Ketamine and Calcium Signaling—A Crosstalk for Neuronal Physiology and Pathology


Abstract
:1. Introduction
2. Ketamine and Neuronal Calcium Homeostasis
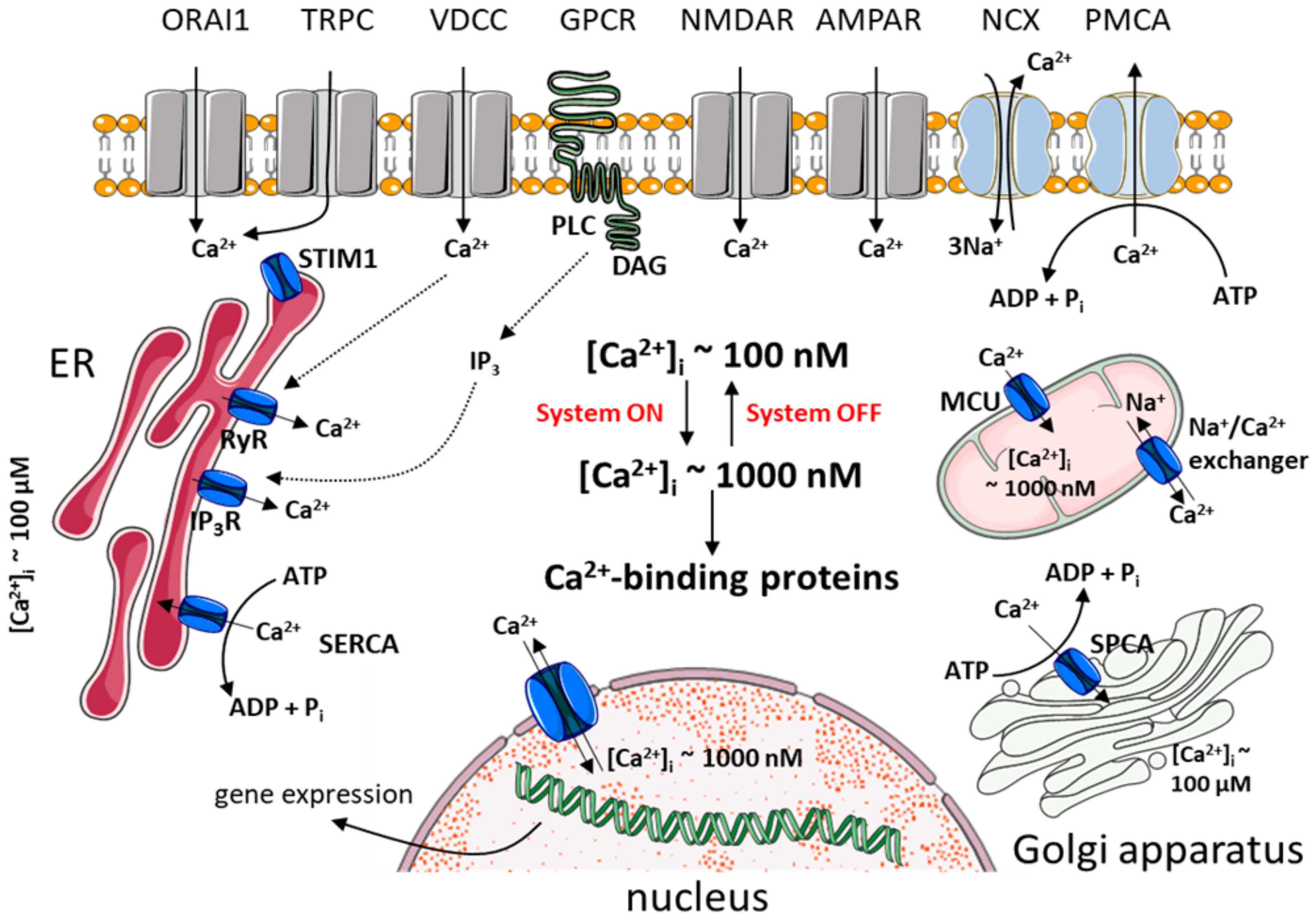
2.1. Ca2+ Homeostasis in Healthy Neurons
2.1.1. Mechanisms that Turn on Ca2+ Signal
2.1.2. Mechanisms that Turn off Ca2+ Signal
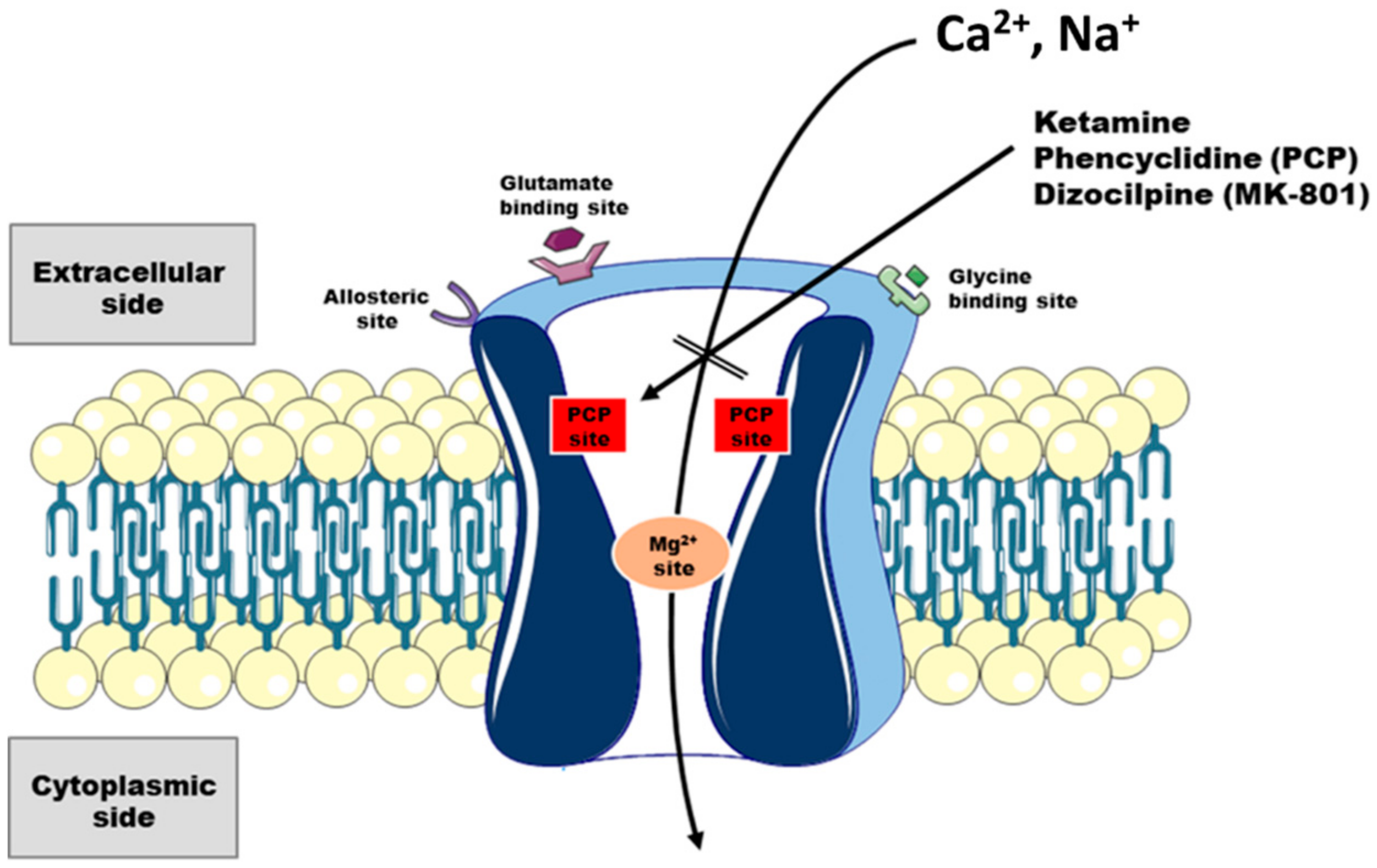
2.2. Interaction of Ketamine with Neuronal Ca2+ Toolkit
3. Ketamine, Calcium and Neuronal Development
3.1. Ketamine and Calcium Oscillations
3.2. Ketamine, Calcium and Enhanced Neuronal Death
3.3. Effects on Neurogenesis
3.3.1. Effects Facilitated by NMDA Receptors
3.3.2. Effects Facilitated by AMPA Receptors
4. Ketamine and Synaptic Transmission
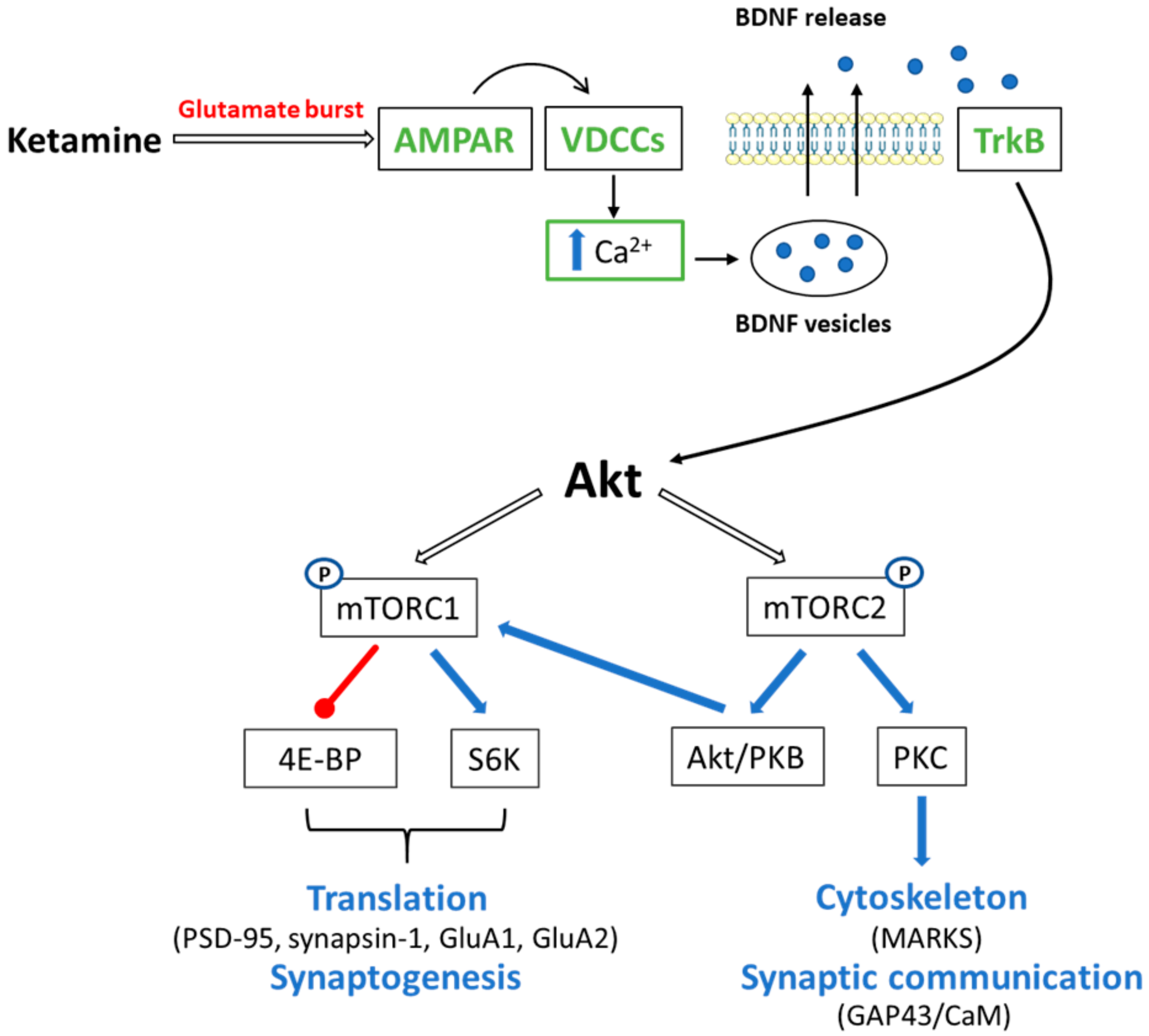
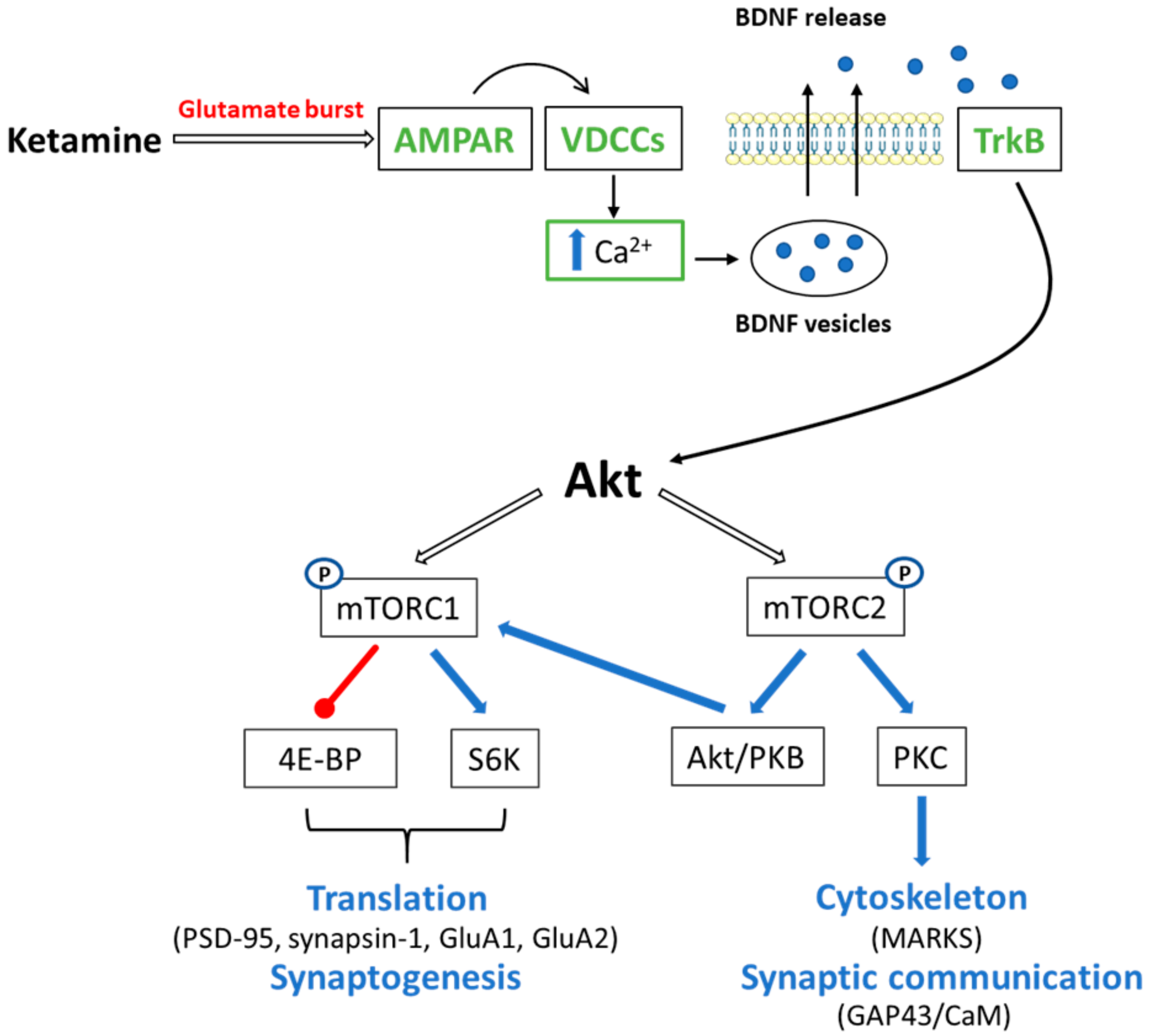
4.1. Ketamine and mTOR Signaling
4.1.1. mTORC1
4.1.2. mTORC2
4.2. Ketamine and Ca2+/CaM Signaling in Memory Processes
5. Concluding Remarks
Funding
Conflicts of Interest
References
- Anis, N.A.; Berry, S.C.; Burton, N.R.; Lodge, D. The dissociative anaesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurones by N-methyl-aspartate. Br. J. Pharmacol. 1983, 79, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Dinis-Oliveira, R.J. Metabolism and metabolomics of ketamine: A toxicological approach. Forensic. Sci. Res. 2017, 2, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Zanos, P.; Gould, T.D. Mechanisms of ketamine action as an antidepressant. Mol. Psychiatry 2018, 23, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Orser, B.A.; Pennefather, P.S.; MacDonald, J.F. Multiple mechanisms of ketamine blockade of N-methyl-D-aspartate receptors. Anesthesiology 1997, 86, 903–917. [Google Scholar] [CrossRef] [PubMed]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Riggs, L.M.; Highland, J.N.; Georgiou, P.; Pereira, E.F.R.; Albuquerque, E.X.; Thomas, C.J.; Zarate, C.A.; et al. Ketamine and Ketamine Metabolite Pharmacology: Insights into Therapeutic Mechanisms. Pharmacol. Rev. 2018, 70, 621–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B.; Charney, D.S. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Zarate, C.A.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 2006, 63, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Nowacka, A.; Borczyk, M. Ketamine applications beyond anesthesia—A literature review. Eur. J. Pharmacol. 2019, 860, 172547. [Google Scholar] [CrossRef]
- Muller, J.; Pentyala, S.; Dilger, J. Ketamine enantiomers in the rapid and sustained antidepressant effects. Ther. Adv. Psychopharmacol. 2016, 6, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Boczek, T.; Radzik, T.; Ferenc, B.; Zylinska, L. The Puzzling Role of Neuron-Specific PMCA Isoforms in the Aging Process. Int. J. Mol. Sci. 2019, 20, 6338. [Google Scholar] [CrossRef] [Green Version]
- Alves, V.S.; Alves-Silva, H.S.; Orts, D.J.B.; Ribeiro-Silva, L.; Arcisio-Miranda, M.; Oliveira, F.A. Calcium Signaling in Neurons and Glial Cells: Role of Cav1 channels. Neuroscience 2019, 421, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Puri, B.K. Calcium Signaling and Gene Expression. Adv. Exp. Med. Biol. 2020, 1131, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E.; Krebs, J. Why Calcium? How Calcium Became the Best Communicator. J. Biol. Chem. 2016, 291, 20849–20857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, C.G.; Takeuchi, K.; Rodenas-Ruano, A.; Takayasu, Y.; Murphy, J.; Bennett, M.V.; Zukin, R.S. Regulation of NMDA receptor Ca2+ signalling and synaptic plasticity. Biochem. Soc. Trans. 2009, 37, 1369–1374. [Google Scholar] [CrossRef] [Green Version]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Kamboj, S.; Ehlers, M.D.; Rosen, K.R.; Fischbach, G.D.; Huganir, R.L. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron 1998, 21, 1067–1078. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Levy, J.M.; Hou, A.; Winters, C.; Azzam, R.; Sousa, A.A.; Leapman, R.D.; Nicoll, R.A.; Reese, T.S. PSD-95 family MAGUKs are essential for anchoring AMPA and NMDA receptor complexes at the postsynaptic density. Proc. Natl. Acad. Sci. USA 2015, 112, E6983–E6992. [Google Scholar] [CrossRef] [Green Version]
- Dolphin, A.C. Voltage-gated calcium channels: Their discovery, function and importance as drug targets. Brain Neurosci. Adv. 2018, 2. [Google Scholar] [CrossRef]
- Brini, M.; Ottolini, D.; Calì, T.; Carafoli, E. Calcium in health and disease. Met. Ions Life Sci. 2013, 13, 81–137. [Google Scholar] [CrossRef]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Pla, A.F.; Maric, D.; Brazer, S.C.; Giacobini, P.; Liu, X.; Chang, Y.H.; Ambudkar, I.S.; Barker, J.L. Canonical transient receptor potential 1 plays a role in basic fibroblast growth factor (bFGF)/FGF receptor-1-induced Ca2+ entry and embryonic rat neural stem cell proliferation. J. Neurosci. 2005, 25, 2687–2701. [Google Scholar] [CrossRef]
