Platelets: Still a Therapeutical Target for Haemostatic Disorders


 and
and 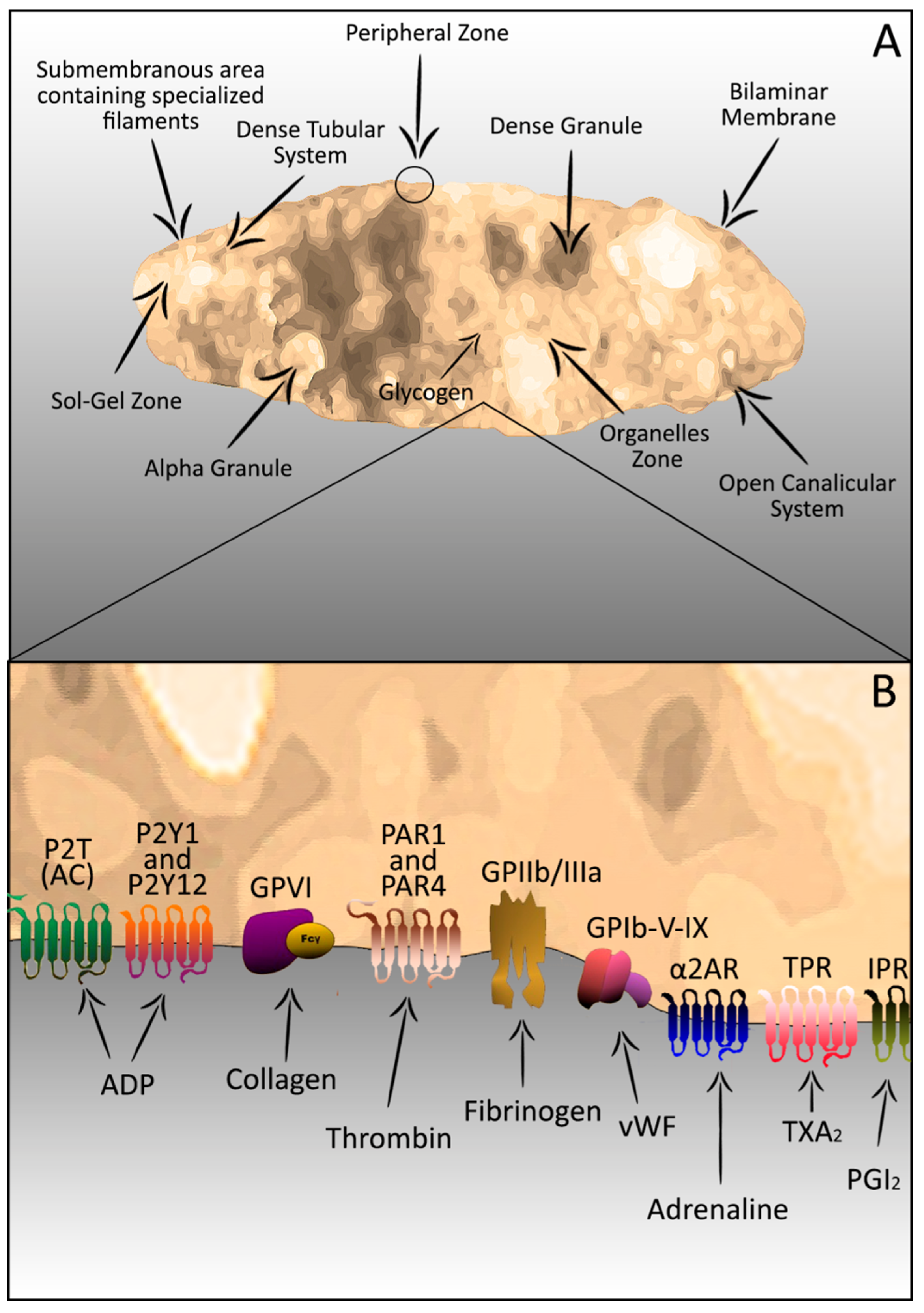{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Platelets–Complex Structure
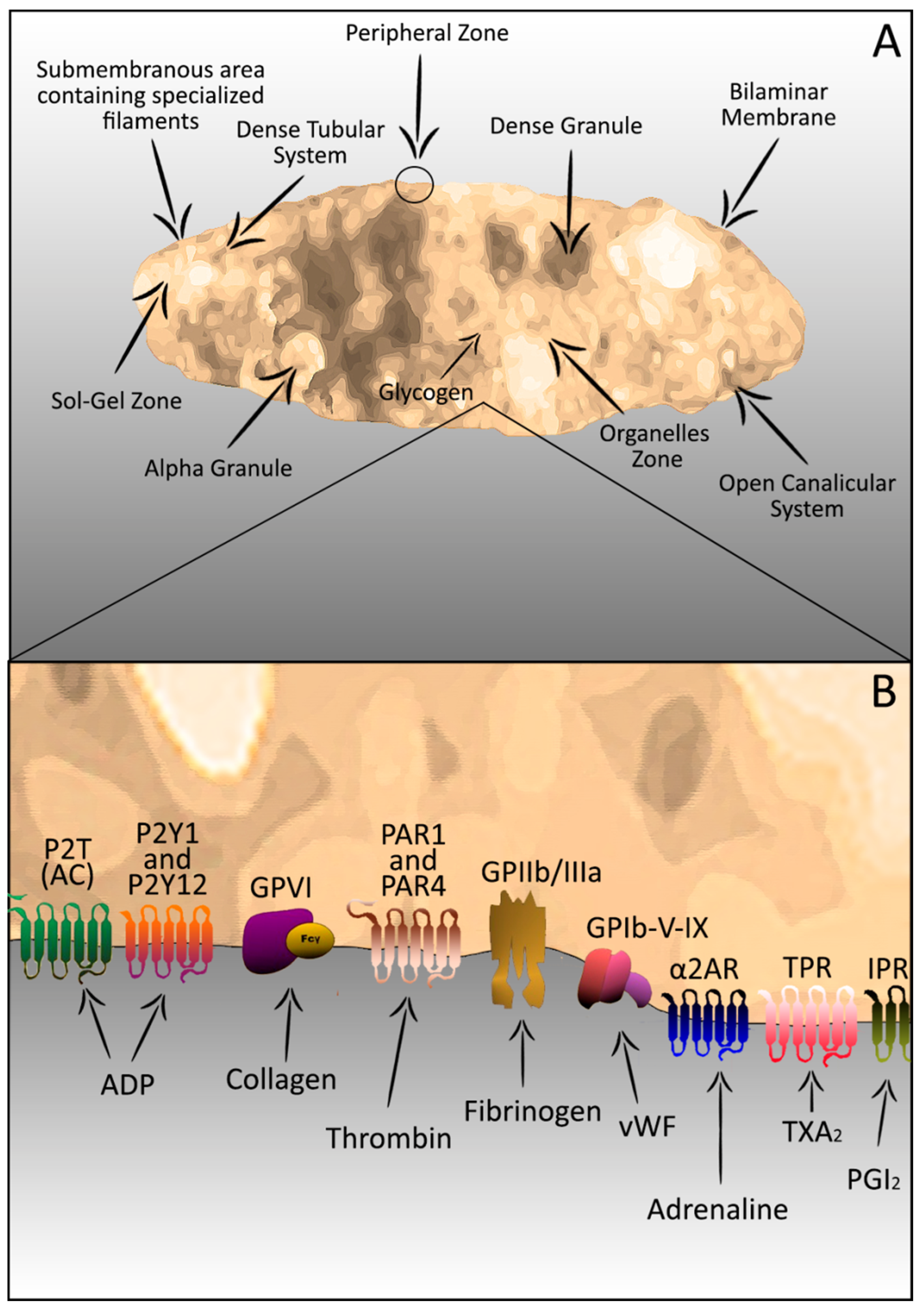
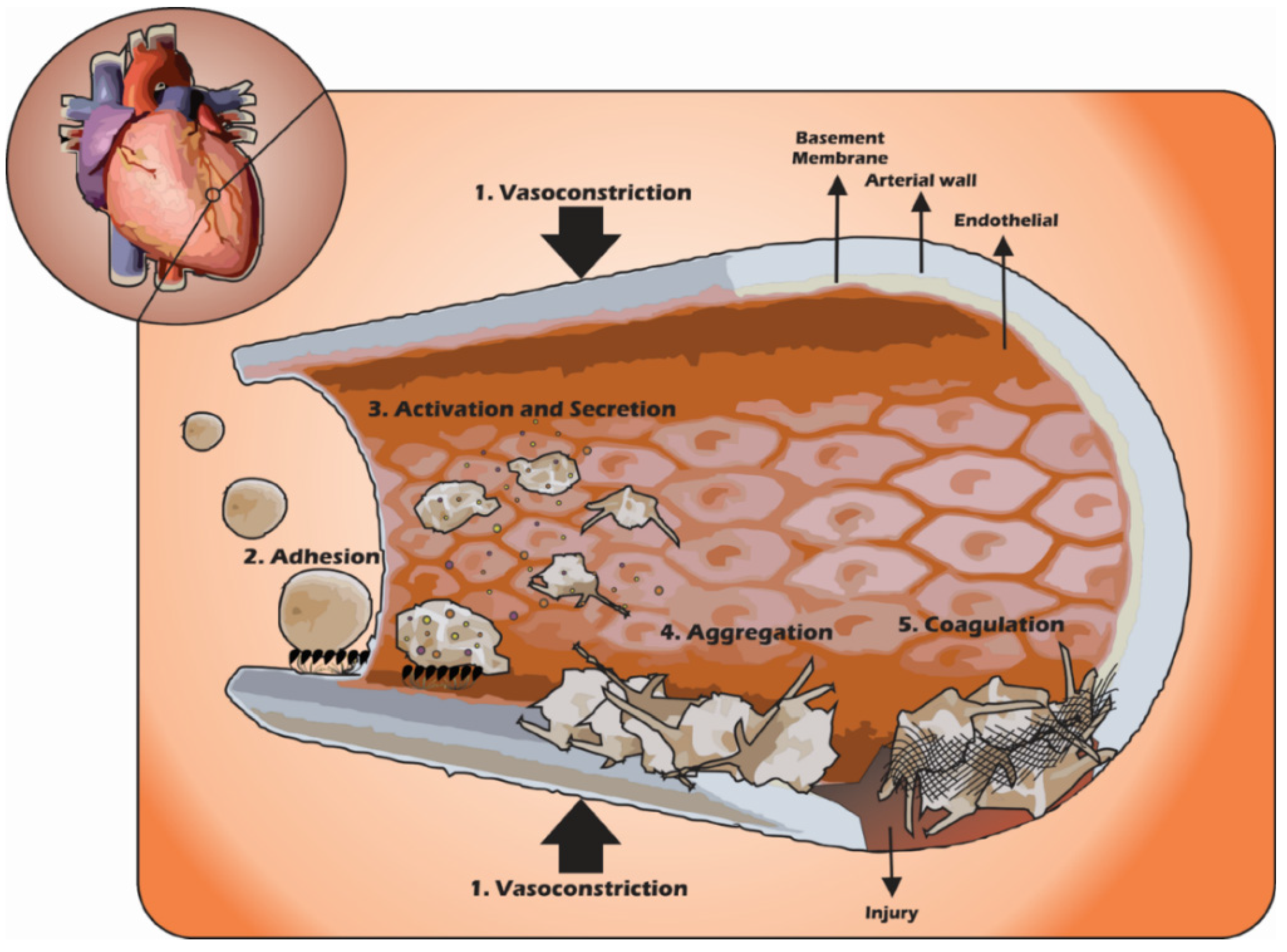
3. Platelets in the Haemostatic Process

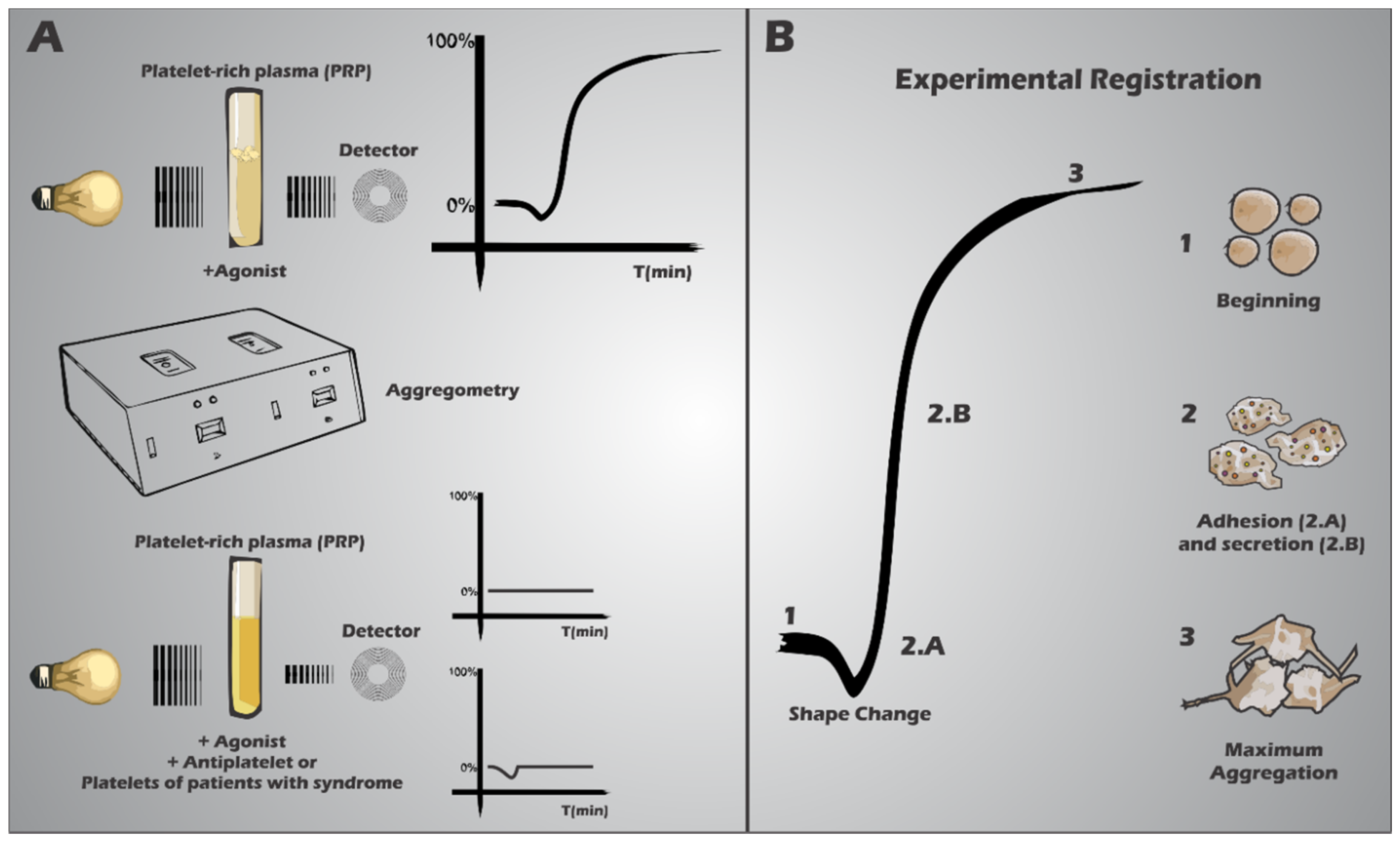
4. Platelet Receptor Defects
5. Granular Disorders
6. Non-Storage Disorders Associated With Systemic Disorders
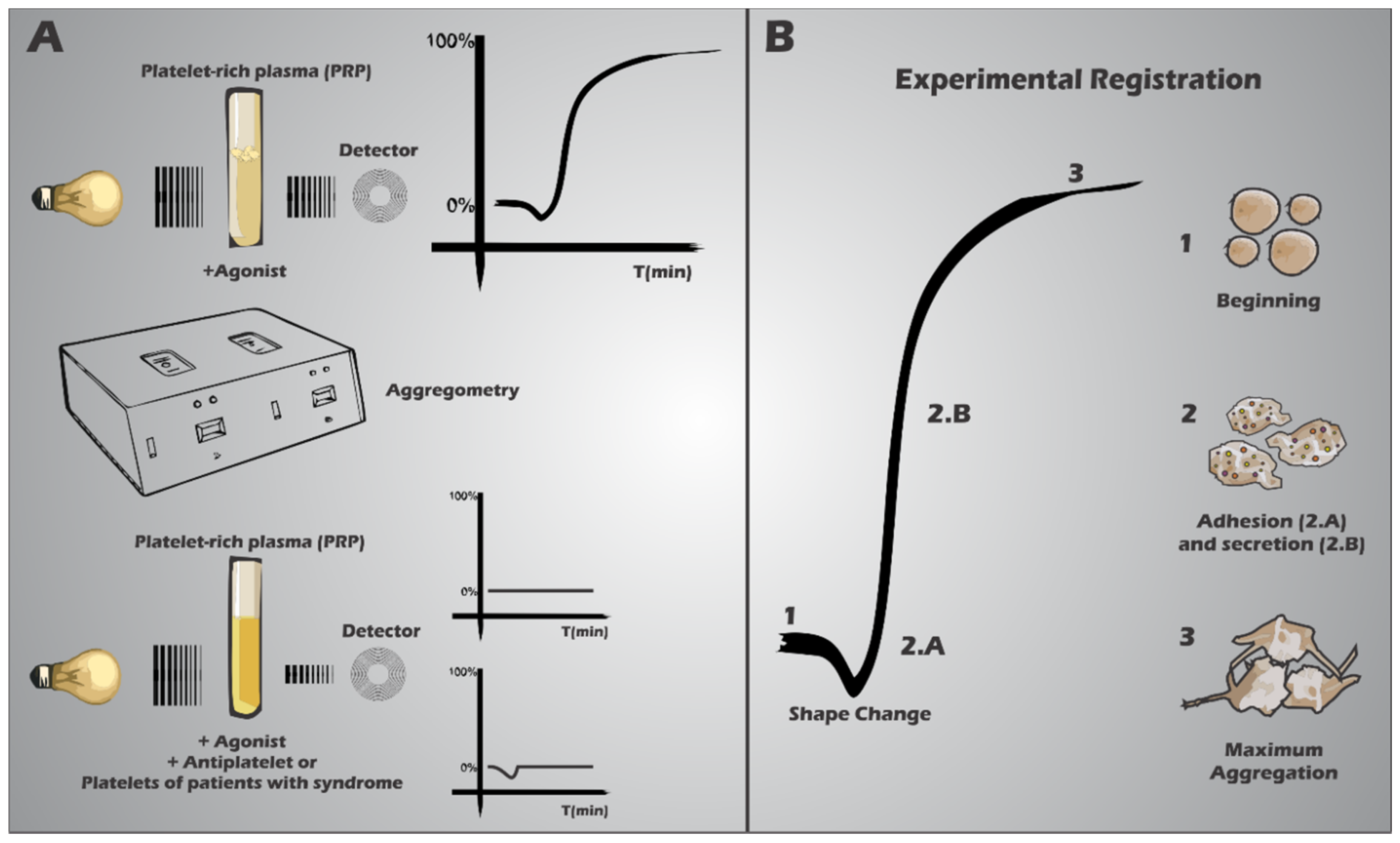
7. Secretion
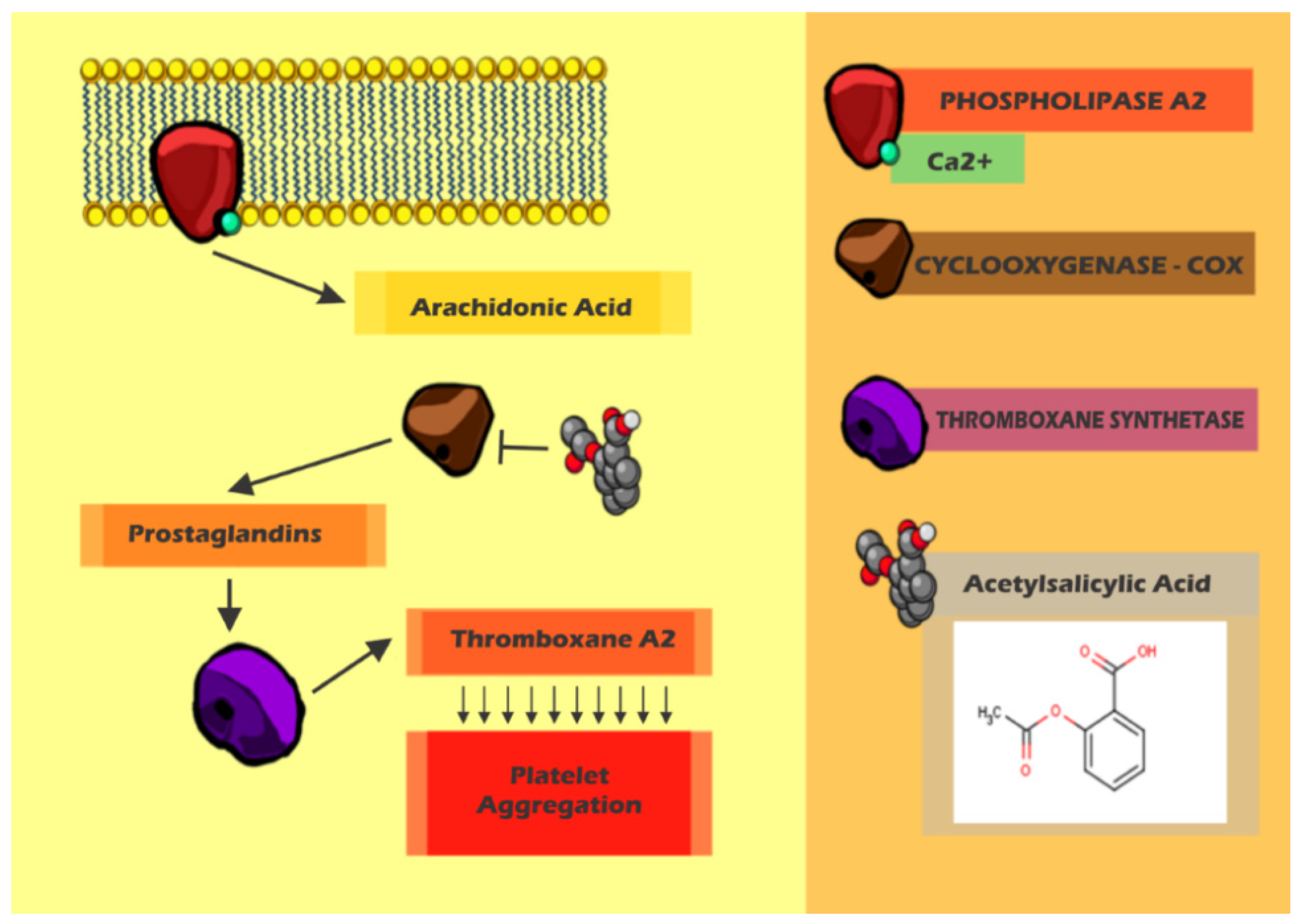
8. Arterial Thrombosis x Treatment

9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Winter, O.; Moser, K.; Mohr, E.; Zotos, D.; Kaminski, H.; Szyska, M.; Roth, K.; Wong, D.M.; Dame, C.; Tarlinton, D.M.; et al. Megakaryocytes constitute a functional component of a plasma cell niche in the bone marrow. Blood 2010, 116, 1867–1875. [Google Scholar]
- Menter, D.G.; Tucker, S.C.; Kopetz, S.; Sood, A.K.; Crissman, J.D.; Honn, K.V. Platelets and cancer: A casual or causal relationship: Revisited. Cancer Metastasis Rev. 2014, 33, 231–269. [Google Scholar]
- Al Ghumlas, A.K.; Gader, A.G.M.A. The blood platelet: An intriguing cell. J. Appl. Hematol. 2013, 4, 1–12. [Google Scholar]
- Walsh, T.G.; Metharom, P.; Berndt, M.C. The functional role of platelets in the regulation of angiogenesis. Platelets 2014. [Google Scholar] [CrossRef]
- Wong, A.K.T. Platelet biology: The role of shear. Expert Rev. Hematol. 2013, 6, 205–212. [Google Scholar]
- Nugent, D.; McMillan, R.; Nichol, J.L.; Slichter, S.J. Pathogenesis of chronic immune thrombocytopenia: Increased platelet destruction and/or decreased platelet production. Br. J. Haematol. 2009, 146, 585–596. [Google Scholar]
- Tsiara, S.; Cooper, N. Eltrombopag for the treatment of chronic immune thrombocytopenia. Clin. Investig. 2011, 1, 295–303. [Google Scholar]
- Jin, R.C.; Loscalzo, J. Vascular nitric oxide: Formation and function. J. Blood Med. 2010, 2010, 147–162. [Google Scholar]
- Rumbaut, R.E.; Thiagarajan, P. Platelet-Vessel Wall Interactions in Hemostasis and Thrombosis— Integrated Systems Physiology: From Molecule to Function to Disease; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Münzer, P.; Borst, O.; Walker, B.; Schmid, E.; Feijge, M.A.H.; Cosemans, J.M.E.M.; Chatterjee, M.; Schmidt, E.-M.; Schmidt, S.; Towhid, S.T.; et al. Acid sphingomyelinase regulates platelet cell membrane scrambling, secretion, and thrombus formation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 61–71. [Google Scholar]
- Bultas, J. Antiplatelet therapy—A pharmacologist’s perspective. Cor Vasa 2013, 55, e86–e94. [Google Scholar]
- Lei, H.; Gui, L.; Xiao, R. The effect of anticoagulants on the quality and biological efficacy of platelet-rich plasma. Clin. Biochem. 2009, 42, 1452–1460. [Google Scholar]
- Bittencourt, C.H.; Bittencourt, P.B.; Neto, O.A.L.; Arenas, G.C.F. The use of platelet-rich plasma in orthopaedic injuries. In Platelet-Rich Plasma; Lana, J.F.S.D., Santana, M.H.A., Belangero, W.D., Luzo, A.C.M., Eds.; Springer: Berlin, Heidelberg, Germany, 2014; pp. 289–313. [Google Scholar]
