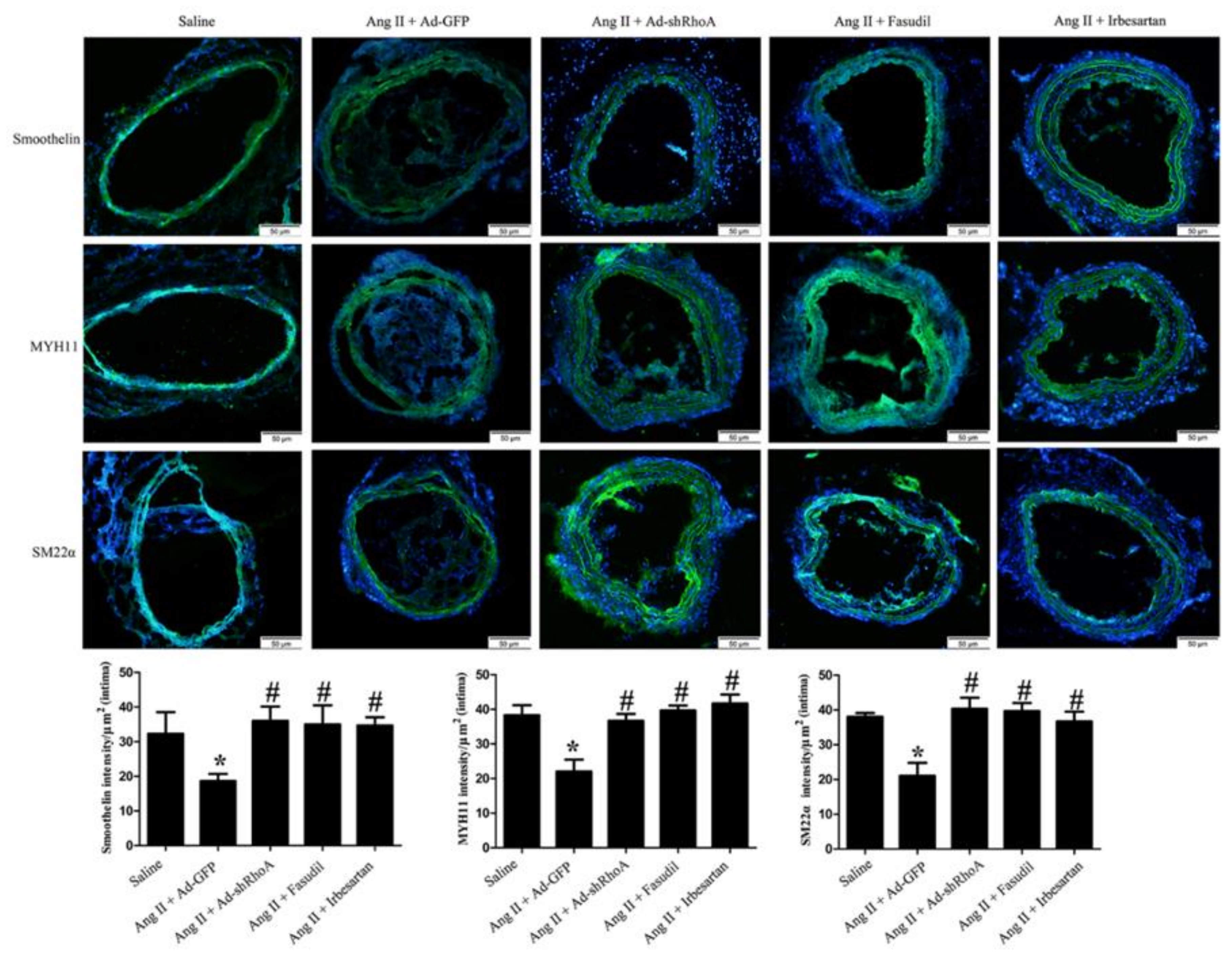
2.4. RhoA/ROCK Pathway Participates in Ang II-Mediated VSMC Migration and Dedifferentiation in Mice
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Wound Healing Assay
4.4. Transwell
4.5. Western Blotting
4.6. Pulldown Assay
4.7. F-actin/G-actin Ratio Assay
4.8. F-actin Morphology
4.9. Animals and Ang II Infusion
4.10. Masson’s Trichrome Staining
4.11. Immunofluorescent Assay
4.12. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Chen, J.; Kitchen, C.M.; Streb, J.W.; Miano, J.M. Myocardin: A component of a molecular switch for smooth muscle differentiation. J. Mol. Cell Cardiol. 2002, 34, 1345–1356. [Google Scholar] [CrossRef]
- Tang, L.; Dai, F.; Liu, Y.; Yu, X.; Huang, C.; Wang, Y.; Yao, W. RhoA/ROCK signaling regulates smooth muscle phenotypic modulation and vascular remodeling via the JNK pathway and vimentin cytoskeleton. Pharmacol. Res. 2018, 133, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Han, Y.; Shen, Y.; Yan, Z.Q.; Zhang, P.; Yao, Q.P.; Shen, B.R.; Gao, L.Z.; Qi, Y.X.; Jiang, Z.L. Endothelial insulin-like growth factor-1 modulates proliferation and phenotype of smooth muscle cells induced by low shear stress. Ann. Biomed. Eng. 2014, 42, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.; Li, C.X.; Cui, X.B.; Tomarev, S.I.; Chen, S.Y. Olfactomedin 2 regulates smooth muscle phenotypic modulation and vascular remodeling through mediating runt-related transcription factor 2 binding to serum response factor. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Zhou, N.; Lee, J.J.; Stoll, S.; Ma, B.; Wiener, R.; Wang, C.; Costa, K.D.; Qiu, H. Inhibition of SRF/myocardin reduces aortic stiffness by targeting vascular smooth muscle cell stiffening in hypertension. Cardiovasc. Res. 2017, 113, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox. Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemarié, C.A.; Schiffrin, E.L. The angiotensin II type 2 receptor in cardiovascular disease. J. Renin. Angiotensin Aldosterone. Syst. 2010, 11, 19–31. [Google Scholar]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Rodríguez, A.; Gómez-Ambrosi, J.; Catalán, V.; Fortuño, A.; Frühbeck, G. Leptin inhibits the proliferation of vascular smooth muscle cells induced by angiotensin II through Nitric Oxide-dependent mechanisms. Mediators Inflamm. 2010, 2010, 105489. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.F.; Ni, G.H.; Chen, M.F.; Li, Y.J.; Wang, Y.J.; Wang, C.L.; Yuan, Q.; Shi, R.Z.; Hu, C.P.; Yang, T.L. Involvement of profilin-1 in angiotensin II-induced vascular smooth muscle cell proliferation. Vascul. Pharmacol. 2011, 55, 34–41. [Google Scholar] [CrossRef]