- Wu, X.; Zagranichnaya, T.K.; Gurda, G.T.; Eves, E.M.; Villereal, M.L. A TRPC1/TRPC3-mediated increase in store-operated calcium entry is required for differentiation of H19-7 hippocampal neuronal cells. J. Biol. Chem. 2004, 279, 43392–43402. [Google Scholar] [CrossRef] [Green Version]
- Sawamura, S.; Hatano, M.; Takada, Y.; Hino, K.; Kawamura, T.; Tanikawa, J.; Nakagawa, H.; Hase, H.; Nakao, A.; Hirano, M.; et al. Screening of Transient Receptor Potential Canonical Channel Activators Identifies Novel Neurotrophic Piperazine Compounds. Mol. Pharmacol. 2016, 89, 348–363. [Google Scholar] [CrossRef] [Green Version]
- Nilius, B.; Owsianik, G. The transient receptor potential family of ion channels. Genome Biol. 2011, 12, 218. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Lidow, M.S. Calcium signaling dysfunction in schizophrenia: A unifying approach. Brain Res. Brain Res. Rev. 2003, 43, 70–84. [Google Scholar] [CrossRef]
- Bojarski, L.; Debowska, K.; Wojda, U. In vitro findings of alterations in intracellular calcium homeostasis in schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 1367–1374. [Google Scholar] [CrossRef]
- Lisek, M.; Boczek, T.; Ferenc, B.; Zylinska, L. Regional brain dysregulation of Ca(2+)-handling systems in ketamine-induced rat model of experimental psychosis. Cell Tissue Res. 2016, 363, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Sinner, B.; Friedrich, O.; Zink, W.; Zausig, Y.; Graf, B.M. The toxic effects of s(+)-ketamine on differentiating neurons in vitro as a consequence of suppressed neuronal Ca2+ oscillations. Anesth. Analg. 2011, 113, 1161–1169. [Google Scholar] [CrossRef]
- Ali, F.; Gerhard, D.M.; Sweasy, K.; Pothula, S.; Pittenger, C.; Duman, R.S.; Kwan, A.C. Ketamine disinhibits dendrites and enhances calcium signals in prefrontal dendritic spines. Nat. Commun. 2020, 11, 72. [Google Scholar] [CrossRef] [Green Version]
- Boczek, T.; Lisek, M.; Kowalski, A.; Pikula, S.; Niewiarowska, J.; Wiktorska, M.; Zylinska, L. Downregulation of PMCA2 or PMCA3 reorganizes Ca(2+) handling systems in differentiating PC12 cells. Cell Calcium 2012, 52, 433–444. [Google Scholar] [CrossRef]
- Fernandes, D.; Zaidi, A.; Bean, J.; Hui, D.; Michaelis, M.L. RNA—Induced silencing of the plasma membrane Ca2+-ATPase 2 in neuronal cells: Effects on Ca2+ homeostasis and cell viability. J. Neurochem. 2007, 102, 454–465. [Google Scholar] [CrossRef]
- Hegedűs, L.; Zámbó, B.; Pászty, K.; Padányi, R.; Varga, K.; Penniston, J.T.; Enyedi, Á. Molecular Diversity of Plasma Membrane Ca. Adv. Exp. Med. Biol. 2020, 1131, 93–129. [Google Scholar] [CrossRef]
- Lisek, M.; Ferenc, B.; Studzian, M.; Pulaski, L.; Guo, F.; Zylinska, L.; Boczek, T. Glutamate Deregulation in Ketamine-Induced Psychosis-A Potential Role of PSD95, NMDA Receptor and PMCA Interaction. Front. Cell NeuroSci. 2017, 11, 181. [Google Scholar] [CrossRef] [Green Version]
- Boczek, T.; Lisek, M.; Ferenc, B.; Zylinska, L. Plasma membrane Ca(2+)-ATPase is a novel target for ketamine action. Biochem. Biophys Res. Commun. 2015, 465, 312–317. [Google Scholar] [CrossRef]
- Cheung, H.M.; Yew, D.T.W. Effects of Perinatal Exposure to Ketamine on the Developing Brain. Front. NeuroSci. 2019, 13, 138. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Li, C.; Wei, W.; Zhang, H.; Ma, D.; Song, X.; Zhou, L. Prenatal ketamine exposure causes abnormal development of prefrontal cortex in rat. Sci. Rep. 2016, 6, 26865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Faria, O.; Gonsalvez, D.G.; Nicholson, M.; Xiao, J. Activity-dependent central nervous system myelination throughout life. J. Neurochem. 2019, 148, 447–461. [Google Scholar] [CrossRef] [Green Version]
- Gafarov, F.M. Neural electrical activity and neural network growth. Neural Netw. 2018, 101, 15–24. [Google Scholar] [CrossRef]
- Kato, D.; Eto, K.; Nabekura, J.; Wake, H. Activity-dependent functions of non-electrical glial cells. J. Biochem. 2018, 163, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Kirischuk, S.; Sinning, A.; Blanquie, O.; Yang, J.W.; Luhmann, H.J.; Kilb, W. Modulation of Neocortical Development by Early Neuronal Activity: Physiology and Pathophysiology. Front. Cell Neurosci. 2017, 11, 379. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, P.; Fritz, N.; Smedler, E.; Malmersjö, S.; Kanatani, S. Calcium signaling in neocortical development. Dev. Neurobiol. 2015, 75, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, C.; Néant, I.; Moreau, M. The calcium: An early signal that initiates the formation of the nervous system during embryogenesis. Front. Mol. Neurosci. 2012, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.S.; Spitzer, N.C. Calcium signaling in neuronal development. Cold Spring Harb. Perspect. Biol. 2011, 3, a004259. [Google Scholar] [CrossRef] [Green Version]
