Resisting Resistance to Immune Checkpoint Therapy: A Systematic Review




Abstract
:1. Introduction
2. Mechanisms of Resistance to Immunotherapy and Ways to Overcome Them
- (1)
- Immune recognition via the alteration of antigen presentation, such as the downregulation of MHC-I, for example [18];
- (2)
- (3)
- (4)
- The DNA damage response [26];
- (5)
- The expression of various different checkpoints in secondary resistance settings to halt T-cell activation, including LAG-3, TIM-3, BTLA, TIGIT, VISTA, etc. [27].
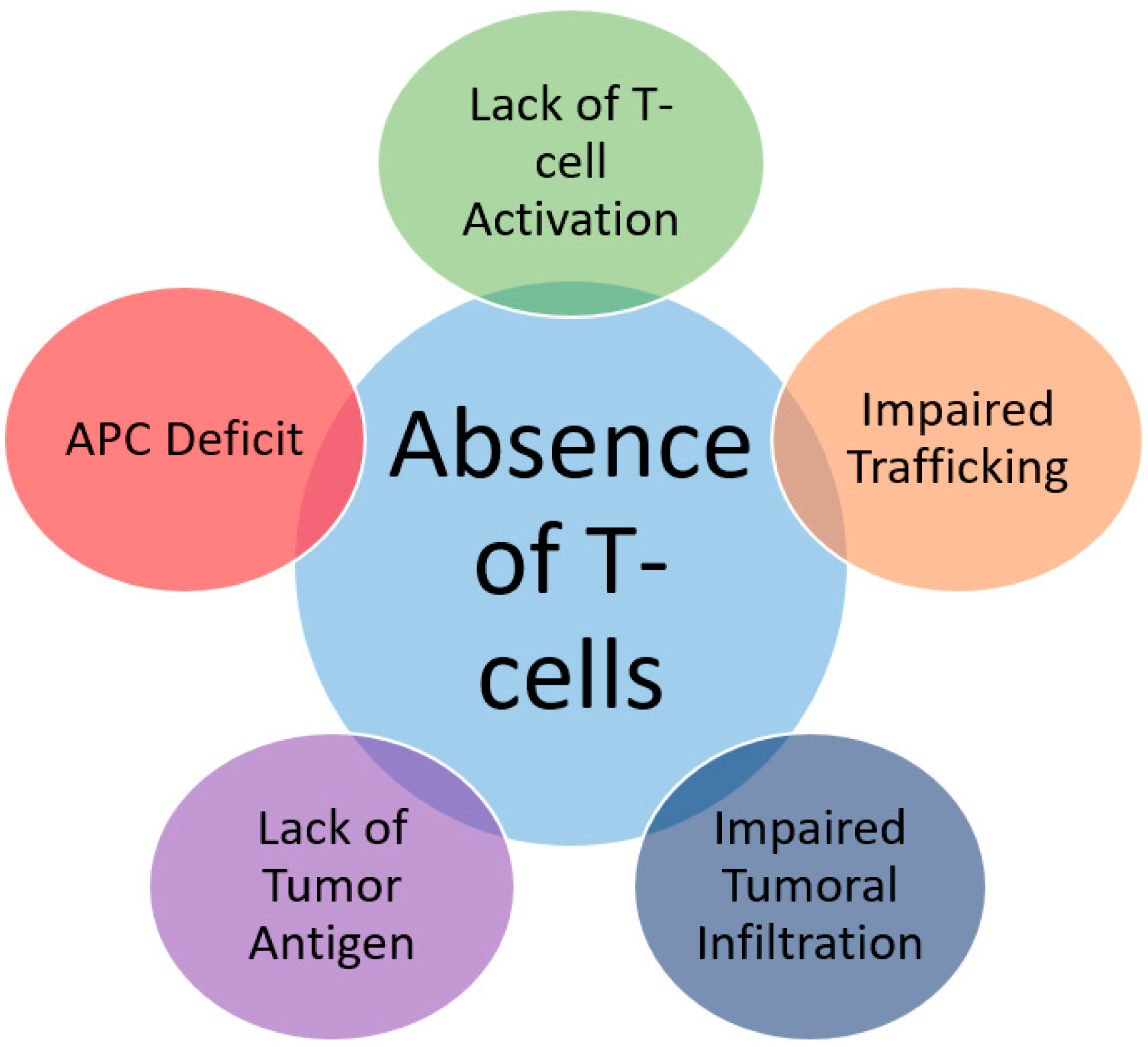
2.1. Inadequate T-Cell Infiltration due to Lack of Tumor Antigens and Absence of Antigen Presentation
2.1.1. Absence of Antigenic Protein
2.1.2. Absence of Antigen Presentation
Beta2-Microglobulin (β2m)
Transporters Associated with Antigen Processing (TAP)
Pattern Recognition Receptors
2.2. Genetic T-Cell Exclusion and Insensibility to T-Cells
2.2.1. MAPK Oncogenic Signaling
2.2.2. Tumor Suppressor Phosphatase and Tensin Homolog (PTEN)
2.2.3. WNT/Beta-Catenin Signaling Pathway
2.2.4. Vascular Endothelial Growth Factor (VEGF)
2.2.5. Transforming Growth Factor-β (TGF-β)
2.2.6. Indoleamine 2,3-Dioxygenase (IDO)
2.2.7. Interferon-γ Receptor Pathway
2.2.8. Enhancer of Zester Homolog-2 (Ezh2)
2.3. Impairment of T-Cell Functionality by Immunosuppressive Signaling Receptors
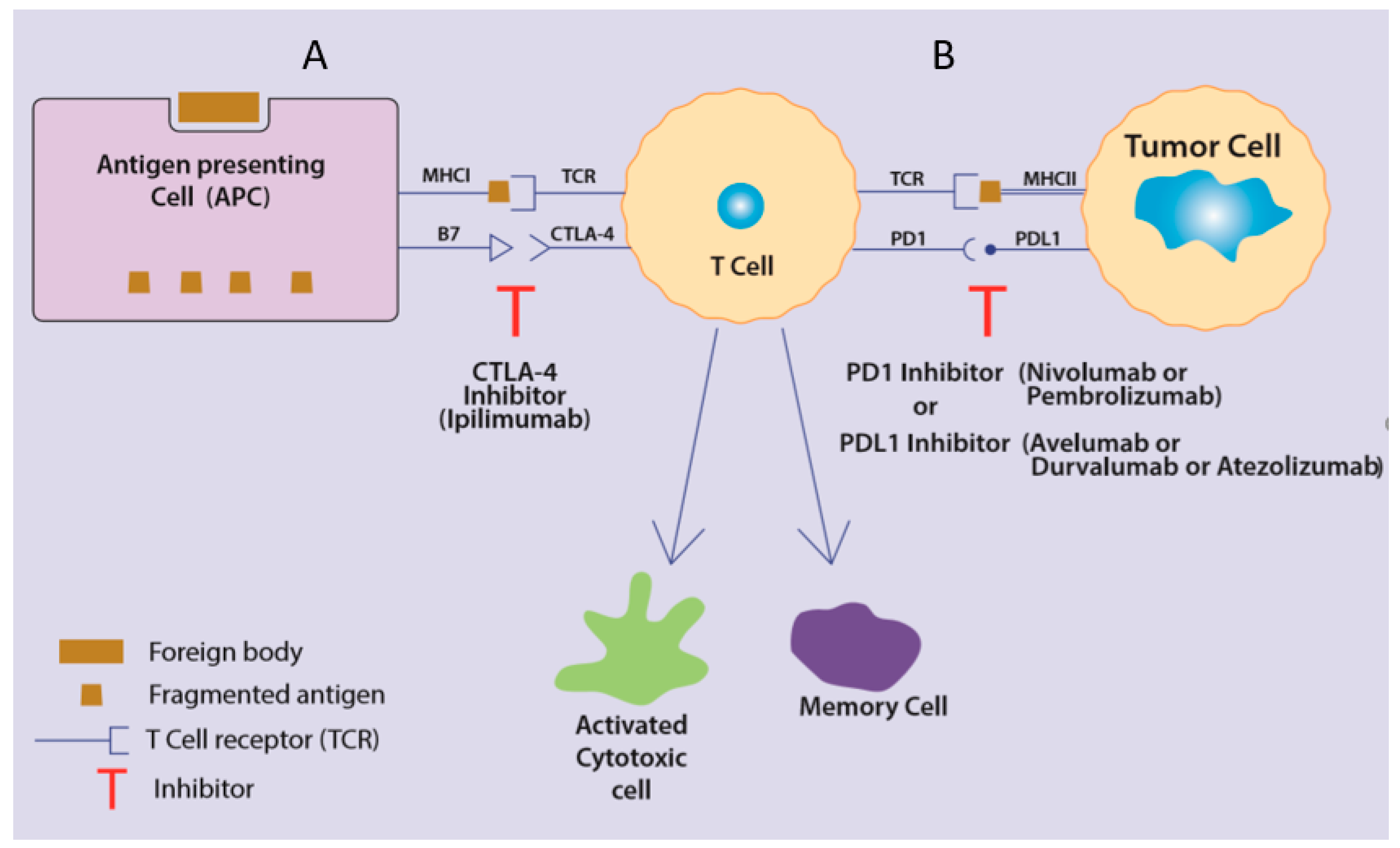
2.3.1. Cytotoxic T-Lymphocyte-Associated Protein 4 (CTLA-4)
2.3.2. Programmed Cell Death Protein-1 (PD-1) and Programmed Cell Death Protein Ligand-1 (PDL-1)
2.3.3. Lymphocyte Activation Gene-3 (LAG-3)
2.3.4. T-Cell Immunoglobulin Mucin-3 (TIM-3)
2.3.5. V-Domain Ig Suppressor of T-Cell Activation (VISTA)
2.3.6. T-Cell Immuno-Receptor with Ig and ITIM Domains (TIGIT)
2.3.7. B and T lymphocyte Attenuator 4 (BTLA-4)
2.4. Lack of Stimulatory Checkpoints Promoting Immune Escape
2.4.1. ICOS
2.4.2. OX40
2.4.3. Glucocorticoid-Induced Tumor Necrosis Factor Receptor (GITR)
2.5. Immunosuppressive Cells within the Tumor Micro-Environment
2.5.1. Tumor Associated Macrophages (TAMs)
2.5.2. Regulatory T-Cells (T Regs)
2.5.3. Myeloid-Derived Suppressor Cells (MDSCs)
2.5.4. Innate Lymphoid Cells (ILCs)
2.6. Immune Metabolism and Resistance to Immunotherapy
2.6.1. Dysregulation of Immune Metabolism at the Cellular Level
Lactate
Hypoxia
Arachidonate Metabolism
Pyruvate Kinase Muscle
Adenosine Pathway
2.6.2. Dysregulation of Immune Metabolism at the Host Level
Obesity
Caloric Deficit
Gender Effect
Microbiota
3. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| APC | Antigen Presenting Cell |
| ACT | Adoptive cell therapy |
| β2m | Beta2-microglobulin |
| BTLA | B and T lymphocyte attenuator |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 |
| CSF1 | Colony stimulating factor 1 |
| CSF1R | CSF1 receptor |
| DCs | Dendritic cells |
| DAMPs | Damage-Associated Molecular Patterns |
| DNMTi | DNA methyl-transferase Inhibitors |
| Ezh2 | Enhancer of zester Homolog-2 |
| FPR-1 | Formyl Peptide Receptor-1 |
| GITR | Glucocorticoid-Induced Tumor Necrosis Factor Receptor |
| HPD | Hyperprogressive Disease |
| HPV | Human papillomavirus 16 |
| HVEM | Herpesvirus entry mediator |
| ICI | Immune checkpoint inhibitor |
| IDO | Indole-amine 2,3-dioxygenase |
| Ig | Immunoglobulin |
| INF | Interferon |
| IL | Interleukin |
| ILCs | Innate lymphoid cells |
| ICOS-L | ICOS-ligand |
| LAG-3 | Lymphocyte Activation Gene-3 |
| MHC-I | Major Histocompatibility Complex-I |
| MAPK | Mitogen-Activated Protein kinase |
| MDSCs | Myeloid-Derived Suppressor Cells |
| NSCLC | Non-small cell lung cancer |
| NK | Natural killer |
| NF-κ B | Nuclear factor-κ B |
| PAMPs | Pathogen-associated molecular patterns |
| PD-1 | Programmed cell death protein-1 |
| PDL-1 | Programmed cell death protein ligand-1 |
| PTEN | Tumor Suppressor Phosphatase and Tensin Homolog |
| PRRs | Pattern Recognition Receptors |
| PI3K | Phosphatidylinositol 3-kinase |
| RCC | Renal cell carcinoma |
| RT | Radiation therapy |
| STAT1 | Signal transducer and activator of transcription 1 |
| TCR | T-cell receptor |
| Treg | Regulatory T-cells |
| TSA | Tumor specific antigen |
| TAMs | Tumor-Associated Macrophages |
| TILs | Tumor Infiltrating Lymphocytes |
| TMB | Tumor Mutational Burden |
| TAP | Transporters Associated with Antigen Processing |
| TME | Tumor microenvironment |
| TGF-β | Transforming growth factor beta |
| TNF-α | Tumor necrosis factor alpha |
| TIM-3 | T-cell Immunoglobulin Mucin-3 |
| TGR | Tumor growth rate |
| VEGF | Vascular endothelial growth factor |
| VISTA | V-domain Ig suppressor of T-cell activation |
| 1MT | 1-methyl-tryptophan |
References
- McCarthy, E.F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. 2006, 26, 154–158. [Google Scholar]
- Ribatti, D. The concept of immune surveillance against tumors. The first theories. Oncotarget 2017, 8, 7175–7180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immuno- surveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Schreiber, R.D. Cancer immunoediting: Antigens, mechanisms, and implications to cancer immunotherapy. Ann. N. Y. Acad. Sci. 2013, 1284, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, R.; Gentry, S.N.; Jackson, T.L. Pathways to tumorigenesis—Modeling mutation acquisition in stem cells and their progeny. Neoplasia 2008, 10, 1170–1182. [Google Scholar] [CrossRef] [Green Version]
- Kim, S. New and emerging factors in tumorigenesis: An overview. Cancer Manag. Res. 2015, 7, 225–239. [Google Scholar] [CrossRef] [Green Version]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases—Elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudino, S.J.; Kumar, P. Cross-Talk Between Antigen Presenting Cells and T Cells Impacts Intestinal Homeostasis, Bacterial Infections, and Tumorigenesis. Front. Immunol. 2019, 10, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Ceeraz, S.; Nowak, E.C.; Burns, C.M.; Noelle, R.J. Immune checkpoint receptors in regulating immune reactivity in rheumatic disease. Arthritis Res. Ther. 2014, 16, 469. [Google Scholar] [CrossRef] [Green Version]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Company, B.-M.S. FDA Approves YERVOY™ (ipilimumab) for the Treatment of Patients with Newly Diagnosed or Previously-Treated Unresectable or Metastatic Melanoma, the Deadliest form of Skin Cancer. 2011. Available online: https://news.bms.com/press-release/rd-news/fda-approves-yervoy-ipilimumab-treatment-patients-newly-diagnosed-or-previousl (accessed on 1 May 2020).
- Wei, S.C.; Anang, N.A.A.; Sharma, R.; Andrews, M.C.; Reuben, A.; Levine, J.H.; Allison, J.P. Combination anti-CTLA-4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc. Natl. Acad. Sci. USA 2019, 116, 22699–22709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Zhang, L.; Yu, J.; Zhang, Y.; Pang, X.; Ma, C.; Qiu, S. Clinical efficacy and safety of anti-PD-1/PD-L1 inhibitors for the treatment of advanced or metastatic cancer: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 2083. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Zitvogel, L. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, S.H.; Keam, B.; Ock, C.Y.; Kim, S.; Han, B.; Kim, J.W.; Kwon, S.K. Prognostic value of the association between MHC class I downregulation and PD-L1 upregulation in head and neck squamous cell carcinoma patients. Sci. Rep. 2019, 9, 7680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Aranda, M.; Redondo, M. Targeting Protein Kinases to Enhance the Response to anti-PD-1/PD-L1 Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2296. [Google Scholar] [CrossRef] [Green Version]