- Flaumenhaft, R. Molecular basis of platelet granule secretion. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1152–1160. [Google Scholar]
- Van Nispen tot Pannerden, H.; de Haas, F.; Geerts, W.; Posthuma, G.; van Dijk, S.; Heijnen, H.F.G. The platelet interior revisited: Electron tomography reveals tubular α-granule subtypes. Blood 2010, 116, 1147–1156. [Google Scholar]
- Furie, B.; Furie, B.C.; Flaumenhaft, R. A journey with platelet P-selectin: The molecular basis of granule secretion, signalling and cell adhesion. Thromb. Haemost. 2001, 86, 214–221. [Google Scholar]
- Brass, L. Understanding and evaluating platelet function. Hematol. Am. Soc. Hematol. Educ. Program 2010, 2010, 387–396. [Google Scholar]
- Veldhuisen, B.; Porcelijn, L.; Ellen van der Schoot, C.; de Haas, M. Molecular typing of human platelet and neutrophil antigens (HPA and HNA). Transfus. Apher. Sci. 2014, 50, 189–199. [Google Scholar]
- Wraith, K.S.; Magwenzi, S.; Aburima, A.; Wen, Y.; Leake, D.; Naseem, K.M. Oxidized low-density lipoproteins induce rapid platelet activation and shape change through tyrosine kinase and Rho kinase-signaling pathways. Blood 2013, 122, 580–589. [Google Scholar]
- Choi, J.-L.; Li, S.; Han, J.-Y. Platelet function tests: A review of progresses in clinical application. BioMed. Res. Int. 2014, 2014, e456569. [Google Scholar]
- Choi, W.; Karim, Z.A.; Whiteheart, S.W. Protein expression in platelets from six species that differ in their open canalicular system. Platelets 2010, 21, 167–175. [Google Scholar]
- Siess, W. Molecular mechanisms of platelet activation. Physiol. Rev. 1989, 69, 58–178. [Google Scholar]
- Ciesienski, K.L.; Caravan, P. Molecular MRI of thrombosis. Curr. Cardiovasc. Imaging Rep. 2010, 4, 77–84. [Google Scholar]
- Versteeg, H.H.; Heemskerk, J.W.M.; Levi, M.; Reitsma, P.H. New fundamentals in hemostasis. Cardiovasc. Res. 2013, 93, 327–358. [Google Scholar]
- Hartwig, J.H. Platelet structure. Platelets 2002, 1, 37–52. [Google Scholar]
- Yan, M.J.; Lesyk, G.; Radziwon-Balicka, A.; Jurasz, P. Pharmacological regulation of platelet factors that influence tumor angiogenesis. Semin. Oncol. 2014, 41, 370–377. [Google Scholar]
- Pierdoná, T.M.; Lima, N.R.; Rodrigues, R.C.M.; Teixeira, J.P.; Gonçalves, R.P.; Fontenele, J.B.; Vasconcelos, S.M.M.; de Barros Viana, G.S.; Leal, L.K.A.M. The Operculina macrocarpa (l.) urb. (jalapa) tincture modulates human blood platelet aggregation. J. Ethnopharmacol. 2014, 151, 151–157. [Google Scholar]
- Badgujar, S.B. Evaluation of hemostatic activity of latex from three Euphorbiaceae species. J. Ethnopharmacol. 2014, 151, 733–739. [Google Scholar]
- Wei, A.H.; Schoenwaelder, S.M.; Andrews, R.K.; Jackson, S.P. New insights into the haemostatic function of platelets. Br. J. Haematol. 2009, 147, 415–430. [Google Scholar]
- Austin, S.K. Haemostasis. Medicine 2009, 37, 133–136. [Google Scholar]
- Zheng, Y.; Chen, J.; López, J.A. Microvascular platforms for the study of platelet-vessel wall interactions. Thromb. Res. 2014, 133, 525–531. [Google Scholar]