- Garaschuk, O.; Hanse, E.; Konnerth, A. Developmental profile and synaptic origin of early network oscillations in the CA1 region of rat neonatal hippocampus. J. Physiol. 1998, 507 Pt 1, 219–236. [Google Scholar] [CrossRef]
- Lin, Y.; Li, L.L.; Nie, W.; Liu, X.; Adler, A.; Xiao, C.; Lu, F.; Wang, L.; Han, H.; Wang, X.; et al. Brain activity regulates loose coupling between mitochondrial and cytosolic Ca. Nat. Commun. 2019, 10, 5277. [Google Scholar] [CrossRef] [Green Version]
- Ali, F.; Kwan, A.C. Interpreting. Neurophotonics 2020, 7, 011402. [Google Scholar] [CrossRef]
- Jiang, S.; Hao, Z.; Li, X.; Bo, L.; Zhang, R.; Wang, Y.; Duan, X.; Kang, R.; Huang, L. Ketamine destabilizes growth of dendritic spines in developing hippocampal neurons in vitro via a Rho-dependent mechanism. Mol. Med. Rep. 2018, 18, 5037–5043. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Liu, Y.; Zhang, P.; Kang, R.; Li, X.; Bo, L.; Dong, Z. In vitro dose-dependent inhibition of the intracellular spontaneous calcium oscillations in developing hippocampal neurons by ketamine. PLoS ONE 2013, 8, e59804. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.L.; Chan, S.L.; Way, W.L.; Trevor, A.J. Distribution in the brain and metabolism of ketamine in the rat after intravenous administration. Anesthesiology 1973, 39, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Allene, C.; Cossart, R. Early NMDA receptor-driven waves of activity in the developing neocortex: Physiological or pathological network oscillations? J. Physiol. 2010, 588, 83–91. [Google Scholar] [CrossRef]
- Salmon, C.K.; Pribiag, H.; Gizowski, C.; Farmer, W.T.; Cameron, S.; Jones, E.V.; Mahadevan, V.; Bourque, C.W.; Stellwagen, D.; Woodin, M.A.; et al. Depolarizing GABA Transmission Restrains Activity-Dependent Glutamatergic Synapse Formation in the Developing Hippocampal Circuit. Front. Cell Neurosci. 2020, 14, 36. [Google Scholar] [CrossRef] [Green Version]
- Sinner, B.; Friedrich, O.; Zink, W.; Martin, E.; Fink, R.H.; Graf, B.M. Ketamine stereoselectively inhibits spontaneous Ca2+-oscillations in cultured hippocampal neurons. Anesth. Analg. 2005, 100, 1660–1666. [Google Scholar] [CrossRef] [Green Version]
- Mestdagh, N.; Wülfert, E. Bicuculline increases Ca2+ transients in rat cerebellar granule cells through non-GABA(A) receptor associated mechanisms. Neurosci. Lett. 1999, 265, 95–98. [Google Scholar] [CrossRef]
- Wang, D.S.; Penna, A.; Orser, B.A. Ketamine Increases the Function of γ-Aminobutyric Acid Type A Receptors in Hippocampal and Cortical Neurons. Anesthesiology 2017, 126, 666–677. [Google Scholar] [CrossRef]
- Ren, Z.; Pribiag, H.; Jefferson, S.J.; Shorey, M.; Fuchs, T.; Stellwagen, D.; Luscher, B. Bidirectional Homeostatic Regulation of a Depression-Related Brain State by Gamma-Aminobutyric Acidergic Deficits and Ketamine Treatment. Biol. Psychiatry 2016, 80, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Lebedeva, Y.A.; Zakharova, A.V.; Sitdikova, G.F.; Zefirov, A.L.; Khazipov, R.N. Ketamine-Midazolam Anesthesia Induces Total Inhibition of Cortical Activity in the Brain of Newborn Rats. Bull. Exp. Biol. Med. 2016, 161, 15–19. [Google Scholar] [CrossRef]
- Groen, M.R.; Paulsen, O.; Pérez-Garci, E.; Nevian, T.; Wortel, J.; Dekker, M.P.; Mansvelder, H.D.; van Ooyen, A.; Meredith, R.M. Development of dendritic tonic GABAergic inhibition regulates excitability and plasticity in CA1 pyramidal neurons. J. Neurophysiol. 2014, 112, 287–299. [Google Scholar] [CrossRef] [Green Version]
- Wilmes, K.A.; Sprekeler, H.; Schreiber, S. Inhibition as a Binary Switch for Excitatory Plasticity in Pyramidal Neurons. PLoS Comput. Biol. 2016, 12, e1004768. [Google Scholar] [CrossRef]
- Peltoniemi, M.A.; Hagelberg, N.M.; Olkkola, K.T.; Saari, T.I. Ketamine: A Review of Clinical Pharmacokinetics and Pharmacodynamics in Anesthesia and Pain Therapy. Clin. Pharm. 2016, 55, 1059–1077. [Google Scholar] [CrossRef]
- Yuryev, M.; Andriichuk, L.; Leiwe, M.; Jokinen, V.; Carabalona, A.; Rivera, C. In vivo two-photon imaging of the embryonic cortex reveals spontaneous ketamine-sensitive calcium activity. Sci. Rep. 2018, 8, 16059. [Google Scholar] [CrossRef] [Green Version]
- Shioda, N.; Fukunaga, K. Physiological and Pathological Roles of CaMKII-PP1 Signaling in the Brain. Int. J. Mol. Sci. 2017, 19, 20. [Google Scholar] [CrossRef] [Green Version]
- Menegon, A.; Verderio, C.; Leoni, C.; Benfenati, F.; Czernik, A.J.; Greengard, P.; Matteoli, M.; Valtorta, F. Spatial and temporal regulation of Ca2+/calmodulin-dependent protein kinase II activity in developing neurons. J. Neurosci. 2002, 22, 7016–7026. [Google Scholar] [CrossRef] [Green Version]