- Takayanagi, T.; Kawai, T.; Forrester, S.J.; Obama, T.; Tsuji, T.; Fukuda, Y.; Elliott, K.J.; Tilley, D.G.; Davisson, R.L.; Park, J.Y.; et al. Role of epidermal growth factor receptor and endoplasmic reticulum stress in vascular remodeling induced by angiotensin II. Hypertension 2015, 65, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.; Qi, Y.; Guan, W.; Meng, G.; Liu, Z.; Zhang, T.; Yao, W. RhoGDI stability is regulated by SUMOylation and ubiquitination via the AT1 receptor and participates in Ang II-induced smooth muscle proliferation and vascular remodeling. Atherosclerosis 2019, 288, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Boureux, A.; Vignal, E.; Faure, S.; Fort, P. Evolution of the Rho family of ras-like GTPases in eukaryotes. Mol. Biol. Evol. 2007, 24, 203–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govek, E.E.; Newey, S.E.; Van Aelst, L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005, 19, 1–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Wu, X.; Surma, M.; Vemula, S.; Zhang, L.; Yang, Y.; Kapur, R.; Wei, L. Distinct roles for ROCK1 and ROCK2 in the regulation of cell detachment. Cell Death Dis. 2013, 4, e483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schofield, A.V.; Bernard, O. Rho-associated coiled-coil kinase (ROCK) signaling and disease. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 301–316. [Google Scholar] [CrossRef]
- Pan, P.; Shen, M.; Yu, H.; Li, Y.; Li, D.; Hou, T. Advances in the development of Rho-associated protein kinase (ROCK) inhibitors. Drug Discov. Today 2013, 18, 1323–1333. [Google Scholar] [CrossRef]
- Yang, L.; Tang, L.; Dai, F.; Meng, G.; Yin, R.; Xu, X.; Yao, W. Raf-1/CK2 and RhoA/ROCK signaling promote TNF-α-mediated endothelial apoptosis via regulating vimentin cytoskeleton. Toxicology 2017, 389, 74–84. [Google Scholar] [CrossRef]
- Bruder-Nascimento, T.; Chinnasamy, P.; Riascos-Bernal, D.F.; Cau, S.B.; Callera, G.E.; Touyz, R.M.; Tostes, R.C.; Sibinga, N.E. Angiotensin II induces Fat1 expression/activation and vascular smooth muscle cell migration via Nox1-dependent reactive oxygen species generation. J. Mol. Cell Cardiol. 2014, 66, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Kirabo, A.; Oh, S.P.; Kasahara, H.; Wagner, K.U.; Sayeski, P.P. Vascular smooth muscle Jak2 deletion prevents angiotensin II-mediated neointima formation following injury in mice. J. Mol. Cell Cardiol. 2011, 50, 1026–1034. [Google Scholar] [CrossRef] [Green Version]