- Roest, M.; Sixma, J.J.; Wu, Y.-P.; Ijsseldijk, M.J.W.; Tempelman, M.; Slootweg, P.J.; de Groot, P.G.; van Zanten, G.H. Platelet adhesion to collagen in healthy volunteers is influenced by variation of both α2β1 density and von Willebrand factor. Blood 2000, 96, 1433–1437. [Google Scholar]
- Shankaran, H.; Alexandridis, P.; Neelamegham, S. Aspects of hydrodynamic shear regulating shear-induced platelet activation and self-association of von Willebrand factor in suspension. Blood 2003, 101, 2637–2645. [Google Scholar]
- Bergmeier, W.; Hynes, R.O. Extracellular matrix proteins in hemostasis and thrombosis. Cold Spring Harb. Perspect. Biol. 2012, 4, a005132. [Google Scholar]
- Jarvis, G.E.; Atkinson, B.T.; Snell, D.C.; Watson, S.P. Distinct roles of GPVI and integrin α2β1 in platelet shape change and aggregation induced by different collagens. Br. J. Pharmacol. 2002, 137, 107–117. [Google Scholar]
- Varga-Szabo, D.; Pleines, I.; Nieswandt, B. Cell adhesion mechanisms in platelets. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 403–412. [Google Scholar]
- Yuan, Y.; Kulkarni, S.; Ulsemer, P.; Cranmer, S.L.; Yap, C.L.; Nesbitt, W.S.; Harper, I.; Mistry, N.; Dopheide, S.M.; Hughan, S.C.; et al. The von Willebrand factor-glycoprotein Ib/V/IX interaction induces actin polymerization and cytoskeletal reorganization in rolling platelets and glycoprotein Ib/V/IX-transfected cells. J. Biol. Chem. 1999, 274, 36241–36251. [Google Scholar]
- Weiss, H.J. Platelet physiology and abnormalities of platelet function (first of two parts). N. Engl. J. Med. 1975, 293, 531–541. [Google Scholar]
- Jurk, K.; Kehrel, B.E. Platelets: Physiology and biochemistry. Semin. Thromb. Hemost. 2005, 31, 381–392. [Google Scholar]
- Reinhart, W.H. Platelets in vascular disease. Clin. Hemorheol. Microcirc. 2013, 53, 71–79. [Google Scholar]
- Erhardtsen, E. To general haemostasis—The evidence-based route. Pathophysiol. Haemost. Thromb. 2002, 32, 47–52. [Google Scholar]
- Kottke-Marchant, K.; Corcoran, G. The laboratory diagnosis of platelet disorders: An algorithmic approach. Arch. Pathol. Lab. Med. 2002, 126, 133–146. [Google Scholar]
- Lind, S.E.; Kurkjian, C.D.; Michaelson, A.D. The bleeding time. In Platelets; Michelson, A.D., Ed.; Elsevier Science: San Diego, CA, USA, 2002; pp. 283–289. [Google Scholar]
- Strukova, S. Blood coagulation-dependent inflammation. Coagulation-dependent inflammation and inflammation-dependent thrombosis. Front. Biosci. J. Virtual Libr. 2006, 11, 59–80. [Google Scholar]
- Kozek-Langenecker, S.A. Perioperative coagulation monitoring. Best Pract. Res. Clin. Anaesthesiol. 2010, 24, 27–40. [Google Scholar]
- Frontroth, J.P. Light transmission aggregometry. In Haemostasis; Monagle, P., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 227–240. [Google Scholar]
- Harrison, P. The role of PFA-100® testing in the investigation and management of haemostatic defects in children and adults. Br. J. Haematol. 2005, 130, 3–10. [Google Scholar]
- Vinholt, P.J.; Hvas, A.-M.; Nybo, M. An overview of platelet indices and methods for evaluating platelet function in thrombocytopenic patients. Eur. J. Haematol. 2014, 92, 367–376. [Google Scholar]