- Ni, L.; Lu, J. Interferon gamma in cancer immunotherapy. Cancer Med. 2018, 7, 4509–4516. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Williams, L.J. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Eng, C. PTEN Mutations Trigger Resistance to Immunotherapy. Trends Mol. Med. 2019, 25, 461–463. [Google Scholar] [CrossRef]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Palaskas, N. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Brea, L.T.; Yu, J. Immune modulatory functions of EZH2 in the tumor microenvironment: Implications in cancer immunotherapy. Am. J. Clin. Exp. Urol. 2019, 7, 85–91. [Google Scholar]
- Trujillo, J.A.; Luke, J.J.; Zha, Y.; Segal, J.P.; Ritterhouse, L.L.; Spranger, S.; Gajewski, T.F. Secondary resistance to immunotherapy associated with β-catenin pathway activation or PTEN loss in metastatic melanoma. J. Immunother. Cancer 2019, 7, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [Green Version]
- De Sousa Linhares, A.; Leitner, J.; Grabmeier-Pfistershammer, K.; Steinberger, P. Not All Immune Checkpoints Are Created Equal. Front. Immunol. 2018, 9, 1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shitara, K.; Nishikawa, H. Regulatory T cells: A potential target in cancer immunotherapy. Ann. N. Y. Acad. Sci. 2018, 1417, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassetta, L.; Kitamura, T. Targeting Tumor-Associated Macrophages as a Potential Strategy to Enhance the Response to Immune Checkpoint Inhibitors. Front. Cell Dev. Biol. 2018, 6, 38. [Google Scholar] [CrossRef]
- Martin-Orozco, E.; Sanchez-Fernandez, A.; Ortiz-Parra, I.; Ayala-San Nicolas, M. WNT Signaling in Tumors: The Way to Evade Drugs and Immunity. Front. Immunol. 2019, 10, 2854. [Google Scholar] [CrossRef]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riaz, N.; Morris, L.; Havel, J.J.; Makarov, V.; Desrichard, A.; Chan, T.A. The role of neoantigens in response to immune checkpoint blockade. Int. Immunol. 2016, 28, 411–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelderman, S.; Kvistborg, P. Tumor antigens in human cancer control. Biochim. Biophys. Acta 2016, 1865, 83–89. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Keskin, D.B.; Reinhold, B.; Lee, S.Y.; Zhang, G.; Lank, S.; O’Connor, D.; Reinherz, E.L. Direct identification of an HPV-16 tumor antigen from cervical cancer biopsy specimens. Front. Immunol. 2011, 2, 75. [Google Scholar] [PubMed] [Green Version]
- Jabbar, B.; Rafique, S.; Salo-Ahen, O.M.; Ali, A.; Munir, M.; Idrees, M.; Rana, M.A. Antigenic Peptide Prediction From E6 and E7 Oncoproteins of HPV Types 16 and 18 for Therapeutic Vaccine Design Using Immunoinformatics and MD Simulation Analysis. Front. Immunol. 2018, 9, 3000. [Google Scholar] [CrossRef] [Green Version]
- Coulie, P.G.; Van den Eynde, B.J.; Van Der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Yarchoan, M.; Johnson, B.A., III; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Fisk, B.; Blevins, T.L.; Wharton, J.T.; Ioannides, C.G. Identification of an immunodominant peptide of HER-2/neu protooncogene recognized by ovarian tumor-specific cytotoxic T lymphocyte lines. J. Exp. Med. 1995, 181, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Miller, M.L. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D. Death-Ligand 1 (PD-L1) Blockade in Patients With Non-Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. J. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Utikal, J. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Anagnostou, V.; Smith, K.; Forde, P.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Luke, J.J.; Bao, R.; Zha, Y.; Hernandez, K.M.; Li, Y.; Gajewski, A.P.; Andrade, J.; Gajewski, T.F. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, E7759–E7768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Velasco, G.; Miao, D.; Voss, M.; Hakimi, A.; Hsieh, J.; Tannir, N.; Tamboli, P.; Appleman, L.; Rathmell, W.; Van Allen, E. Tumor Mutational Load and Immune Parameters across Metastatic Renal Cell Carcinoma Risk Groups. Cancer Immunol. Res. 2016, 4, 820–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chodon, T.; Comin-Anduix, B.; Chmielowski, B.; Koya, R.C.; Wu, Z.; Auerbach, M.; Ng, C.; Avramis, E.; Seja, E.; Villanueva, A. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin. Cancer Res. 2014, 20, 2457–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Somaiah, N.; Block, M.S.; Kim, J.W.; Shapiro, G.I.; Do, K.T.; Hwu, P.; Eder, J.P.; Jones, R.L.; Lu, H.; Ter Meulen, J.H. First-in-Class, First-in-Human Study Evaluating LV305, a Dendritic-Cell Tropic Lentiviral Vector, in Sarcoma and Other Solid Tumors Expressing NY-ESO-1. Clin. Cancer Res. 2019, 25, 5808–5817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Ranganathan, R.; Eruslanov, E.; Kim, S.; Newick, K.; O’Brien, S.; Lo, A.; Liu, X.; Zhao, Y.; Albelda, S.M. Blockade of Programmed Death 1 Augments the Ability of Human T Cells Engineered to Target NY-ESO-1 to Control Tumor Growth after Adoptive Transfer. Clin. Cancer Res. 2016, 22, 436–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenblatt, J.; Glotzbecker, B.; Mills, H.; Vasir, B.; Tzachanis, D.; Levine, J.D.; Joyce, R.M.; Wellenstein, K.; Keefe, W.; Schickler, M. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J. Immunother. 2011, 34, 409–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, P.; Paluch, B.E.; Matsuzaki, J.; James, S.R.; Collamat-Lai, G.; Karbach, J.; Nemeth, M.J.; Taverna, P.; Karpf, A.R.; Griffiths, E.A. Immunomodulatory action of SGI-110, a hypomethylating agent, in acute myeloid leukemia cells and xenografts. Leuk. Res. 2014, 38, 1332–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Restifo, N.P.; Marincola, F.M.; Kawakami, Y.; Taubenberger, J.; Yannelli, J.R.; Rosenberg, S.A. Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J. Natl. Cancer Inst. 1996, 88, 100–108. [Google Scholar] [CrossRef]
- Porgador, A.; Mandelboim, O.; Restifo, N.P.; Strominger, J.L. Natural killer cell lines kill autologous beta2-microglobulin-deficient melanoma cells: Implications for cancer immunotherapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13140–13145. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Li, Y.; Liu, Y.Q.; Li, L.; Liu, J.; Shen, X.; Shen, G.X.; Tu, Y.T. Expression of transporters associated with antigen processing and human leucocyte antigen class I in malignant melanoma and its association with prognostic factors. Br. J. Dermatol. 2008, 158, 88–94. [Google Scholar] [CrossRef]
- Vitale, M.; Rezzani, R.; Rodella, L.; Zauli, G.; Grigolato, P.; Cadei, M.; Hicklin, D.J.; Ferrone, S. HLA class I antigen and transporter associated with antigen processing (TAP1 and TAP2) down-regulation in high-grade primary breast carcinoma lesions. Cancer Res. 1998, 58, 737–742. [Google Scholar]
- Momburg, F.; Müllbacher, A.; Lobigs, M. Modulation of transporter associated with antigen processing (TAP)-mediated peptide import into the endoplasmic reticulum by flavivirus infection. J. Virol. 2001, 75, 5663–5671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turvey, S.E.; Broide, D.H. Innate immunity. J. Allergy Clin. Immunol. 2010, 125 (Suppl. S2), S24–S32. [Google Scholar] [CrossRef] [PubMed]