- Moraes, J.A.; Frony, A.C.; Dias, A.M.; Renovato-Martins, M.; Rodrigues, G.; Marcinkiewicz, C.; Assreuy, J.; Barja-Fidalgo, C. Alpha1beta1 and integrin-linked kinase interact and modulate angiotensin II effects in vascular smooth muscle cells. Atherosclerosis 2015, 243, 477–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arita, R.; Hata, Y.; Nakao, S.; Kita, T.; Miura, M.; Kawahara, S.; Zandi, S.; Almulki, L.; Tayyari, F.; Shimokawa, H.; et al. Rho kinase inhibition by fasudil ameliorates diabetes-induced microvascular damage. Diabetes 2008, 58, 215–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.B.; Wan, M.Y.; Wang, P.Y.; Zhang, C.X.; Xu, D.Y.; Liao, X.; Sun, H.J. Chicoric acid prevents PDGF-BB-induced VSMC dedifferentiation, proliferation and migration by suppressing ROS/NFκB/mTOR/P70S6K signaling cascade. Redox. Biol. 2018, 14, 656–668. [Google Scholar] [CrossRef]
- Liu, W.; Liu, X.; Luo, M.; Liu, X.; Luo, Q.; Tao, H.; Wu, D.; Lu, S.; Jin, J.; Zhao, Y.; et al. dNK derived IFN- g mediates VSMC migration and apoptosis via the induction of LncRNA MEG3: A role in uterovascular transformation. Placenta 2017, 50, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Nour-Eldine, W.; Ghantous, C.M.; Zibara, K.; Dib, L.; Issaa, H.; Itani, H.A.; El-Zein, N.; Zeidan, A. Adiponectin attenuates angiotensin II-induced vascular smooth muscle cell remodeling through nitric oxide and the RhoA/ROCK pathway. Front. Pharmacol. 2016, 7, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.B.; Wang, H.P.; Tang, Z.H.; Cheng, H.; Du, Q.; Wang, Y.B.; Feng, W.B.; Li, K.X.; Cai, W.W.; Qiu, L.Y.; et al. Nesfatin-1 functions as a switch for phenotype transformation and proliferation of VSMCs in hypertensive vascular remodeling. Biochim. Biophys. 2018, 1864, 2154–2168. [Google Scholar] [CrossRef]
- Moriki, N.; Ito, M.; Seko, T.; Kureishi, Y.; Okamoto, R.; Nakakuki, T.; Kongo, M.; Isaka, N.; Kaibuchi, K.; Nakano, T. RhoA activation in vascular smooth muscle cells from stroke-prone spontaneously hypertensive rats. Hypertens. Res. 2004, 27, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Shen, Z.; Du, L.; He, J.; Chen, S.; Zhang, J.; Luan, Y.; Fu, G. Fn14 is regulated via the RhoA pathway and mediates nuclear factor-kappaB activation by Angiotensin II. Am. J. Transl. Res. 2016, 8, 5386–5398. [Google Scholar]
- Liu, L.J.; Yao, F.J.; Lu, G.H.; Xu, C.G.; Xu, Z.; Tang, K.; Cheng, Y.J.; Gao, X.R.; Wu, S.H. The role of the Rho/ROCK pathway in AngII and TGF-β1-induced atrial remodeling. PLoS ONE 2016, 11, e0161625. [Google Scholar]
- Xie-Zukauskas, H.; Das, J.; Short, B.L.; Gutkind, J.S.; Ray, P.E. Heparin inhibits Angiotensin II-induced vasoconstriction on isolated mouse mesenteric resistance arteries through Rho-Aand PKA-dependent pathways. Vascul. Pharmacol. 2013, 58, 313–318. [Google Scholar] [CrossRef] [Green Version]