- Koscielny, J.; Ziemer, S.; Radtke, H.; Schmutzler, M.; Pruss, A.; Sinha, P.; Salama, A.; Kiesewetter, H.; Latza, R. A practical concept for preoperative identification of patients with impaired primary hemostasis. Clin. Appl. Thromb. 2004, 10, 195–204. [Google Scholar]
- Koscielny, J.; von Tempelhoff, G.-F.; Ziemer, S.; Radtke, H.; Schmutzler, M.; Sinha, P.; Salama, A.; Kiesewetter, H.; Latza, R. A practical concept for preoperative management of patients with impaired primary hemostasis. Clin. Appl. Thromb. 2004, 10, 155–166. [Google Scholar]
- Janssen, P.W.; ten Berg, J.M.; Hackeng, C.M. The use of platelet function testing in PCI and CABG patients. Blood Rev. 2014, 28, 109–121. [Google Scholar]
- FERRER-MARIN, F.; Chavda, C.; Lampa, M.; Michelson, A.D.; Frelinger, A.L.; SOLA-VISNER, M. Effects of in vitro adult platelet transfusions on neonatal hemostasis. J. Thromb. Haemost. 2011, 9, 1020–1028. [Google Scholar]
- Panzer, S.; Jilma, P. Methods for testing platelet function for transfusion medicine. Vox Sang. 2011, 101, 1–9. [Google Scholar]
- Refaai, M.A.; Laposata, M. Platelet aggregation. In Platelets; Michelson, A.D., Ed.; Elsevier Science: San Diego, CA, USA, 2002; pp. 291–296. [Google Scholar]
- Hathaway, W.E.; Goodnight, S.H. Hereditary platelet function defects. Disord. Haemost. Thromb. Clin. Guide N. Y. Mc Grew-Hile Inc. (Disorders of haemostasis and thrombosis: a clinical guide. New York: Mc Grew-Hile) 1993, 94–102. [Google Scholar]
- Slichter, S.J. Evidence-based platelet transfusion guidelines. ASH Educ. Program Book 2007, 2007, 172–178. [Google Scholar]
- Moncharmont, P.; Barday, G.; Meyer, F. Red blood cell alloimmunisation after platelet transfusion: A 5-year study. Blood Transfus. 2014, 12, s147–s148. [Google Scholar]
- Julmy, F.; Ammann, R.A.; Fontana, S.; Taleghani, B.M.; Hirt, A.; Leibundgut, K. Transfusion efficacy of apheresis platelet concentrates irradiated at the day of transfusion is significantly superior compared to platelets irradiated in advance. Transfus. Med. Hemotherapy 2014, 41, 176–181. [Google Scholar]
- Noh, J.-Y.; Weiss, M.J.; Poncz, M. Personalized platelet transfusions: One step closer to the clinic. Cell Stem Cell 2014, 14, 425–426. [Google Scholar]
- Franchini, M.; Lippi, G.; Veneri, D.; Targher, G.; Zaffanello, M.; Guidi, G.C. Inherited platelet disorders. Clin. Chim. Acta Int. J. Clin. Chem. 2008, 387, 1–8. [Google Scholar]
- Franchini, M.; Favaloro, E.J.; Lippi, G. Glanzmann thrombasthenia: An update. Clin. Chim. Acta Int. J. Clin. Chem. 2010, 411, 1–6. [Google Scholar]
- Pontara, E.; Gresele, P.; Cattini, M.G.; Daidone, V.; Barbon, G.; Girolami, A.; Zanon, E.; Casonato, A. Spontaneous hemarthrosis in combined Glanzmann thrombasthenia and type 2N von Willebrand disease. Blood Coagul. Fibrinolysis 2014, 25, 401–404. [Google Scholar]
- Kannan, M. Role of conformation sensitive gel electrophoresis in identifying mutations in Glanzmann’s thrombasthenia patients. 2014, 1, 104. [Google Scholar]