- Ten Broeke, T.; Wubbolts, R.; Stoorvogel, W. MHC class II antigen presentation by dendritic cells regulated through endosomal sorting. Cold Spring Harb. Perspect. Biol. 2013, 5, a016873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgdorf, S.; Kautz, A.; Böhnert, V.; Knolle, P.A.; Kurts, C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 2007, 316, 612–616. [Google Scholar] [CrossRef]
- Burgdorf, S.; Kurts, C. Endocytosis mechanisms and the cell biology of antigen presentation. Curr. Opin. Immunol. 2008, 20, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeway Jr, C.A.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [Green Version]
- Reis e Sousa, CDendritic cells in a mature age. Nat. Rev. Immunol. 2006, 6, 476–483. [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 2009, 9, 353–363. [Google Scholar] [CrossRef]
- Vacchelli, E.; Ma, Y.; Baracco, E.E.; Sistigu, A.; Enot, D.P.; Pietrocola, F.; Yang, H.; Adjemian, S.; Chaba, K.; Semeraro, M. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 2015, 350, 972–978. [Google Scholar] [CrossRef]
- Shekarian, T.; Valsesia-Wittmann, S.; Brody, J.; Michallet, M.; Depil, S.; Caux, C.; Marabelle, A. Pattern recognition receptors: Immune targets to enhance cancer immunotherapy. Ann. Oncol. 2017, 28, 1756–1766. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.J.; Kasper, S.; Vokes, E.E.; Even, C. Nivolumab vs investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck: 2-year long-term survival update of CheckMate 141 with analyses by tumor PD-L1 expression. Oral Oncol. 2018, 81, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. Cancer Immunotherapy, Part 2: Efficacy, Safety, and Other Clinical Considerations. P T 2017, 42, 452–463. [Google Scholar] [PubMed]
- Sharabi, A.B.; Lim, M.; DeWeese, T.L.; Drake, C.G. Radiation and checkpoint blockade immunotherapy: Radiosensitisation and potential mechanisms of synergy. Lancet Oncol. 2015, 16, e498–e509. [Google Scholar] [CrossRef]
- Eriksson, D.; Stigbrand, T. Radiation-induced cell death mechanisms. Tumour Biol. 2010, 31, 363–372. [Google Scholar] [CrossRef]
- Chajon, E.; Castelli, J.; Marsiglia, H.; De Crevoisier, R. The synergistic effect of radiotherapy and immunotherapy: A promising but not simple partnership. Crit. Rev. Oncol. Hematol. 2017, 111, 124–132. [Google Scholar] [CrossRef]
- Zhang, P.; Su, D.-M.; Liang, M.; Fu, J. Chemopreventive agents induce programmed death-1-ligand 1 (PD-L1) surface expression in breast cancer cells and promote PD-L1-mediated T cell apoptosis. Mol. Immunol. 2008, 45, 1470–1476. [Google Scholar] [CrossRef]
- Corso, C.D.; Ali, A.N.; Diaz, R. Radiation-induced tumor neoantigens: Imaging and therapeutic implications. Am. J. Cancer Res. 2011, 1, 390–412. [Google Scholar]
- Demaria, S.; Kawashima, N.; Yang, A.M.; Devitt, M.L.; Babb, J.S.; Allison, J.P.; Formenti, S.C. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. 2005, 11 Pt 1, 728–734. [Google Scholar]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef]
- Kepp, O.; Galluzzi, L.; Martins, I.; Schlemmer, F.; Adjemian, S.; Michaud, M.; Sukkurwala, A.Q.; Menger, L.; Zitvogel, L.; Kroemer, G. Molecular determinants of immunogenic cell death elicited by anticancer chemotherapy. Cancer Metastasis Rev. 2011, 30, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.; Middleton, G. The interplay of immunotherapy and chemotherapy: Harnessing potential synergies. Cancer Immunol. Res. 2015, 3, 436–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Peng, W.; Xu, C.; Lou, Y.; Zhang, M.; Wargo, J.A.; Chen, J.Q.; Li, H.S.; Watowich, S.S.; Yang, Y. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin. Cancer Res. 2013, 19, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Hu-Lieskovan, S.; Mok, S.; Moreno, B.H.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci. Transl. Med. 2015, 7, 279ra41. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.-Y.; Hopson, C. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 2015, 21, 1639–1651. [Google Scholar] [CrossRef] [Green Version]
- Bendell, J.C.; Kim, T.W.; Goh, B.C.; Wallin, J.; Oh, D.-Y.; Han, S.-W.; Lee, C.B.; Hellmann, M.D.; Desai, J.; Lewin, J.H. Clinical activity and safety of cobimetinib (cobi) and atezolizumab in colorectal cancer (CRC). J. Clin. Oncol. 2016, 34, 3502. [Google Scholar] [CrossRef]
- Ribas, A.; Butler, M.; Lutzky, J.; Lawrence, D.P.; Robert, C.; Miller, W.; Linette, G.P.; Ascierto, P.A.; Kuzel, T.; Algazi, A.P. Phase I study combining anti-PD-L1 (MEDI4736) with BRAF (dabrafenib) and/or MEK (trametinib) inhibitors in advanced melanoma. J. Clin. Oncol. 2015, 33, 3003. [Google Scholar] [CrossRef]
- Kaur, S.; Uddin, S.; Platanias, L.C. The PI3’ kinase pathway in interferon signaling. J. Interferon Cytokine Res. 2005, 25, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, E.; Kawazoe, A.; Kuboki, Y.; Komatsu, Y.; Nishina, T.; Hara, H.; Yuki, S.; Shitara, K.; Bando, H.; Kotani, D. Multicenter phase I/II trial of BBI608 and pembrolizumab combination in patients with metastatic colorectal cancer (SCOOP Study): EPOC1503. J. Clin. Oncol. 2018, 36, 3530. [Google Scholar] [CrossRef]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.-L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Ohm, J.E.; Carbone, D.P. VEGF as a mediator of tumor-associated immunodeficiency. Immunol. Res. 2001, 23, 263–272. [Google Scholar] [CrossRef]
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.; Lawrence, D.; Lezcano, C.; Wu, X.; Zhou, J.; Sasada, T.; Zeng, W.; Giobbie-Hurder, A.; Atkins, M.; Ibrahim, N. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol. Res. 2014, 2, 632–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, M.; Jiao, D.; Qin, S.; Chu, Q.; Wu, K.; Li, A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol. Cancer 2019, 18, 60. [Google Scholar] [CrossRef]
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Yi, M.; Jiao, Y.; Chu, Q.; Wu, K. Blocking TGF-β Signaling To Enhance The Efficacy Of Immune Checkpoint Inhibitor. Onco Targets Ther. 2019, 12, 9527–9538. [Google Scholar] [CrossRef] [Green Version]
- Eberlein, C.; Kendrew, J.; McDaid, K.; Alfred, A.; Kang, J.; Jacobs, V.; Ross, S.; Rooney, C.; Smith, N.; Rinkenberger, J. A human monoclonal antibody 264RAD targeting αvβ6 integrin reduces tumour growth and metastasis, and modulates key biomarkers in vivo. Oncogene 2013, 32, 4406–4416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I study of GC1008 (fresolimumab): A human anti-transforming growth factor-beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Berlin, J.D.; Cosaert, J.; Kauh, J.; Chan, E.; Piha-Paul, S.A.; Amaya, A.; Tang, S.; Driscoll, K.; Kimbung, R. A phase 1 study of anti-TGFβ receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2017, 79, 673–680. [Google Scholar] [CrossRef] [Green Version]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef]