- Viberg, H.; Pontén, E.; Eriksson, P.; Gordh, T.; Fredriksson, A. Neonatal ketamine exposure results in changes in biochemical substrates of neuronal growth and synaptogenesis, and alters adult behavior irreversibly. Toxicology 2008, 249, 153–159. [Google Scholar] [CrossRef]
- De Roo, M.; Klauser, P.; Briner, A.; Nikonenko, I.; Mendez, P.; Dayer, A.; Kiss, J.Z.; Muller, D.; Vutskits, L. Anesthetics rapidly promote synaptogenesis during a critical period of brain development. PLoS ONE 2009, 4, e7043. [Google Scholar] [CrossRef]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Ureshino, R.P.; Erustes, A.G.; Bassani, T.B.; Wachilewski, P.; Guarache, G.C.; Nascimento, A.C.; Costa, A.J.; Smaili, S.S.; Pereira, G.J.D.S. The Interplay between Ca. Int. J. Mol. Sci. 2019, 20, 6004. [Google Scholar] [CrossRef] [Green Version]
- Bosnjak, Z.J.; Yan, Y.; Canfield, S.; Muravyeva, M.Y.; Kikuchi, C.; Wells, C.W.; Corbett, J.A.; Bai, X. Ketamine induces toxicity in human neurons differentiated from embryonic stem cells via mitochondrial apoptosis pathway. Curr. Drug Saf. 2012, 7, 106–119. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Shen, F.Y.; Zou, R.; Zheng, J.J.; Yu, X.; Wang, Y.W. Ketamine-induced apoptosis in the mouse cerebral cortex follows similar characteristic of physiological apoptosis and can be regulated by neuronal activity. Mol. Brain 2017, 10, 24. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Li, Y.; Zhao, J.; Li, L.; Wang, Y.; Zhang, Y.; Chen, Y.; Liu, W.; Gao, L. Administration of Ketamine Causes Autophagy and Apoptosis in the Rat Fetal Hippocampus and in PC12 Cells. Front. Cell Neurosci. 2018, 12, 21. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Guo, L.; Patterson, T.A.; Dial, S.; Li, Q.; Sadovova, N.; Zhang, X.; Hanig, J.P.; Paule, M.G.; Slikker, W.; et al. Gene expression profiling in the developing rat brain exposed to ketamine. Neuroscience 2010, 166, 852–863. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Patterson, T.A.; Sadovova, N.; Zhang, X.; Liu, S.; Zou, X.; Hanig, J.P.; Paule, M.G.; Slikker, W.; Wang, C. Ketamine-induced neuronal damage and altered N-methyl-D-aspartate receptor function in rat primary forebrain culture. Toxicol. Sci. 2013, 131, 548–557. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liu, F.; Patterson, T.A.; Paule, M.G.; Slikker, W. Relationship between ketamine-induced developmental neurotoxicity and NMDA receptor-mediated calcium influx in neural stem cell-derived neurons. Neurotoxicology 2017, 60, 254–259. [Google Scholar] [CrossRef]
- Ito, H.; Uchida, T.; Makita, K. Ketamine causes mitochondrial dysfunction in human induced pluripotent stem cell-derived neurons. PLoS ONE 2015, 10, e0128445. [Google Scholar] [CrossRef] [Green Version]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Xavier, J.M.; Rodrigues, C.M.; Solá, S. Mitochondria: Major Regulators of Neural Development. Neuroscientist 2016, 22, 346–358. [Google Scholar] [CrossRef]
- Behrens, M.M.; Ali, S.S.; Dao, D.N.; Lucero, J.; Shekhtman, G.; Quick, K.L.; Dugan, L.L. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science 2007, 318, 1645–1647. [Google Scholar] [CrossRef] [Green Version]
- Ikonomidou, C.; Bosch, F.; Miksa, M.; Bittigau, P.; Vöckler, J.; Dikranian, K.; Tenkova, T.I.; Stefovska, V.; Turski, L.; Olney, J.W. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999, 283, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Slikker, W.; Liu, F.; Rainosek, S.W.; Patterson, T.A.; Sadovova, N.; Hanig, J.P.; Paule, M.G.; Wang, C. Ketamine-Induced Toxicity in Neurons Differentiated from Neural Stem Cells. Mol. Neurobiol. 2015, 52, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Varbanov, H.; Dityatev, A. Regulation of extrasynaptic signaling by polysialylated NCAM: Impact for synaptic plasticity and cognitive functions. Mol. Cell Neurosci. 2017, 81, 12–21. [Google Scholar] [CrossRef]
- Murray, H.C.; Low, V.F.; Swanson, M.E.; Dieriks, B.V.; Turner, C.; Faull, R.L.; Curtis, M.A. Distribution of PSA-NCAM in normal, Alzheimer’s and Parkinson’s disease human brain. Neuroscience 2016, 330, 359–375. [Google Scholar] [CrossRef]
- Dityatev, A.; Dityateva, G.; Sytnyk, V.; Delling, M.; Toni, N.; Nikonenko, I.; Muller, D.; Schachner, M. Polysialylated neural cell adhesion molecule promotes remodeling and formation of hippocampal synapses. J. Neurosci. 2004, 24, 9372–9382. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Pralong, W.F.; Schulz, M.F.; Rougon, G.; Aubry, J.M.; Pagliusi, S.; Robert, A.; Kiss, J.Z. Functional N-methyl-D-aspartate receptors in O-2A glial precursor cells: A critical role in regulating polysialic acid-neural cell adhesion molecule expression and cell migration. J. Cell Biol. 1996, 135, 1565–1581. [Google Scholar] [CrossRef] [Green Version]