- Karanth, L.; Kanagasabai, S.; Abas, A.B. Maternal and foetal outcomes following natural vaginal versus caesarean section (c-section) delivery in carriers and women with bleeding disorders. Cochrane Libr. 2014. [Google Scholar] [CrossRef]
- Seligsohn, U. Treatment of inherited platelet disorders. Haemophilia 2012, 18, 161–165. [Google Scholar]
- Vadász, D.; Sztriha, L.K.; Sas, K.; Vécsei, L. Aspirin and clopidogrel resistance: Possible mechanisms and clinical relevance. Part II: Potential causes and laboratory tests. Ideggyógy. Szle. 2013, 66, 15–22. [Google Scholar]
- Panchadhyayee, P.; Saha, A.; Saha, K.; Ta, R.K.; Barma, P. Hermansky-Pudlak syndrome. Muller J. Med. Sci. Res. 2014, 5, 74. [Google Scholar]
- Jelenska, M.; Kopeć, M.; Breddin, K. On the retraction of collagen and fibrin induced by normal, defective and modified platelets. Haemostasis 1985, 15, 169–175. [Google Scholar]
- Vanhoorelbeke, K.; Schlammadinger, A.; Delville, J.P.; Handsaeme, J.; Vandecasteele, G.; Vauterin, S.; Pradier, O.; Wijns, W.; Deckmyn, H. Occurrence of the Asn45Ser mutation in the GPIX gene in a Belgian patient with Bernard Soulier syndrome. Platelets 2001, 12, 114–120. [Google Scholar]
- White, B.N.; Cox, A.C.; Taylor, F.B., Jr. The procoagulant effect of platelets on conversion of prothrombin to thrombin in nonanticoagulated plasma. J. Lab. Clin. Med. 1980, 95, 827–841. [Google Scholar]
- Thielen, N.; Huizing, M.; Krabbe, J.G.; White, J.G.; Jansen, T.J.; Merle, P.A.; Gahl, W.A.; Zweegman, S. Hermansky-Pudlak syndrome: The importance of molecular subtyping. J. Thromb. Haemost. 2010, 8, 1643–1645. [Google Scholar]
- Gunay-Aygun, M.; Huizing, M.; Gahl, W.A. Molecular defects that affect platelet dense granules. Semin. Thromb. Hemost. 2004, 30, 537–547. [Google Scholar]
- Braun, C.J.; Boztug, K.; Paruzynski, A.; Witzel, M.; Schwarzer, A.; Rothe, M.; Modlich, U.; Beier, R.; Göhring, G.; Steinemann, D. Gene therapy for Wiskott-Aldrich syndrome—Long-term efficacy and genotoxicity. Sci. Transl. Med. 2014, 6, 227ra33. [Google Scholar]
- Cui, S.H.; Tanabe, F.; Terunuma, H.; Iwatani, Y.; Nunoi, H.; Agematsu, K.; Komiyama, A.; Nomura, A.; Hara, T.; Onodera, T.; Iwata, T.; Ito, M. A thiol proteinase inhibitor, E-64-d, corrects the abnormalities in concanavalin A cap formation and the lysosomal enzyme activity in leucocytes from patients with Chediak-Higashi syndrome by reversing the down-regulated protein kinase C activity. Clin. Exp. Immunol. 2001, 125, 283–290. [Google Scholar]
- Marone, G.; Albini, F.; di Martino, L.; Quattrin, S.; Poto, S.; Condorelli, M. The Wiskott-Aldrich syndrome: Studies of platelets, basophils and polymorphonuclear leucocytes. Br. J. Haematol. 1986, 62, 737–745. [Google Scholar]
- Orange, J.S.; Stone, K.D.; Turvey, S.E.; Krzewski, K. The Wiskott-Aldrich syndrome. Cell. Mol. Life Sci. 2004, 61, 2361–2385. [Google Scholar]
- Fuster, V.; Badimon, L.; Badimon, J.J.; Chesebro, J.H. The pathogenesis of coronary artery disease and the acute coronary syndromes. N. Engl. J. Med. 1992, 326, 242–250. [Google Scholar]
- Michelson, A.D. Antiplatelet therapies for the treatment of cardiovascular disease. Nat. Rev. Drug Discov. 2010, 9, 154–169. [Google Scholar]