- Van Baren, N.; Van den Eynde, B. Tryptophan-degrading enzymes in tumoral immune resistance. Front. Immunol. 2015, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Bjoern, J.; Iversen, T.Z.; Nitschke, N.J.; Andersen, M.H.; Svane, I.M. Safety, immune and clinical responses in metastatic melanoma patients vaccinated with a long peptide derived from indoleamine 2,3-dioxygenase in combination with ipilimumab. Cytotherapy 2016, 18, 1043–1055. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Kim, Y.J. Subverting the adaptive immune resistance mechanism to improve clinical responses to immune checkpoint blockade therapy. Oncoimmunology 2014, 3, e954868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Zingg, D.; Arenas-Ramirez, N.; Sahin, D.; Rosalia, R.A.; Antunes, A.T.; Haeusel, J.; Sommer, L.; Boyman, O. The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. Cell Rep. 2017, 20, 854–867. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Mihm, M.C.; Soiffer, R.J.; Haluska, F.G.; Butler, M.; Seiden, M.V.; Davis, T.; Henry-Spires, R.; MacRae, S.; Willman, A. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc. Natl. Acad. Sci. USA 2003, 100, 4712–4717. [Google Scholar] [CrossRef] [Green Version]
- Phan, G.Q.; Yang, J.C.; Sherry, R.M.; Hwu, P.; Topalian, S.L.; Schwartzentruber, D.J.; Restifo, N.P.; Haworth, L.R.; Seipp, C.A.; Freezer, L.J. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 2003, 100, 8372–8377. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [Green Version]
- Antonia, S.J.; López-Martin, J.A.; Bendell, J.; Ott, P.A.; Taylor, M.; Eder, J.P.; Jäger, D.; Pietanza, M.C.; Le, D.T.; de Braud, F. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): A multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 883–895. [Google Scholar] [CrossRef] [Green Version]
- Dosset, M.; Vargas, T.R.; Lagrange, A.; Boidot, R.; Végran, F.; Roussey, A.; Chalmin, F.; Dondaine, L.; Paul, C.; Marie-Joseph, E.L. PD-1/PD-L1 pathway: An adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. Oncoimmunology 2018, 7, e1433981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Chen, W.; Xu, Z.P.G.; Gu, W. PD-L1 Distribution and Perspective for Cancer Immunotherapy-Blockade, Knockdown, or Inhibition. Front. Immunol. 2019, 10, 2022. [Google Scholar] [CrossRef] [Green Version]
- Ju, X.; Zhang, H.; Zhou, Z.; Wang, Q. Regulation of PD-L1 expression in cancer and clinical implications in immunotherapy. Am. J. Cancer Res. 2020, 10, 1–11. [Google Scholar] [PubMed]
- Li, X.; Lian, Z.; Wang, S.; Xing, L.; Yu, J. Interactions between EGFR and PD-1/PD-L1 pathway: Implications for treatment of NSCLC. Cancer Lett. 2018, 418, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.-J. Combination of Osimertinib with Durvalumab in Epidermal Growth Factor Receptor-Mutant Non-Small Cell Lung Cancer: Is There Room for Reinvestigation? J. Thorac. Oncol. 2019, 14, 766–767. [Google Scholar] [CrossRef] [PubMed]
- Gettinger, S.; Hellmann, M.D.; Chow, L.Q.; Borghaei, H.; Antonia, S.; Brahmer, J.R.; Goldman, J.W.; Gerber, D.E.; Juergens, R.A.; Shepherd, F.A. Nivolumab Plus Erlotinib in Patients With EGFR-Mutant Advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1363–1372. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Chen, X.; Li, M.; Liu, J.; Qi, X.; Yang, W.; Zhang, H.; Cai, Z.; Dai, Y.; Ouyang, X. Programmed Death-Ligand 1 Expression Predicts Tyrosine Kinase Inhibitor Response and Better Prognosis in a Cohort of Patients With Epidermal Growth Factor Receptor Mutation-Positive Lung Adenocarcinoma. Clin. Lung Cancer 2015, 16, e25–e35. [Google Scholar] [CrossRef]
- Champiat, S.; Dercle, L.; Ammari, S.; Massard, C.; Hollebecque, A.; Postel-Vinay, S.; Chaput, N.; Eggermont, A.; Marabelle, A.; Soria, J.-C. Hyperprogressive Disease Is a New Pattern of Progression in Cancer Patients Treated by Anti-PD-1/PD-L1. Clin. Cancer Res. 2017, 23, 1920–1928. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, F.; Zhong, M.; Yarden, Y.; Fu, L. The biomarkers of hyperprogressive disease in PD-1/PD-L1 blockage therapy. Mol. Cancer 2020, 19, 81. [Google Scholar] [CrossRef]
- Nag, S.; Zhang, X.; Srivenugopal, K.; Wang, M.-H.; Wang, W.; Zhang, R. Targeting MDM2-p53 interaction for cancer therapy: Are we there yet? Curr. Med. Chem. 2014, 21, 553–574. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Goodman, A.; Walavalkar, V.; Barkauskas, D.A.; Sharabi, A.; Kurzrock, R. Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin. Cancer Res. 2017, 23, 4242–4250. [Google Scholar] [CrossRef] [Green Version]
- Champiat, S.; Ferrara, R.; Massard, C.; Besse, B.; Marabelle, A.; Soria, J.-C.; Ferté, C. Hyperprogressive disease: Recognizing a novel pattern to improve patient management. Nat. Rev. Clin. Oncol. 2018, 15, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Nakamura, Y.; Mishima, S.; Kawazoe, A.; Kuboki, Y.; Bando, H.; Kojima, T.; Doi, T.; Ohtsu, A.; Yoshino, T. Predictive factors for hyperprogressive disease during nivolumab as anti-PD1 treatment in patients with advanced gastric cancer. Gastric Cancer 2019, 22, 793–802. [Google Scholar] [CrossRef] [Green Version]
- Peters, S.; Cappuzzo, F.; Horn, L.; Paz-Ares, L.; Borghaei, H.; Barlesi, F.; Steins, M.; Felip, E.; Spigel, D.; Dorange, C. OA03.05 Analysis of Early Survival in Patients with Advanced Non-Squamous NSCLC Treated with Nivolumab vs Docetaxel in CheckMate 057. J. Thorac. Oncol. 2017, 12, S253. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, R.; Mezquita, L.; Texier, M.; Lahmar, J.; Audigier-Valette, C.; Tessonnier, L.; Mazieres, J.; Zalcman, G.; Brosseau, S.; Le Moulec, S. Hyperprogressive Disease in Patients With Advanced Non-Small Cell Lung Cancer Treated With PD-1/PD-L1 Inhibitors or With Single-Agent Chemotherapy. JAMA Oncol. 2018, 4, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Grosso, J.F.; Goldberg, M.V.; Getnet, D.; Bruno, T.C.; Yen, H.-R.; Pyle, K.J.; Hipkiss, E.; Vignali, D.A.; Pardoll, D.M.; Drake, C.G. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J. Immunol. 2009, 182, 6659–6669. [Google Scholar] [CrossRef] [Green Version]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Lines, J.L.; Pantazi, E.; Mak, J.; Sempere, L.F.; Wang, L.; O’Connell, S.; Ceeraz, S.; Suriawinata, A.A.; Yan, S.; Ernstoff, M.S. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014, 74, 1924–1932. [Google Scholar] [CrossRef] [Green Version]
- Le Mercier, I.; Chen, W.; Lines, J.L.; Day, M.; Li, J.; Sergent, P.; Noelle, R.J.; Wang, L. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res. 2014, 74, 1933–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boles, K.S.; Vermi, W.; Facchetti, F.; Fuchs, A.; Wilson, T.J.; Diacovo, T.G.; Cella, M.; Colonna, M. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur. J. Immunol. 2009, 39, 695–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhu, H.-X.; Yao, Y.; Bian, Z.-H.; Zheng, Y.-J.; Li, L.; Moutsopoulos, H.M.; Gershwin, M.E.; Lian, Z.-X. Immune checkpoint molecules. Possible future therapeutic implications in autoimmune diseases. J. Autoimmun. 2019, 104, 102333. [Google Scholar] [CrossRef] [PubMed]
- Burton, B.R.; Britton, G.J.; Fang, H.; Verhagen, J.; Smithers, B.; Sabatos-Peyton, C.A.; Carney, L.J.; Gough, J.; Strobel, S.; Wraith, D.C. Sequential transcriptional changes dictate safe and effective antigen-specific immunotherapy. Nat. Commun. 2014, 5, 4741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.; Tan, T.G.; Sefik, E.; Yajnik, V. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 2014, 40, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Lozano, E.; Dominguez-Villar, M.; Kuchroo, V.; Hafler, D.A. The TIGIT/CD226 axis regulates human T cell function. J. Immunol. 2012, 188, 3869–3875. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.-H.T.; Maurer, M.; Korman, A.J. TIGIT and PD-1 impair tumor antigen-specific CD8⁺ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, C.S.; Özgüroğlu, M.; Bang, Y.-J.; Di Bartolomeo, M.; Mandalà, M.; Ryu, M.-h.; Fornaro, L.; Olesinski, T.; Caglevic, C.; Chung, H.C. Pembrolizumab versus paclitaxel for previously treated patients with PD-L1–positive advanced gastric or gastroesophageal junction cancer (GC): Update from the phase III KEYNOTE-061 trial. J. Clin. Oncol. 2020, 38, 4503. [Google Scholar] [CrossRef]