- Brusés, J.L.; Rutishauser, U. Regulation of neural cell adhesion molecule polysialylation: Evidence for nontranscriptional control and sensitivity to an intracellular pool of calcium. J. Cell Biol. 1998, 140, 1177–1186. [Google Scholar] [CrossRef] [Green Version]
- Bustamante, J.; Acosta, L.; Karadayian, A.G.; Lores-Arnaiz, S. Ketamine induced cell death can be mediated by voltage dependent calcium channels in PC12 cells. Exp. Mol. Pathol 2019, 111, 104318. [Google Scholar] [CrossRef]
- Yan, J.; Huang, Y.; Lu, Y.; Chen, J.; Jiang, H. Repeated administration of ketamine can induce hippocampal neurodegeneration and long-term cognitive impairment via the ROS/HIF-1α pathway in developing rats. Cell Physiol. Biochem. 2014, 33, 1715–1732. [Google Scholar] [CrossRef] [PubMed]
- Yamakage, M.; Hirshman, C.A.; Croxton, T.L. Inhibitory effects of thiopental, ketamine, and propofol on voltage-dependent Ca2+ channels in porcine tracheal smooth muscle cells. Anesthesiology 1995, 83, 1274–1282. [Google Scholar] [CrossRef]
- Toth, A.B.; Shum, A.K.; Prakriya, M. Regulation of neurogenesis by calcium signaling. Cell Calcium 2016, 59, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Keilhoff, G.; Bernstein, H.G.; Becker, A.; Grecksch, G.; Wolf, G. Increased neurogenesis in a rat ketamine model of schizophrenia. Biol. Psychiatry 2004, 56, 317–322. [Google Scholar] [CrossRef]
- Adesnik, H.; Li, G.; During, M.J.; Pleasure, S.J.; Nicoll, R.A. NMDA receptors inhibit synapse unsilencing during brain development. Proc. Natl. Acad. Sci. USA 2008, 105, 5597–5602. [Google Scholar] [CrossRef] [Green Version]
- Luk, K.C.; Kennedy, T.E.; Sadikot, A.F. Glutamate promotes proliferation of striatal neuronal progenitors by an NMDA receptor-mediated mechanism. J. Neurosci. 2003, 23, 2239–2250. [Google Scholar] [CrossRef] [Green Version]
- Lerea, L.S.; Butler, L.S.; McNamara, J.O. NMDA and non-NMDA receptor-mediated increase of c-fos mRNA in dentate gyrus neurons involves calcium influx via different routes. J. NeuroSci. 1992, 12, 2973–2981. [Google Scholar] [CrossRef]
- De Bartolomeis, A.; Sarappa, C.; Buonaguro, E.F.; Marmo, F.; Eramo, A.; Tomasetti, C.; Iasevoli, F. Different effects of the NMDA receptor antagonists ketamine, MK-801, and memantine on postsynaptic density transcripts and their topography: Role of Homer signaling, and implications for novel antipsychotic and pro-cognitive targets in psychosis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 46, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.M.; Ai, N.; Ke, M.; Tan, Y.; Huang, Z.; Li, Y.; Lu, J.H.; Ge, W.; Su, H. Roles of Nitric Oxide Synthase Isoforms in Neurogenesis. Mol. Neurobiol. 2018, 55, 2645–2652. [Google Scholar] [CrossRef]
- Fritzen, S.; Schmitt, A.; Köth, K.; Sommer, C.; Lesch, K.P.; Reif, A. Neuronal nitric oxide synthase (NOS-I) knockout increases the survival rate of neural cells in the hippocampus independently of BDNF. Mol. Cell Neurosci. 2007, 35, 261–271. [Google Scholar] [CrossRef]
- Jin, X.; Yu, Z.F.; Chen, F.; Lu, G.X.; Ding, X.Y.; Xie, L.J.; Sun, J.T. Neuronal Nitric Oxide Synthase in Neural Stem Cells Induces Neuronal Fate Commitment via the Inhibition of Histone Deacetylase 2. Front. Cell Neurosci. 2017, 11, 66. [Google Scholar] [CrossRef] [Green Version]
- Tellios, V.; Maksoud, M.J.E.; Xiang, Y.Y.; Lu, W.Y. Nitric Oxide Critically Regulates Purkinje Neuron Dendritic Development Through a Metabotropic Glutamate Receptor Type 1-Mediated Mechanism. Cerebellum 2020, 19, 510–526. [Google Scholar] [CrossRef]
- Gui, L.; Zhu, J.; Lu, X.; Sims, S.M.; Lu, W.Y.; Stathopulos, P.B.; Feng, Q. S-Nitrosylation of STIM1 by Neuronal Nitric Oxide Synthase Inhibits Store-Operated Ca. J. Mol. Biol. 2018, 430, 1773–1785. [Google Scholar] [CrossRef] [PubMed]
- Nagy, D.; Stoiljkovic, M.; Menniti, F.S.; Hajós, M. Differential Effects of an NR2B NAM and Ketamine on Synaptic Potentiation and Gamma Synchrony: Relevance to Rapid-Onset Antidepressant Efficacy. Neuropsychopharmacology 2016, 41, 1486–1494. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Lee, B.; Liu, R.J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.Y.; Aghajanian, G.; Duman, R.S. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, J.S.; Kee, N.; Wojtowicz, J.M. Effects of adult neurogenesis on synaptic plasticity in the rat dentate gyrus. J. Neurophysiol. 2001, 85, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Choo, A.M.; Geddes-Klein, D.M.; Hockenberry, A.; Scarsella, D.; Mesfin, M.N.; Singh, P.; Patel, T.P.; Meaney, D.F. NR2A and NR2B subunits differentially mediate MAP kinase signaling and mitochondrial morphology following excitotoxic insult. Neurochem. Int. 2012, 60, 506–516. [Google Scholar] [CrossRef] [Green Version]