- Chiu, W.C.; Juang, J.M.; Chang, S.N.; Wu, C.K.; Tsai, C.T.; Tseng, Y.Z.; Chiang, F.T. Angiotensin II regulates the LARG/RhoA/MYPT1 axis in rat vascular smooth muscle in vitro. Acta. Pharmacol. Sin. 2012, 33, 1502–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garvey, S.M.; Sinden, D.S.; Schoppee Bortz, P.D.; Wamhoff, B.R. Cyclosporine up-regulates kru¨ppel-like Factor-4 (KLF4) in vascular smooth muscle cells and drives phenotypic modulation In Vivo. J. Pharmacol. Exp. Ther. 2010, 333, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Bishop, A.L.; Hall, A. Rho GTPases and their effector proteins. Biochem. J. 2000, 348, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Loredo, A.Y.; Lopez, E.; Lopez-Colome, A.M. Thrombin promotes actin stress fiber formation in RPE through Rho/ROCK-mediated MLC phosphorylation. J. Cell. Physiol. 2011, 226, 414–423. [Google Scholar] [CrossRef]
- Jordan, S.N.; Canman, J.C. Rho GTPases in animal cell cytokinesis: An occupation by the one percent. Cytoskeleton 2012, 69, 919–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.H.; Wu, C.C.; Hsu, S.H.; Liou, J.Y.; Li, Y.W.; Wu, K.K.; Lai, Y.K.; Yen, B.L. The role of RhoA kinase inhibition in human placenta-derived multipotent cells on neural phenotype and cell survival. Biomaterials 2013, 34, 3223–3230. [Google Scholar] [CrossRef] [PubMed]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, Y.; Liang, X.; Dai, F.; Guan, H.; Sun, J.; Yao, W. RhoA/ROCK Pathway Activation is Regulated by AT1 Receptor and Participates in Smooth Muscle Migration and Dedifferentiation via Promoting Actin Cytoskeleton Polymerization. Int. J. Mol. Sci. 2020, 21, 5398. https://doi.org/10.3390/ijms21155398
Qi Y, Liang X, Dai F, Guan H, Sun J, Yao W. RhoA/ROCK Pathway Activation is Regulated by AT1 Receptor and Participates in Smooth Muscle Migration and Dedifferentiation via Promoting Actin Cytoskeleton Polymerization. International Journal of Molecular Sciences. 2020; 21(15):5398. https://doi.org/10.3390/ijms21155398
Chicago/Turabian StyleQi, Yan, Xiuying Liang, Fan Dai, Haijing Guan, Jingwen Sun, and Wenjuan Yao. 2020. "RhoA/ROCK Pathway Activation is Regulated by AT1 Receptor and Participates in Smooth Muscle Migration and Dedifferentiation via Promoting Actin Cytoskeleton Polymerization" International Journal of Molecular Sciences 21, no. 15: 5398. https://doi.org/10.3390/ijms21155398
APA StyleQi, Y., Liang, X., Dai, F., Guan, H., Sun, J., & Yao, W. (2020). RhoA/ROCK Pathway Activation is Regulated by AT1 Receptor and Participates in Smooth Muscle Migration and Dedifferentiation via Promoting Actin Cytoskeleton Polymerization. International Journal of Molecular Sciences, 21(15), 5398. https://doi.org/10.3390/ijms21155398