- Nieswandt, B.; Aktas, B.; Moers, A.; Sachs, U.J.H. Platelets in atherothrombosis: Lessons from mouse models. J. Thromb. Haemost. 2005, 3, 1725–1736. [Google Scholar]
- Ruggeri, Z.M. Mechanisms initiating platelet thrombus formation. Thromb. Haemost. 1997, 78, 611–616. [Google Scholar]
- Schäfer, A.; Eigenthaler, M.; Bauersachs, J. Platelet activation in heart failure. Clin. Lab. 2004, 50, 559–566. [Google Scholar]
- Sobieszczyk, P.; Fishbein, M.C.; Goldhaber, S.Z. Acute pulmonary embolism: Don’t ignore the platelet. Circulation 2002, 106, 1748–1749. [Google Scholar]
- Warrier, I.; Lusher, J.M. Congenital thrombocytopenias. Curr. Opin. Hematol. 1995, 2, 395–401. [Google Scholar]
- Watala, C. Blood platelet reactivity and its pharmacological modulation in (people with) diabetes mellitus. Curr. Pharm. Des. 2005, 11, 2331–2365. [Google Scholar]
- Wisler, J.W.; Becker, R.C. Emerging paradigms in arterial thrombosis. J. Thromb. Thrombolysis 2014, 37, 4–11. [Google Scholar]
- Gawaz, M.; Neumann, F.J.; Schomig, A. Evaluation of platelet membrane glycoproteins in coronary artery disease: Consequences for diagnosis and therapy. Circulation 1999, 99, E1–E11. [Google Scholar]
- Gregg, D.; Goldschmidt-Clermont, P.J. Cardiology patient page. Platelets and cardiovascular disease. Circulation 2003, 108, e88–e90. [Google Scholar]
- Ross, R. The pathogenesis of atherosclerosis: A perspective for the 1990s. 1993, 362, 801–809. [Google Scholar]
- Jackson, S.P.; Nesbitt, W.S.; Westein, E. Dynamics of platelet thrombus formation. J. Thromb. Haemost. 2009, 7, 17–20. [Google Scholar]
- Dvorak, M.; Vlasin, M.; Dvorakova, M.; Rauser, P.; Lexmaulova, L.; Gregor, Z.; Staffa, R. Heparin and its derivatives in the treatment of arterial thrombosis: A review. Vet. Med. 2010, 55, 523–546. [Google Scholar]
- Elg, M.; Gustafsson, D.; Carlsson, S. Antithrombotic effects and bleeding time of thrombin inhibitors and warfarin in the rat. Thromb. Res. 1999, 94, 187–197. [Google Scholar]
- Floyd, C.N.; Ferro, A. Mechanisms of aspirin resistance. Pharmacol. Ther. 2014, 141, 69–78. [Google Scholar]
- Angiolillo, D.J.; Datto, C.; Raines, S.; Yeomans, N.D. Impact of concomitant low-dose aspirin on the safety and tolerability of naproxen and esomeprazole magnesium delayed-release tablets in patients requiring chronic nonsteroidal anti-inflammatory drug therapy: An analysis from 5 phase III studies. J. Thromb. Thrombolysis 2013, 38, 1–13. [Google Scholar]
- Choi, J.-T.; Shin, K.-A.; Kim, Y.-K. Prevalence of aspirin resistance and clinical characteristics in patients with cerebral infarction. Korean Soc. Biomed. Lab. Sci. 2013, 19, 233–238. [Google Scholar]
- Cleland, J.G. For debate: Preventing atherosclerotic events with aspirin. BMJ 2002, 324, 103. [Google Scholar]
- Kunadian, V.; Sinclair, H.; Sutton, A.; Dangas, G.D. Aspirin, platelet P2Y12 receptor inhibitors, and other oral antiplatelets: Comparative pharmacology and role in elective PCI. Interv. Cardiol. Clin. 2013, 2, 527–535. [Google Scholar]
- Curtin, R.; Cox, D.; Fitzgerald, D. Clopidogrel and ticlopidine. In Platelets; Michelson, A.D., Ed.; Elsevier Science: San Diego, CA, USA, 2002; pp. 787–801. [Google Scholar]