- Watanabe, N.; Gavrieli, M.; Sedy, J.R.; Yang, J.; Fallarino, F.; Loftin, S.K.; Hurchla, M.A.; Zimmerman, N.; Sim, J.; Zang, X. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat. Immunol. 2003, 4, 670–679. [Google Scholar] [CrossRef]
- Murphy, T.L.; Murphy, K.M. Slow down and survive: Enigmatic immunoregulation by BTLA and HVEM. Annu. Rev. Immunol. 2010, 28, 389–411. [Google Scholar] [CrossRef]
- Sedy, J.R.; Gavrieli, M.; Potter, K.G.; Hurchla, M.A.; Lindsley, R.C.; Hildner, K.; Scheu, S.; Pfeffer, K.; Ware, C.F.; Murphy, T.L. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat. Immunol. 2005, 6, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.C.; Steinberg, M.W.; Oborne, L.M.; Macauley, M.G.; Fukuyama, S.; Sanjo, H.; D’Souza, C.; Norris, P.S.; Pfeffer, K.; Murphy, K.M. Unconventional ligand activation of herpesvirus entry mediator signals cell survival. Proc. Natl. Acad. Sci. USA 2009, 106, 6244–6249. [Google Scholar] [CrossRef] [Green Version]
- Cai, G.; Freeman, G.J. The CD160, BTLA, LIGHT/HVEM pathway: A bidirectional switch regulating T-cell activation. Immunol. Rev. 2009, 229, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Haymaker, C.L.; Wu, R.C.; Ritthipichai, K.; Bernatchez, C.; Forget, M.-A.; Chen, J.Q.; Liu, H.; Wang, E.; Marincola, F.; Hwu, P. BTLA marks a less-differentiated tumor-infiltrating lymphocyte subset in melanoma with enhanced survival properties. Oncoimmunology 2015, 4, e1014246. [Google Scholar] [CrossRef] [Green Version]
- Stecher, C.; Battin, C.; Leitner, J.; Zettl, M.; Grabmeier-Pfistershammer, K.; Höller, C.; Zlabinger, G.J.; Steinberger, P. PD-1 Blockade Promotes Emerging Checkpoint Inhibitors in Enhancing T Cell Responses to Allogeneic Dendritic Cells. Front. Immunol. 2017, 8, 572. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Pagliano, O.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Olive, D.; Kuchroo, V.; Zarour, H.M. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. 2012, 72, 887–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aicher, A.; Hayden-Ledbetter, M.; Brady, W.A.; Pezzutto, A.; Richter, G.; Magaletti, D.; Buckwalter, S.; Ledbetter, J.A.; Clark, E.A. Characterization of human inducible costimulator ligand expression and function. J. Immunol. 2000, 164, 4689–4696. [Google Scholar] [CrossRef] [Green Version]
- Coyle, A.J.; Lehar, S.; Lloyd, C.; Tian, J.; Delaney, T.; Manning, S.; Nguyen, T.; Burwell, T.; Schneider, H.; Gonzalo, J.A.; et al. The CD28-related molecule ICOS is required for effective T cell-dependent immune responses. Immunity 2000, 13, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Gigoux, M.; Lovato, A.; Leconte, J.; Leung, J.; Sonenberg, N.; Suh, W.-K. Inducible costimulator facilitates T-dependent B cell activation by augmenting IL-4 translation. Mol. Immunol. 2014, 59, 46–54. [Google Scholar] [CrossRef]
- Burugu, S.; Dancsok, A.R.; Nielsen, T.O. Emerging targets in cancer immunotherapy. Semin. Cancer Biol. 2018, 52 Pt 2, 39–52. [Google Scholar] [CrossRef]
- Lee, H.; Kim, J.H.; Yang, S.Y.; Kong, J.; Oh, M.; Jeong, D.H.; Chung, J.-I.; Bae, K.B.; Shin, J.Y.; Hong, K.H. Peripheral blood gene expression of B7 and CD28 family members associated with tumor progression and microscopic lymphovascular invasion in colon cancer patients. J. Cancer Res. Clin. Oncol. 2010, 136, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Quezada, S.A.; Sepulveda, M.A.; Sharma, P.; Allison, J.P. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J. Exp. Med. 2014, 211, 715–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, T.; He, Q.; Sharma, P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy. Cancer Res. 2011, 71, 5445–5454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamphorst, A.O.; Pillai, R.N.; Yang, S.; Nasti, T.H.; Akondy, R.S.; Wieland, A.; Sica, G.L.; Yu, K.; Koenig, L.; Patel, N.T. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. USA 2017, 114, 4993–4998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donini, C.; D’Ambrosio, L.; Grignani, G.; Aglietta, M.; Sangiolo, D. Next generation immune-checkpoints for cancer therapy. J. Thorac. Dis. 2018, 10 (Suppl. S13), S1581–S1601. [Google Scholar] [CrossRef]
- Croft, M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu. Rev. Immunol. 2010, 28, 57–78. [Google Scholar] [CrossRef] [Green Version]
- Mallett, S.; Fossum, S.; Barclay, A.N. Characterization of the MRC OX40 antigen of activated CD4 positive T lymphocytes—A molecule related to nerve growth factor receptor. Embo J. 1990, 9, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Ruby, C.E.; Yates, M.A.; Hirschhorn-Cymerman, D.; Chlebeck, P.; Wolchok, J.D.; Houghton, A.N.; Offner, H.; Weinberg, A.D. Cutting Edge: OX40 agonists can drive regulatory T cell expansion if the cytokine milieu is right. J. Immunol. 2009, 183, 4853–4857. [Google Scholar] [CrossRef] [Green Version]
- So, T.; Song, J.; Sugie, K.; Altman, A.; Croft, M. Signals from OX40 regulate nuclear factor of activated T cells c1 and T cell helper 2 lineage commitment. Proc. Natl. Acad. Sci. USA 2006, 103, 3740–3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polesso, F.; Weinberg, A.D.; Moran, A.E. Late-Stage Tumor Regression after PD-L1 Blockade Plus a Concurrent OX40 Agonist. Cancer Immunol. Res. 2019, 7, 269–281. [Google Scholar] [CrossRef]
- Alves Costa Silva, C.; Facchinetti, F.; Routy, B.; Derosa, L. New pathways in immune stimulation: Targeting OX40. ESMO Open 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, P.R.; Song, J.; Gramaglia, I.; Killeen, N.; Croft, M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity 2001, 15, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; So, T.; Cheng, M.; Tang, X.; Croft, M. Sustained survivin expression from OX40 costimulatory signals drives T cell clonal expansion. Immunity 2005, 22, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Linch, S.N.; McNamara, M.J.; Redmond, W.L. Redmond, OX40 Agonists and Combination Immunotherapy: Putting the Pedal to the Metal. Front. Oncol. 2015, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Kjaergaard, J.; Tanaka, J.; Kim, J.A.; Rothchild, K.; Weinberg, A.; Shu, S. Therapeutic efficacy of OX-40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer Res. 2000, 60, 5514–5521. [Google Scholar]
- Weinberg, A.D.; Rivera, M.-M.; Prell, R.; Morris, A.; Ramstad, T.; Vetto, J.T.; Urba, W.J.; Alvord, G.; Bunce, C.; Shields, J. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J. Immunol. 2000, 164, 2160–2169. [Google Scholar] [CrossRef]
- Aspeslagh, S.; Postel-Vinay, S.; Rusakiewicz, S.; Soria, J.-C.; Zitvogel, L.; Marabelle, A. Rationale for anti-OX40 cancer immunotherapy. Eur. J. Cancer 2016, 52, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Gurney, A.; Marsters, S.; Huang, A.; Pitti, R.; Mark, M.; Baldwin, D.; Gray, A.; Dowd, P.; Brush, J.; Heldens, S. Identification of a new member of the tumor necrosis factor family and its receptor, a human ortholog of mouse GITR. Curr. Biol. 1999, 9, 215–218. [Google Scholar] [CrossRef] [Green Version]