- Sobczyk, A.; Scheuss, V.; Svoboda, K. NMDA receptor subunit-dependent [Ca2+] signaling in individual hippocampal dendritic spines. J. Neurosci. 2005, 25, 6037–6046. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Liu, H.J.; Qi, L.; Tao, C.L.; Wang, Y.J.; Shen, Z.; Tian, C.L.; Lau, P.M.; Bi, G.Q. Structure and plasticity of silent synapses in developing hippocampal neurons visualized by super-resolution imaging. Cell Discov 2020, 6, 8. [Google Scholar] [CrossRef] [Green Version]
- Groc, L.; Gustafsson, B.; Hanse, E. AMPA signalling in nascent glutamatergic synapses: There and not there! Trends Neurosci. 2006, 29, 132–139. [Google Scholar] [CrossRef]
- Collo, G.; Cavalleri, L.; Chiamulera, C.; Merlo Pich, E. Ketamine increases the expression of GluR1 and GluR2 α-amino-3-hydroxy-5-methy-4-isoxazole propionate receptor subunits in human dopaminergic neurons differentiated from induced pluripotent stem cells. Neuroreport 2019, 30, 207–212. [Google Scholar] [CrossRef]
- Zhang, K.; Yamaki, V.N.; Wei, Z.; Zheng, Y.; Cai, X. Differential regulation of GluA1 expression by ketamine and memantine. Behav. Brain Res. 2017, 316, 152–159. [Google Scholar] [CrossRef]
- Pałucha-Poniewiera, A. The role of glutamatergic modulation in the mechanism of action of ketamine, a prototype rapid-acting antidepressant drug. Pharmacol. Rep. 2018, 70, 837–846. [Google Scholar] [CrossRef]
- Kavalali, E.T.; Monteggia, L.M. How does ketamine elicit a rapid antidepressant response? Curr. Opin. Pharmacol. 2015, 20, 35–39. [Google Scholar] [CrossRef] [Green Version]
- Lepack, A.E.; Fuchikami, M.; Dwyer, J.M.; Banasr, M.; Duman, R.S. BDNF release is required for the behavioral actions of ketamine. Int. J. Neuropsychopharmacol. 2014, 18, pyu033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deisseroth, K.; Singla, S.; Toda, H.; Monje, M.; Palmer, T.D.; Malenka, R.C. Excitation-neurogenesis coupling in adult neural stem/progenitor cells. Neuron 2004, 42, 535–552. [Google Scholar] [CrossRef] [Green Version]
- Zheng, F.; Wang, H. NMDA-mediated and self-induced bdnf exon IV transcriptions are differentially regulated in cultured cortical neurons. Neurochem. Int. 2009, 54, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Björkholm, C.; Monteggia, L.M. BDNF—A key transducer of antidepressant effects. Neuropharmacology 2016, 102, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Réus, G.Z.; Abaleira, H.M.; Titus, S.E.; Arent, C.O.; Michels, M.; da Luz, J.R.; dos Santos, M.A.; Carlessi, A.S.; Matias, B.I.; Bruchchen, L.; et al. Effects of ketamine administration on the phosphorylation levels of CREB and TrKB and on oxidative damage after infusion of MEK inhibitor. Pharmacol. Rep. 2016, 68, 177–184. [Google Scholar] [CrossRef]
- Li, X.; Guo, C.; Li, Y.; Li, L.; Wang, Y.; Zhang, Y.; Chen, Y.; Liu, W.; Gao, L. Ketamine administered pregnant rats impair learning and memory in offspring via the CREB pathway. Oncotarget 2017, 8, 32433–32449. [Google Scholar] [CrossRef] [Green Version]
- Parise, E.M.; Alcantara, L.F.; Warren, B.L.; Wright, K.N.; Hadad, R.; Sial, O.K.; Kroeck, K.G.; Iñiguez, S.D.; Bolaños-Guzmán, C.A. Repeated ketamine exposure induces an enduring resilient phenotype in adolescent and adult rats. Biol. Psychiatry 2013, 74, 750–759. [Google Scholar] [CrossRef] [Green Version]
- Moda-Sava, R.N.; Murdock, M.H.; Parekh, P.K.; Fetcho, R.N.; Huang, B.S.; Huynh, T.N.; Witztum, J.; Shaver, D.C.; Rosenthal, D.L.; Alway, E.J.; et al. Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 2019, 364, eaat8078. [Google Scholar] [CrossRef]
- Liu, F.; Paule, M.G.; Ali, S.; Wang, C. Ketamine-induced neurotoxicity and changes in gene expression in the developing rat brain. Curr. Neuropharmacol. 2011, 9, 256–261. [Google Scholar] [CrossRef] [Green Version]
- Upton, D.H.; Popovic, K.; Fulton, R.; Kassiou, M. Anaesthetic-dependent changes in gene expression following acute and chronic exposure in the rodent brain. Sci. Rep. 2020, 10, 9366. [Google Scholar] [CrossRef]
- Phoumthipphavong, V.; Barthas, F.; Hassett, S.; Kwan, A.C. Longitudinal Effects of Ketamine on Dendritic Architecture In Vivo in the Mouse Medial Frontal Cortex. eNeuro 2016, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, T.H.; Gardier, A.M. Fast-acting antidepressant activity of ketamine: Highlights on brain serotonin, glutamate, and GABA neurotransmission in preclinical studies. Pharmacol. Ther. 2019, 199, 58–90. [Google Scholar] [CrossRef]
- Kavalali, E.T.; Monteggia, L.M. Synaptic mechanisms underlying rapid antidepressant action of ketamine. Am. J. Psychiatry 2012, 169, 1150–1156. [Google Scholar] [CrossRef]