- Eisert, W.G. Dipyridamole. In Platelets; Michelson, A.D., Ed.; Elsevier Science: San Diego, CA, USA, 2002; pp. 803–815. [Google Scholar]
- Farré, A.L.; Caramelo, C.; Casado, S. Nuevos mecanismos antiagregantes y vasodilatadores inducidos por la aspirina. Nefrología 1995, 16, 315–318. [Google Scholar]
- Geiger, J. Inhibitors of platelet signal transduction as anti-aggregatory drugs. Expert Opin. Investig. Drugs 2001, 10, 865–890. [Google Scholar]
- Zhang, Y. Apixaban for oral antithrombotic therapy: Is a new era coming? Mol. Cell. Ther. 2014, 2, 4. [Google Scholar]
- Catella-Lawson, F.; Reilly, M.P.; Kapoor, S.C.; Cucchiara, A.J.; DeMarco, S.; Tournier, B.; Vyas, S.N.; FitzGerald, G.A. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N. Engl. J. Med. 2001, 345, 1809–1817. [Google Scholar]
- Bates, E.R.; Lau, W.C. Controversies in antiplatelet therapy for patients with cardiovascular disease. Circulation 2005, 111, e267–e271. [Google Scholar]
- Pedersen, A.K.; FitzGerald, G.A. Dose-related kinetics of aspirin. Presystemic acetylation of platelet cyclooxygenase. N. Engl. J. Med. 1984, 311, 1206–1211. [Google Scholar]
- Altman, R.; Rivas, A.J.; Gonzalez, C.D. Bleeding tendency in dual antiplatelet therapy with aspirin/clopidogrel: Rescue of the template bleeding time in a single-center prospective study. Thromb. J. 2012, 10, 3. [Google Scholar]
- Cate, J.W.; Vries, S.I. The effect of aspirin on the bleeding time. Acta Med. Scand. 1972, 191, 215–217. [Google Scholar]
- Mielke, C.H., Jr. Influence of aspirin on platelets and the bleeding time. Am. J. Med. 1983, 74, 72–78. [Google Scholar]
- Wu, K.K.; Matijevic-Aleksic, N. Molecular aspects of thrombosis and antithrombotic drugs. Crit. Rev. Clin. Lab. Sci. 2005, 42, 249–277. [Google Scholar]
- Raju, N.C.; Eikelboom, J.W.; Hirsh, J. Platelet ADP-receptor antagonists for cardiovascular disease: Past, present and future. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 766–780. [Google Scholar]
- Weitz, J.I.; Eikelboom, J.W.; Samama, M. New antithrombotic drugs. Antithrombotic therapy and prevention of thrombosis. CHEST J. 2012, 141, e120. [Google Scholar]
- Angiolillo, D.J.; Ferreiro, J.L. Antiplatelet and anticoagulant therapy for atherothrombotic disease: The role of current and emerging agents. Am. J. Cardiovasc. Drugs 2013, 13, 233–250. [Google Scholar]
- Koster, A.; Chew, D.; Merkle, F.; Gruendel, M.; Jurmann, M.; Kuppe, H.; Oertel, R. Extracorporeal elimination of large concentrations of tirofiban by zero-balanced ultrafiltration during cardiopulmonary bypass: An in vitro investigation. Anesth. Analg. 2004, 99, 989–992. [Google Scholar]
- Vincentelli, A.; Jude, B.; Belisle, S. Antithrombotic therapy in cardiac surgery. Can. J. Anesth. 2006, 53, S89–S102. [Google Scholar]
- Warkentin, T.E.; Greinacher, A.; Koster, A. Heparin-induced thrombocytopenia in patients with ventricular assist devices: Are new prevention strategies required? Ann. Thorac. Surg. 2009, 87, 1633–1640. [Google Scholar]
- Warkentin, T.E. Agents for the treatment of heparin-induced thrombocytopenia. Hematol. Oncol. Clin. North Am. 2010, 24, 755–775. [Google Scholar]