- Ronchetti, S.; Zollo, O.; Bruscoli, S.; Agostini, M.; Bianchini, R.; Nocentini, G.; Ayroldi, E.; Riccardi, C. GITR, a member of the TNF receptor superfamily, is costimulatory to mouse T lymphocyte subpopulations. Eur. J. Immunol. 2004, 34, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.A.; Murphy, J.T.; Wolchok, J.D. Modulation of GITR for cancer immunotherapy. Curr. Opin. Immunol. 2012, 24, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Knee, D.A.; Hewes, B.; Brogdon, J.L. Rationale for anti-GITR cancer immunotherapy. Eur. J. Cancer 2016, 67, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aida, K.; Miyakawa, R.; Suzuki, K.; Narumi, K.; Udagawa, T.; Yamamoto, Y.; Chikaraishi, T.; Yoshida, T.; Aoki, K. Suppression of Tregs by anti-glucocorticoid induced TNF receptor antibody enhances the antitumor immunity of interferon-α gene therapy for pancreatic cancer. Cancer Sci. 2014, 105, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Xu, X.; Zhang, B.; Zhang, R.; Ji, H.; Wang, X. Combined PD-1 blockade and GITR triggering induce a potent antitumor immunity in murine cancer models and synergizes with chemotherapeutic drugs. J. Transl. Med. 2014, 12, 36. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.D.; Schaer, D.A.; Liu, C.; Li, Y.; Hirschhorn-Cymmerman, D.; Kim, S.C.; Diab, A.; Rizzuto, G.; Duan, F.; Perales, M.A. Agonist anti-GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation. PLoS ONE 2010, 5, e10436. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, K.P.; Autio, K.A.; Golan, T.; Dobrenkov, K.; Chartash, E.; Li, X.N.; Wnek, R.; Long, G.V. Phase 1 study of MK-4166, an anti-human glucocorticoid-induced tumor necrosis factor receptor (GITR) antibody, as monotherapy or with pembrolizumab (pembro) in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2019, 37, 9509. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Li, X.; Liu, R.; Su, X.; Pan, Y.; Han, X.; Shao, C.; Shi, Y. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol. Cancer 2019, 18, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [Green Version]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Vargas, F.A.; Furness, A.J.; Solomon, I.; Joshi, K.; Mekkaoui, L.; Lesko, M.H.; Rota, E.M.; Dahan, R.; Georgiou, A.; Sledzinska, A. Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors. Immunity 2017, 46, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Verma, A.; Mathur, R.; Farooque, A.; Kaul, V.; Gupta, S.; Dwarakanath, B.S. T-Regulatory Cells in Tumor Progression And Therapy. Cancer Manag. Res. 2019, 11, 10731–10747. [Google Scholar] [CrossRef] [Green Version]
- Shrimali, R.K.; Ahmad, S.; Verma, V.; Zeng, P.; Ananth, S.; Gaur, P.; Gittelman, R.M.; Yusko, E.; Sanders, C.; Robins, H. Concurrent PD-1 Blockade Negates the Effects of OX40 Agonist Antibody in Combination Immunotherapy through Inducing T-cell Apoptosis. Cancer Immunol. Res. 2017, 5, 755–766. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Yang, L.; Huang, J.; Ren, X.; Gorska, A.E.; Chytil, A.; Aakre, M.; Carbone, D.P.; Matrisian, L.M.; Richmond, A.; Lin, P.C. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008, 13, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.; Cagnon, L.; Costa-Nunes, C.M.; Baumgaertner, P.; Montandon, N.; Leyvraz, L.; Michielin, O.; Romano, E.; Speiser, D.E. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. 2014, 63, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, A.; Routkevitch, D.; Jackson, C.; Lim, M. Targeting Myeloid Cells in Combination Treatments for Glioma and Other Tumors. Front. Immunol. 2019, 10, 1715. [Google Scholar] [CrossRef] [Green Version]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra67. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Strauss, O.; Kokkinou, E.; Bruchard, M.; Tripathi, K.P.; Schlums, H.; Carrasco, A.; Mazzurana, L.; Konya, V.; Villablanca, E.J. Cytokines regulate the antigen-presenting characteristics of human circulating and tissue-resident intestinal ILCs. Nat. Commun. 2020, 11, 2049. [Google Scholar] [CrossRef]
- Crinier, A.; Vivier, E.; Bléry, M. Helper-like innate lymphoid cells and cancer immunotherapy. Semin. Immunol. 2019, 41, 101274. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Tsang, J.C.; Wang, C.; Clare, S.; Wang, J.; Chen, X.; Brandt, C.; Kane, L.; Campos, L.S.; Lu, L. Single-cell RNA-seq identifies a PD-1(hi) ILC progenitor and defines its development pathway. Nature 2016, 539, 102–106. [Google Scholar] [CrossRef]
- Taylor, S.; Huang, Y.; Mallett, G.; Stathopoulou, C.; Felizardo, T.C.; Sun, M.-A.; Martin, E.L.; Zhu, N.; Woodward, E.L.; Elias, M.S. PD-1 regulates KLRG1(+) group 2 innate lymphoid cells. J. Exp. Med. 2017, 214, 1663–1678. [Google Scholar] [CrossRef] [Green Version]
- Mariotti, F.R.; Quatrini, L.; Munari, E.; Vacca, P.; Moretta, L. Innate Lymphoid Cells: Expression of PD-1 and Other Checkpoints in Normal and Pathological Conditions. Front. Immunol. 2019, 10, 910. [Google Scholar] [CrossRef]
- Tumino, N.; Martini, S.; Munari, E.; Scordamaglia, F.; Besi, F.; Mariotti, F.R.; Bogina, G.; Mingari, M.C.; Vacca, P.; Moretta, L. Presence of innate lymphoid cells in pleural effusions of primary and metastatic tumors: Functional analysis and expression of PD-1 receptor. Int. J. Cancer 2019, 145, 1660–1668. [Google Scholar] [CrossRef]
- Vacca, P.; Pesce, S.; Greppi, M.; Fulcheri, E.; Munari, E.; Olive, D.; Mingari, M.C.; Moretta, A.; Moretta, L.; Marcenaro, E. PD-1 is expressed by and regulates human group 3 innate lymphoid cells in human decidua. Mucosal Immunol. 2019, 12, 624–631. [Google Scholar] [CrossRef]
- Bal, S.M.; Golebski, K.; Spits, H. Plasticity of innate lymphoid cell subsets. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Ma, Z.; Li, W.; Yoshiya, S.; Xu, Y.; Hata, M.; El-Darawish, Y.; Markova, T.; Yamanishi, K.; Yamanishi, H.; Tahara, H. Augmentation of Immune Checkpoint Cancer Immunotherapy with IL18. Clin. Cancer Res. 2016, 22, 2969–2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, M.F.; Cozzo, A.J.; Pfeil, A.J.; Etigunta, S.K.; Hursting, S.D. Cell Intrinsic and Systemic Metabolism in Tumor Immunity and Immunotherapy. Cancers (Basel) 2020, 12, 852. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.K. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef] [Green Version]
- Malinarich, F.; Duan, K.; Hamid, R.A.; Bijin, A.; Lin, W.X.; Poidinger, M.; Fairhurst, A.-M.; Connolly, J.E. High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J. Immunol. 2015, 194, 5174–5186. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, P.S.; Chamoto, K.; Kumar, A.; Honjo, T. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8(+) T Cells and Facilitates Anti-PD-1 Therapy. Cancer Immunol. Res. 2018, 6, 1375–1387. [Google Scholar] [CrossRef]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, R.J.; Su, L.J.; Pinckney, J.; Critton, D.; Boyer, E.; Krishnakumar, A.; Corbett, M.; Rankin, A.L.; Dibella, R.; Campbell, L. VISTA is an acidic pH-selective ligand for PSGL-1. Nature 2019, 574, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef]