- Suárez-Santiago, J.E.; Orozco-Suárez, S.; Vega-García, A.; Bautista-Orozco, L.; Picazo, O. Repeated ketamine administration induces recognition memory impairment together with morphological changes in neurons from ventromedial prefrontal cortex, dorsal striatum, and hippocampus. Behav. Pharmacol. 2020, 31, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Sial, O.K.; Parise, E.M.; Parise, L.F.; Gnecco, T.; Bolaños-Guzmán, C.A. Ketamine: The final frontier or another depressing end? Behav. Brain Res. 2020, 383, 112508. [Google Scholar] [CrossRef]
- Ryskalin, L.; Lazzeri, G.; Flaibani, M.; Biagioni, F.; Gambardella, S.; Frati, A.; Fornai, F. mTOR-Dependent Cell Proliferation in the Brain. BioMed Res. Int. 2017, 2017, 7082696. [Google Scholar] [CrossRef]
- Hoeffer, C.A.; Klann, E. mTOR signaling: At the crossroads of plasticity, memory and disease. Trends Neurosci. 2010, 33, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockaert, J.; Marin, P. mTOR in Brain Physiology and Pathologies. Physiol. Rev. 2015, 95, 1157–1187. [Google Scholar] [CrossRef]
- Vilar, M.; Mira, H. Regulation of Neurogenesis by Neurotrophins during Adulthood: Expected and Unexpected Roles. Front. Neurosci. 2016, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Miao, L.; Liang, F.; Huang, H.; Teng, X.; Li, S.; Nuriddinov, J.; Selzer, M.E.; Hu, Y. The mTORC1 effectors S6K1 and 4E-BP play different roles in CNS axon regeneration. Nat. Commun. 2014, 5, 5416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Liu, R.J.; Dwyer, J.M.; Banasr, M.; Lee, B.; Son, H.; Li, X.Y.; Aghajanian, G.; Duman, R.S. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol. Psychiatry 2011, 69, 754–761. [Google Scholar] [CrossRef] [Green Version]
- Takei, N.; Inamura, N.; Kawamura, M.; Namba, H.; Hara, K.; Yonezawa, K.; Nawa, H. Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J. Neurosci. 2004, 24, 9760–9769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duman, R.S.; Deyama, S.; Fogaça, M.V. Role of BDNF in the pathophysiology and treatment of depression: Activity-dependent effects distinguish rapid-acting antidepressants. Eur. J. Neurosci. 2019. [Google Scholar] [CrossRef]
- Deyama, S.; Duman, R.S. Neurotrophic mechanisms underlying the rapid and sustained antidepressant actions of ketamine. Pharmacol. Biochem. Behav. 2020, 188, 172837. [Google Scholar] [CrossRef]
- Cheng, Q.; Song, S.H.; Augustine, G.J. Calcium-Dependent and Synapsin-Dependent Pathways for the Presynaptic Actions of BDNF. Front. Cell Neurosci. 2017, 11, 75. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic, J.N.; Czernik, A.J.; Fienberg, A.A.; Greengard, P.; Sihra, T.S. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat. Neurosci. 2000, 3, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.D.; Pozzo-Miller, L. Intracellular Ca2+ stores and Ca2+ influx are both required for BDNF to rapidly increase quantal vesicular transmitter release. Neural Plast. 2012, 2012, 203536. [Google Scholar] [CrossRef] [Green Version]
- Yoo, K.S.; Lee, K.; Oh, J.Y.; Lee, H.; Park, H.; Park, Y.S.; Kim, H.K. Postsynaptic density protein 95 (PSD-95) is transported by KIF5 to dendritic regions. Mol. Brain 2019, 12, 97. [Google Scholar] [CrossRef] [Green Version]
- Aleksandrova, L.R.; Phillips, A.G.; Wang, Y.T. Antidepressant effects of ketamine and the roles of AMPA glutamate receptors and other mechanisms beyond NMDA receptor antagonism. J. Psychiatry Neurosci. 2017, 42, 222–229. [Google Scholar] [CrossRef] [Green Version]
- Buonarati, O.R.; Hammes, E.A.; Watson, J.F.; Greger, I.H.; Hell, J.W. Mechanisms of postsynaptic localization of AMPA-type glutamate receptors and their regulation during long-term potentiation. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Zhu, P.J.; Zhang, S.; Zhou, H.; Stoica, L.; Galiano, M.; Krnjević, K.; Roman, G.; Costa-Mattioli, M. mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat. Neurosci. 2013, 16, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Thomanetz, V.; Angliker, N.; Cloëtta, D.; Lustenberger, R.M.; Schweighauser, M.; Oliveri, F.; Suzuki, N.; Rüegg, M.A. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J. Cell Biol. 2013, 201, 293–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sossin, W.S. Isoform specificity of protein kinase Cs in synaptic plasticity. Learn. Mem. 2007, 14, 236–246. [Google Scholar] [CrossRef] [Green Version]
- Sweatt, J.D.; Atkins, C.M.; Johnson, J.; English, J.D.; Roberson, E.D.; Chen, S.J.; Newton, A.; Klann, E. Protected-site phosphorylation of protein kinase C in hippocampal long-term potentiation. J. Neurochem. 1998, 71, 1075–1085. [Google Scholar] [CrossRef] [Green Version]
- Trovò, L.; Ahmed, T.; Callaerts-Vegh, Z.; Buzzi, A.; Bagni, C.; Chuah, M.; Vandendriessche, T.; D’Hooge, R.; Balschun, D.; Dotti, C.G. Low hippocampal PI(4,5)P₂ contributes to reduced cognition in old mice as a result of loss of MARCKS. Nat. Neurosci. 2013, 16, 449–455. [Google Scholar] [CrossRef]