- Hayden, M.; Pignone, M.; Phillips, C.; Mulrow, C. Aspirin for the primary prevention of cardiovascular events: A summary of the evidence for the U.S. preventive services task force. Ann. Intern. Med. 2002, 136, 161–172. [Google Scholar]
- Hankey, G.J.; Eikelboom, J.W. Aspirin resistance. Lancet 2006, 367, 606–617. [Google Scholar]
- Hennekens, C.H.; Schror, K.; Weisman, S.; FitzGerald, G.A. Terms and conditions: Semantic complexity and aspirin resistance. Circulation 2004, 110, 1706–1708. [Google Scholar]
- Mukherjee, D.; Nissen, S.E.; Topol, E.J. Risk of cardiovascular events associated with selective COX-2 inhibitors. J. Am. Med. Assoc. 2001, 286, 954–959. [Google Scholar]
- Altman, R.; Luciardi, H.L.; Muntaner, J.; Herrera, R.N. The antithrombotic profile of aspirin. Aspirin resistance, or simply failure? Thromb. J. 2004, 2, 1. [Google Scholar]
- Patrono, C.; García Rodríguez, L.A.; Landolfi, R.; Baigent, C. Low-dose aspirin for the prevention of atherothrombosis. N. Engl. J. Med. 2005, 353, 2373–2383. [Google Scholar]
- Floyd, C.N.; Goodman, T.; Becker, S.; Chen, N.; Mustafa, A.; Schofield, E.; Campbell, J.; Ward, M.; Sharma, P.; Ferro, A. Increased platelet expression of glycoprotein IIIa following aspirin treatment in aspirin-resistant but not aspirin-sensitive subjects. Br. J. Clin. Pharmacol. 2014, 78, 320–328. [Google Scholar]
- Gallego-Fabrega, C.; Krupinski, J.; Fernandez-Cadenas, I. Drug resistance and secondary treatment of ischaemic stroke: The genetic componente of the response to acetylsalicylic acid and clopidogrel (In Spanish). Neurología 2014, in press. [Google Scholar]
- Qureshi, Z.; Hobson, A.R. Clopidogrel “resistance”: Where are we now? Cardiovasc. Ther. 2013, 31, 3–11. [Google Scholar]
- Knoepp, S.M.; Laposata, M. Aspirin resistance: Moving forward with multiple definitions, different assays, and a clinical imperative. Am. J. Clin. Pathol. 2005, 123, S125–S132. [Google Scholar]
- Linden, M.D.; Frelinger, A.L., 3rd; Barnard, M.R.; Przyklenk, K.; Furman, M.I.; Michelson, A.D. Application of flow cytometry to platelet disorders. Semin. Thromb. Hemost. 2004, 30, 501–511. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geraldo, R.B.; Sathler, P.C.; Lourenço, A.L.; Saito, M.S.; Cabral, L.M.; Rampelotto, P.H.; Castro, H.C. Platelets: Still a Therapeutical Target for Haemostatic Disorders. Int. J. Mol. Sci. 2014, 15, 17901-17919. https://doi.org/10.3390/ijms151017901
Geraldo RB, Sathler PC, Lourenço AL, Saito MS, Cabral LM, Rampelotto PH, Castro HC. Platelets: Still a Therapeutical Target for Haemostatic Disorders. International Journal of Molecular Sciences. 2014; 15(10):17901-17919. https://doi.org/10.3390/ijms151017901
Chicago/Turabian StyleGeraldo, Reinaldo Barros, Plínio Cunha Sathler, André Luiz Lourenço, Max Seidy Saito, Lucio M. Cabral, Pabulo Henrique Rampelotto, and Helena Carla Castro. 2014. "Platelets: Still a Therapeutical Target for Haemostatic Disorders" International Journal of Molecular Sciences 15, no. 10: 17901-17919. https://doi.org/10.3390/ijms151017901