- Ohashi, T.; Aoki, M.; Tomita, H.; Akazawa, T.; Sato, K.; Kuze, B.; Mizuta, K.; Hara, A.; Nagaoka, H.; Inoue, N. M2-like macrophage polarization in high lactic acid-producing head and neck cancer. Cancer Sci. 2017, 108, 1128–1134. [Google Scholar] [CrossRef] [Green Version]
- Triner, D.; Shah, Y.M. Hypoxia-inducible factors: A central link between inflammation and cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef]
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; DeNardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010, 70, 7465–7475. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Clambey, E.T.; McNamee, E.N.; Westrich, J.A.; Glover, L.E.; Campbell, E.L.; Jedlicka, P.; de Zoeten, E.F.; Cambier, J.C.; Stenmark, K.R.; Colgan, S.P. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. USA 2012, 109, E2784–E2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, V.S.; Hafez, E.A.A. Synopsis of arachidonic acid metabolism: A review. J. Adv. Res. 2018, 11, 23–32. [Google Scholar] [CrossRef]
- Zelenay, S.; Van Der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, J.; Lu, X.; Hu, Y.; Piao, C.; Wu, X.; Liu, X.; Huang, C.; Wang, Y.; Li, D.; Liu, J. Prostaglandin E(2) and PD-1 mediated inhibition of antitumor CTL responses in the human tumor microenvironment. Oncotarget 2017, 8, 89802–89810. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Xu, Z.; Li, H. NSAIDs Use and Reduced Metastasis in Cancer Patients: Results from a meta-analysis. Sci. Rep. 2017, 7, 1875. [Google Scholar] [CrossRef]
- Dong, G.; Mao, Q.; Xia, W.; Xu, Y.; Wang, J.; Xu, L.; Jiang, F. PKM2 and cancer: The function of PKM2 beyond glycolysis. Oncol. Lett. 2016, 11, 1980–1986. [Google Scholar] [CrossRef] [Green Version]
- Palsson-McDermott, E.M.; Dyck, L.; Zasłona, Z.; Menon, D.; McGettrick, A.F.; Mills, K.H.; O’Neill, L.A. Pyruvate Kinase M2 Is Required for the Expression of the Immune Checkpoint PD-L1 in Immune Cells and Tumors. Front. Immunol. 2017, 8, 1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, R.; Shirai, T.; Namkoong, H.; Zhang, H.; Berry, G.J.; Wallis, B.B.; Schaefgen, B.; Harrison, D.G.; Tremmel, J.A.; Giacomini, J.C. Pyruvate controls the checkpoint inhibitor PD-L1 and suppresses T cell immunity. J. Clin. Investig. 2017, 127, 2725–2738. [Google Scholar] [CrossRef]
- Liu, W.-R.; Tian, M.-X.; Yang, L.-X.; Lin, Y.-L.; Jin, L.; Ding, Z.-B.; Shen, Y.-H.; Peng, Y.-F.; Gao, D.-M.; Zhou, J. PKM2 promotes metastasis by recruiting myeloid-derived suppressor cells and indicates poor prognosis for hepatocellular carcinoma. Oncotarget 2015, 6, 846–861. [Google Scholar] [CrossRef] [Green Version]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7, 277ra30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnemann, C.; Schildberg, F.A.; Schurich, A.; Diehl, L.; Hegenbarth, S.I.; Endl, E.; Lacher, S.; Müller, C.E.; Frey, J.; Simeoni, L. Adenosine regulates CD8 T-cell priming by inhibition of membrane-proximal T-cell receptor signalling. Immunology 2009, 128, e728–e737. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Pommey, S.; Smyth, M.J.; Stagg, J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin. Cancer Res. 2013, 19, 5626–5635. [Google Scholar] [CrossRef] [Green Version]
- Overman, M.J.; LoRusso, P.; Strickler, J.H.; Patel, S.P.; Clarke, S.J.; Noonan, A.M.; Prasanna, T.; Amin, M.A.; Nemunaitis, J.J.; Desai, J. Safety, efficacy and pharmacodynamics (PD) of MEDI9447 (oleclumab) alone or in combination with durvalumab in advanced colorectal cancer (CRC) or pancreatic cancer (panc). J. Clin. Oncol. 2018, 36, 4123. [Google Scholar] [CrossRef]
- De Oliveira Leal, V.; Mafra, D. Adipokines in obesity. Clin. Chim. Acta 2013, 419, 87–94. [Google Scholar] [CrossRef]
- Turer, A.; Scherer, P. Adiponectin: Mechanistic insights and clinical implications. Diabetologia 2012, 55, 2319–2326. [Google Scholar] [CrossRef] [Green Version]
- McArdle, M.A.; Finucane, O.M.; Connaughton, R.M.; McMorrow, A.M.; Roche, H.M. Mechanisms of obesity-induced inflammation and insulin resistance: Insights into the emerging role of nutritional strategies. Front. Endocrinol. (Lausanne) 2013, 4, 52. [Google Scholar] [CrossRef] [Green Version]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef]
- Rassy, E.E.; Ghosn, M.; Rassy, N.A.; Assi, T.; Robert, C. Do immune checkpoint inhibitors perform identically in patients with weight extremes? Immunotherapy 2018, 10, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H. The hyperleptinemia of obesity-regulator of caloric surpluses. Cell 2004, 117, 145–146. [Google Scholar] [CrossRef] [Green Version]
- Clements, V.; Long, T.; Long, R.; Figley, C.; Ostrand-Rosenberg, S. Frontline Science: High fat diet and leptin promote tumor progression by inducing myeloid-derived suppressor cells. J. Leukoc. Biol. 2018, 103, 395–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlesinger, S.; Siegert, S.; Koch, M.; Walter, J.; Heits, N.; Hinz, S.; Jacobs, G.; Hampe, J.; Schafmayer, C.; Nöthlings, U. Postdiagnosis body mass index and risk of mortality in colorectal cancer survivors: A prospective study and meta-analysis. Cancer Causes Control 2014, 25, 1407–1418. [Google Scholar] [CrossRef]
- Lennon, H.; Sperrin, M.; Badrick, E.; Renehan, A.G. The Obesity Paradox in Cancer: A Review. Curr. Oncol. Rep. 2016, 18, 56. [Google Scholar] [CrossRef] [PubMed]
- Amptoulach, S.; Gross, G.; Kalaitzakis, E. Differential impact of obesity and diabetes mellitus on survival after liver resection for colorectal cancer metastases. J. Surg. Res. 2015, 199, 378–385. [Google Scholar] [CrossRef]
- Donnelly, D.; Bajaj, S.; Yu, J.; Hsu, M.; Balar, A.; Pavlick, A.; Weber, J.; Osman, I.; Zhong, J. The complex relationship between body mass index and response to immune checkpoint inhibition in metastatic melanoma patients. J. Immunother. Cancer 2019, 7, 222. [Google Scholar] [CrossRef] [Green Version]
- Galli, G.; Corsetto, P.; Ferrara, R.; Prelaj, A.; Proto, C.; Signorelli, D.; Zilembo, N.; De Toma, A.; Pagani, F.; Randon, G. Impact of cholesterolemia and body mass index on outcome of metastatic non small cell lung cancer treated with immunotherapy. J. Clin. Oncol. 2019, 37, e20691. [Google Scholar] [CrossRef]
- Turbitt, W.J.; Xu, Y.; Sosnoski, D.M.; Collins, S.D.; Meng, H.; Mastro, A.M.; Rogers, C.J. Physical Activity Plus Energy Restriction Prevents 4T1.2 Mammary Tumor Progression, MDSC Accumulation, and an Immunosuppressive Tumor Microenvironment. Cancer Prev. Res. (Phila) 2019, 12, 493–506. [Google Scholar] [CrossRef]
- Messaoudi, I.; Warner, J.; Fischer, M.; Park, B.; Hill, B.; Mattison, J.; Lane, M.A.; Roth, G.S.; Ingram, D.K.; Picker, L.J. Delay of T cell senescence by caloric restriction in aged long-lived nonhuman primates. Proc. Natl. Acad. Sci. USA 2006, 103, 19448–19453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Tao, S.; Chen, Z.; Koliesnik, I.O.; Calmes, P.G.; Hoerr, V.; Han, B.; Gebert, N.; Zörnig, M.; Löffler, B. Dietary restriction improves repopulation but impairs lymphoid differentiation capacity of hematopoietic stem cells in early aging. J. Exp. Med. 2016, 213, 535–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-E.; Rayyan, M.; Liao, A.; Edery, I.; Pletcher, S.D. Acute Dietary Restriction Acts via TOR, PP2A, and Myc Signaling to Boost Innate Immunity in Drosophila. Cell Rep. 2017, 20, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Farazi, M.; Nguyen, J.; Goldufsky, J.; Linnane, S.; Lukaesko, L.; Weinberg, A.D.; Ruby, C.E. Caloric restriction maintains OX40 agonist-mediated tumor immunity and CD4 T cell priming during aging. Cancer Immunol. Immunother. 2014, 63, 615–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbiano, S.; Suárez-Zamorano, N.; Rigo, D.; Veyrat-Durebex, C.; Dokic, A.S.; Colin, D.J.; Trajkovski, M. Caloric Restriction Leads to Browning of White Adipose Tissue through Type 2 Immune Signaling. Cell Metab. 2016, 24, 434–446. [Google Scholar] [CrossRef]