- Holahan, M.R. A Shift from a Pivotal to Supporting Role for the Growth-Associated Protein (GAP-43) in the Coordination of Axonal Structural and Functional Plasticity. Front. Cell Neurosci. 2017, 11, 266. [Google Scholar] [CrossRef] [Green Version]
- Yamniuk, A.P.; Vogel, H.J. Calmodulin’s flexibility allows for promiscuity in its interactions with target proteins and peptides. Mol. Biotechnol. 2004, 27, 33–57. [Google Scholar] [CrossRef]
- Denny, J.B. Molecular mechanisms, biological actions, and neuropharmacology of the growth-associated protein GAP-43. Curr. Neuropharmacol. 2006, 4, 293–304. [Google Scholar] [CrossRef] [Green Version]
- El Amri, M.; Fitzgerald, U.; Schlosser, G. MARCKS and MARCKS-like proteins in development and regeneration. J. Biomed. Sci. 2018, 25, 43. [Google Scholar] [CrossRef]
- Brudvig, J.J.; Weimer, J.M. X MARCKS the spot: Myristoylated alanine-rich C kinase substrate in neuronal function and disease. Front. Cell Neurosci. 2015, 9, 407. [Google Scholar] [CrossRef] [Green Version]
- Nicoll, R.A. A Brief History of Long-Term Potentiation. Neuron 2017, 93, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Purkey, A.M.; Dell’Acqua, M.L. Phosphorylation-Dependent Regulation of Ca. Front. Synaptic. Neurosci. 2020, 12, 8. [Google Scholar] [CrossRef]
- Diering, G.H.; Huganir, R.L. The AMPA Receptor Code of Synaptic Plasticity. Neuron 2018, 100, 314–329. [Google Scholar] [CrossRef] [Green Version]
- Yoshii, A.; Constantine-Paton, M. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev. Neurobiol. 2010, 70, 304–322. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Ge, T.; Leng, Y.; Pan, Z.; Fan, J.; Yang, W.; Cui, R. The Role of Neural Plasticity in Depression: From Hippocampus to Prefrontal Cortex. Neural Plast. 2017, 2017, 6871089. [Google Scholar] [CrossRef] [Green Version]
- Aleksandrova, L.R.; Wang, Y.T.; Phillips, A.G. Ketamine and its metabolite, (2R,6R)-HNK, restore hippocampal LTP and long-term spatial memory in the Wistar-Kyoto rat model of depression. Mol. Brain 2020, 13, 92. [Google Scholar] [CrossRef]
- Tizabi, Y.; Bhatti, B.H.; Manaye, K.F.; Das, J.R.; Akinfiresoye, L. Antidepressant-like effects of low ketamine dose is associated with increased hippocampal AMPA/NMDA receptor density ratio in female Wistar-Kyoto rats. Neuroscience 2012, 213, 72–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adaikkan, C.; Taha, E.; Barrera, I.; David, O.; Rosenblum, K. Calcium/Calmodulin-Dependent Protein Kinase II and Eukaryotic Elongation Factor 2 Kinase Pathways Mediate the Antidepressant Action of Ketamine. Biol. Psychiatry 2018, 84, 65–75. [Google Scholar] [CrossRef]
- Tang, X.H.; Zhang, G.F.; Xu, N.; Duan, G.F.; Jia, M.; Liu, R.; Zhou, Z.Q.; Yang, J.J. Extrasynaptic CaMKIIα is involved in the antidepressant effects of ketamine by downregulating GluN2B receptors in an LPS-induced depression model. J. Neuroinflamm. 2020, 17, 181. [Google Scholar] [CrossRef]
- Na, K.S.; Kim, Y.K. Increased use of ketamine for the treatment of depression: Benefits and concerns. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 104, 110060. [Google Scholar] [CrossRef]
- Short, B.; Fong, J.; Galvez, V.; Shelker, W.; Loo, C.K. Side-effects associated with ketamine use in depression: A systematic review. Lancet Psychiatry 2018, 5, 65–78. [Google Scholar] [CrossRef]
- Niesters, M.; Martini, C.; Dahan, A. Ketamine for chronic pain: Risks and benefits. Br. J. Clin. Pharmacol. 2014, 77, 357–367. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Genes Upregulated | Genes Downregulated | |
|---|---|---|
| cortex | Cacna1c, Orai1, PMCA4, SPCA1, MCU | PMCA2, SERCA3, NCX3 |
| cerebellum | Cacna1d, Cacna1h, PMCA1, SERCA3, NCX1 | Cacna1b, STIM1, PMCA3, MCU |
| hippocampus | Cacna1d, SERCA3, NCX3 | TRPC1, Cacna1b, PMCA3, MCU |
| striatum | TRPC1, PMCA1, SERCA3, MCU | TRPC6, Cacna1b, Cacna1c, Cacna1d, STIM1, PMCA3, PMCA4, SPCA2, NCX2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lisek, M.; Zylinska, L.; Boczek, T. Ketamine and Calcium Signaling—A Crosstalk for Neuronal Physiology and Pathology. Int. J. Mol. Sci. 2020, 21, 8410. https://doi.org/10.3390/ijms21218410
Lisek M, Zylinska L, Boczek T. Ketamine and Calcium Signaling—A Crosstalk for Neuronal Physiology and Pathology. International Journal of Molecular Sciences. 2020; 21(21):8410. https://doi.org/10.3390/ijms21218410
Chicago/Turabian StyleLisek, Malwina, Ludmila Zylinska, and Tomasz Boczek. 2020. "Ketamine and Calcium Signaling—A Crosstalk for Neuronal Physiology and Pathology" International Journal of Molecular Sciences 21, no. 21: 8410. https://doi.org/10.3390/ijms21218410
APA StyleLisek, M., Zylinska, L., & Boczek, T. (2020). Ketamine and Calcium Signaling—A Crosstalk for Neuronal Physiology and Pathology. International Journal of Molecular Sciences, 21(21), 8410. https://doi.org/10.3390/ijms21218410