- Di Biase, S.; Lee, C.; Brandhorst, S.; Manes, B.; Buono, R.; Cheng, C.-W.; Cacciottolo, M.; Martin-Montalvo, A.; de Cabo, R.; Wei, M. Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell 2016, 30, 136–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lévesque, S.; Le Naour, J.; Pietrocola, F.; Paillet, J.; Kremer, M.; Castoldi, F.; Baracco, E.E.; Wang, Y.; Vacchelli, E.; Stoll, G. A synergistic triad of chemotherapy, immune checkpoint inhibitors, and caloric restriction mimetics eradicates tumors in mice. Oncoimmunology 2019, 8, e1657375. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Jing, Y.; Li, L.; Mills, G.B.; Diao, L.; Liu, H.; Han, L. Sex-associated molecular differences for cancer immunotherapy. Nat. Commun. 2020, 11, 1779. [Google Scholar] [CrossRef] [Green Version]
- Conforti, F.; Pala, L.; Bagnardi, V.; De Pas, T.; Martinetti, M.; Viale, G.; Gelber, R.D.; Goldhirsch, A. Cancer immunotherapy efficacy and patients’ sex: A systematic review and meta-analysis. Lancet Oncol. 2018, 19, 737–746. [Google Scholar] [CrossRef]
- Wu, Y.; Ju, Q.; Jia, K.; Yu, J.; Shi, H.; Wu, H.; Jiang, M. Correlation between sex and efficacy of immune checkpoint inhibitors (PD-1 and CTLA-4 inhibitors). Int. J. Cancer 2018, 143, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Cowley, L.A.; Liu, X.-S. Sex Differences in Cancer Immunotherapy Efficacy, Biomarkers, and Therapeutic Strategy. Molecules 2019, 24, 3214. [Google Scholar] [CrossRef] [Green Version]
- Grassadonia, A.; Sperduti, I.; Vici, P.; Iezzi, L.; Brocco, D.; Gamucci, T.; Pizzuti, L.; Maugeri-Saccà, M.; Marchetti, P.; Cognetti, G. Effect of Gender on the Outcome of Patients Receiving Immune Checkpoint Inhibitors for Advanced Cancer: A Systematic Review and Meta-Analysis of Phase III Randomized Clinical Trials. J. Clin. Med. 2018, 7, 542. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Ayyoub, M.; Routy, B.; Kroemer, G. Microbiome and Anticancer Immunosurveillance. Cell 2016, 165, 276–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankel, A.E.; Coughlin, L.A.; Kim, J.; Froehlich, T.W.; Xie, Y.; Frenkel, E.P.; Koh, A.Y. Metagenomic Shotgun Sequencing and Unbiased Metabolomic Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune Checkpoint Therapy Efficacy in Melanoma Patients. Neoplasia 2017, 19, 848–855. [Google Scholar] [CrossRef]
- Chalabi, M.; Cardona, A.; Nagarkar, D.R.; Dhawahir Scala, A.; Gandara, D.R.; Rittmeyer, A.; Albert, M.L.; Powles, T.; Kok, M.; Herrera, F.G. Efficacy of chemotherapy and atezolizumab in patients with non-small-cell lung cancer receiving antibiotics and proton pump inhibitors: Pooled post hoc analyses of the OAK and POPLAR trials. Ann. Oncol. 2020, 31, 525–531. [Google Scholar] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, M.A.; Goodrich, J.K.; Maxan, M.-E.; Freedberg, D.E.; Abrams, J.A.; Poole, A.C.; Sutter, J.L.; Welter, D.; Ley, R.E.; Bell, J.T. Proton pump inhibitors alter the composition of the gut microbiota. Gut 2016, 65, 749–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Types | Mechanism of Resistance | Means to Overcome It |
|---|---|---|
| Immunosuppressive cells | Tumor-Associated Macrophages (TAMs) | Immune checkpoint inhibitor (ICI )+ CSF1R blockade |
| Regulatory T-cells (Tregs) | Anti-PD-1 + Anti-CD25 | |
| Myeloid-derived Suppressor cells (MDSCs) | Anti-CXCR2 | |
| Innate Lymphoid Cells and Tumor Micro-Environment | IL-15, anti-IL-12, anti-IL13, anti-IL-23 | |
| Absence of antigen presentation | Beta2-Microglobulin (β2M) | Adoptive natural killer (NK) cells |
| Transporters Associated with Antigen Processing (TAP) | ||
| Pattern Recognition Receptors (PRR) | PRR agonists | |
| Genetic T-cell exclusion and insensibility to T-cells | MAPK Oncogenic Signaling | MAPK inhibitors |
| Tumor Suppressor Phosphate and Tensin Homolog (PTEN) | Anti-PD-1 + Anti-PI3K | |
| WNT/β -Catenin Signaling Pathway | Anti-PD-1 + BBI608 | |
| Vascular Endothelial Growth Factor (VEGF) | Anti-VEGF + ACT, Anti-VEGF + Anti-CTLA-4 | |
| Transforming Growth Factor- β (TGF-β) | Anti-PD-L1 + Anti- TGF- β | |
| Indoleamine 2,3-dioxygenase (IDO) | Anti-CTLA-4 + 1MT, IDO inhibitor + ICI, Peptide vaccination + Anti-CTLA-4 | |
| Interferon-gamma Receptor Pathway | Anti-PD-1 + cell-based vaccine | |
| Enhancer of zester homolog-2 (Ezh2) | Anti-CTLA-4 + GSK503 | |
| Impaired T-cell functionality by immunosuppresive signaling receptors | Lymphocyte Activation Gene-3 (LAG-3) | LAG Ig + peptide vaccine, Anti-LAG3 + Anti-PD-1 |
| T-cell Immunoglobulin Mucin-3 (TIM-3) | Anti-TIM-3 + Anti-PD-1 | |
| V-domain Ig Suppressor of T-cell Activation (VISTA) | Anti-VISTA + peptide cancer vaccine | |
| T-cell Immunoreceptor with Ig and ITIM domains (TIGIT) | Anti-TIGIT + Anti-PD-1 | |
| B and T Lymphocyte Attenuator-4 (BTLA-4) | Anti-BTLA + Anti-PD-1 | |
| Lack of stimulatory checkpoints | ICOS | ICOS stimulators + ICI |
| OX40 | OX40 stimulators + ICI | |
| Glucocorticoid-Induced TNF Receptor (GITR) | GITR stimulators + ICI |
| Types | Receptor Partner and Ligands | Action |
|---|---|---|
| Stimulatory | ICOS/ICOS-L | Increase T-cell activation and cytotoxicity |
| GITR/GITR-L | Increase T-cell activation and decrease Treg cell functions | |
| CD28/CD80,86 | Increase T-cell activation | |
| CD27/CD70 | Increase naïve T-cell proliferation and cytotoxic T-cell differentiation | |
| CD40/CD40L | Increase T-cell and APC activation | |
| OX40/OX40-L | Increase T-cell activation and T reg dysfunction/depletion | |
| Inhibitory | PD1/PDL-1 | Decrease T-cell activation and increase T reg proliferation |
| CTLA-4/CD80,86 | Decrease T-cell activation and increase T reg proliferation | |
| TIM-3/Galectin-9 | Decrease T-cell activation and increase T-cell apoptosis and Treg function | |
| LAG-3/MHC-II | Increase T-cell expansion and T reg cell functions | |
| VSIG-3/VISTA | Decrease the activity of cytotoxic T-cells, stimulate production of Treg | |
| BTLA-4/HVEM | Decrease T-cell activation | |
| TIGIT/CD155, CD112, PVR | Block T-cell activation, increased tolerance of DCs | |
| CD47/SIRP alpha | Decrease APC presentation |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haibe, Y.; El Husseini, Z.; El Sayed, R.; Shamseddine, A. Resisting Resistance to Immune Checkpoint Therapy: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 6176. https://doi.org/10.3390/ijms21176176
Haibe Y, El Husseini Z, El Sayed R, Shamseddine A. Resisting Resistance to Immune Checkpoint Therapy: A Systematic Review. International Journal of Molecular Sciences. 2020; 21(17):6176. https://doi.org/10.3390/ijms21176176
Chicago/Turabian StyleHaibe, Yolla, Ziad El Husseini, Rola El Sayed, and Ali Shamseddine. 2020. "Resisting Resistance to Immune Checkpoint Therapy: A Systematic Review" International Journal of Molecular Sciences 21, no. 17: 6176. https://doi.org/10.3390/ijms21176176