Comparison of Techniques to Control Ice Nucleation during Lyophilization



Abstract
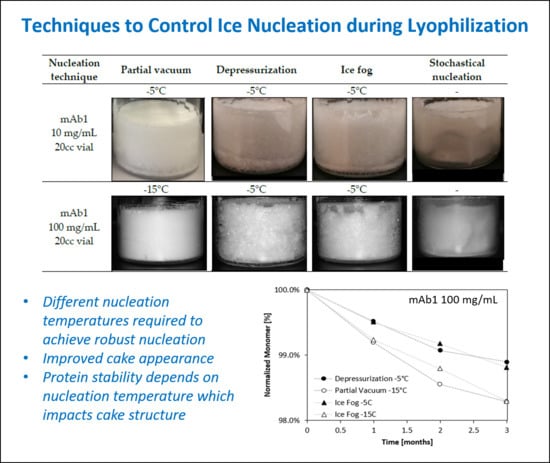
:
1. Introduction
2. Materials and Methods
2.1. Materials Formulations
2.2. Lyophilization
2.2.1. Depressurization Method
2.2.2. Partial Vacuum Method
2.2.3. Ice Fog Method
2.3. Solid State Characterization
2.3.1. Specific Surface Area
2.3.2. Residual Moisture Content (by Volumetric Karl Fischer)
2.3.3. Residual Moisture Content (by Coulometric Karl Fischer)
2.3.4. Reconstitution Time
2.3.5. Visual Inspection
2.3.6. Cake Imaging by Cake Embedding and Microcomputed Tomography (µCT)
2.4. Protein stability
2.4.1. Size Exclusion Chromatography (mAb)
2.4.2. Size Exclusion Chromatography (Enzyme)
2.4.3. Ion Exchange Chromatography (mAb)
2.5. Statistical Analyses
3. Results
3.1. Nucleation Temperatures
3.2. mAb1 at 10 mg/mL
3.3. mAb1 at 100 mg/mL
3.4. Enzyme at 2.5 mg/mL
4. Discussion
4.1. Comparability of Product Attributes
4.2. Nucleation Robustness
4.3. Operational and Installation Considerations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) mAb1 10 mg/mL (20 cc Configuration) | |||||||||||||
| Nucleation Technique/ | Partial Vacuum | Depressurization | |||||||||||
| Nucleation Temperature (°C) | −5 | −5 | |||||||||||
| Storage Temperature (°C)/ Time (months) | 5 | 25 | 40 | 5 | 25 | 40 | |||||||
| (a) Monomer (%) by SE-HPLC | |||||||||||||
| initial | 99.7 | - | - | 99.7 | - | - | |||||||
| 1 | 99.6 | - | 99.6 | 99.7 | 99.7 | 99.7 | |||||||
| 3 | 99.7 | - | 99.7 | 99.7 | 99.7 | 99.7 | |||||||
| 6 | 99.6 | - | - | 99.6 | 99.7 | 99.6 | |||||||
| 12 | 99.7 | - | 99.7 | 99.7 | - | - | |||||||
| 24 | - | - | - | 99.7 | - | - | |||||||
| (b) aa102 iso-Aspartate (%) by IEC | |||||||||||||
| initial | 69.6 | - | - | 69.5 | - | - | |||||||
| 1 | 69.4 | - | 67.5 | 69.5 | 69.4 | 68.3 | |||||||
| 3 | 69.2 | - | 66.7 | 73.3 | 73.3 | 67.1 | |||||||
| 6 | 67.5 | - | - | 68.8 | 68.4 | 67.7 | |||||||
| 12 | 68.6 | - | 68.6 | 68.6 | - | - | |||||||
| 24 | - | - | - | 68.5 | - | - | |||||||
| (B) mAb1 100 mg/mL (20 cc Configuration) | |||||||||||||
| Nucl. Technique/ | Partial Vacuum | Depressurization | Ice Fog | STN (1% rm) | STN (2% rm) | STN(3% rm) | STN (4%rm) | ||||||
| Nucl. Temp. (°C) | −15 | −5 | −5 | −15 | - | - | - | - | |||||
| Storage Temp. (°C)/ Time (months) | 5 | 25 | 40 | 5 | 25 | 40 | 40 | 40 | 40 | 40 | 40 | 40 | |
| (a) Monomer (%) by SE-HPLC | |||||||||||||
| initial | 99.5 | - | - | 99.6 | - | - | 66.4 | 99.5 | 99.3 | 99.3 | 99.3 | 99.4 | |
| 1 | 99.5 | 99.3 | 98.8 | 99.6 | 99.5 | 99.1 | 65.0 | 98.8 | 98.3 | 98.6 | 98.7 | 99.4 | |
| 2 | - | - | 98.1 | - | - | 98.6 | 63.7 | 98.3 | 97.8 | 98.1 | 98.3 | 99.4 | |
| 3 | 99.3 | 99.1 | 97.8 | 99.5 | 99.3 | 98.5 | 63.1 | 97.8 | 97.5 | 97.9 | 98.1 | 99.4 | |
| 6 | 99.4 | 98.9 | - | 99.4 | 99.1 | - | 59.2 | 97.0 | 96.6 | 97.2 | 97.4 | 98.9 | |
| 12 | 99.3 | - | - | 99.4 | - | - | 56.3 | - | 95.1 | 96.0 | 96.4 | 99.4 | |
| 24 | 99.0 | - | - | 99.4 | - | - | - | - | 92.7 | 92.0 | 94.3 | 64.1 | |
| (b) aa102 iso-Aspartate (%) by IEC | |||||||||||||
| initial | 69.6 | - | - | 69.5 | - | - | 68.0 | 67.2 | 66.1 | 66.1 | 66.1 | 66.4 | |
| 1 | 69.5 | 69.3 | 68.6 | 69.5 | 69.4 | 69.0 | 67.8 | 66.8 | 64.9 | 65.1 | 65.2 | 65.0 | |
| 2 | - | - | 67.5 | - | - | 67.8 | 67.5 | 66.4 | 64.3 | 64.6 | 64.7 | 63.7 | |
| 3 | 73.2 | 73.3 | 71.4 | 73.3 | 73.2 | 71.8 | 66.9 | 65.6 | 63.5 | 63.9 | 64.1 | 63.1 | |
| 6 | 68.7 | 68.0 | 68.6 | 68.2 | - | 66.5 | 64.6 | 62.1 | 62.2 | 62.0 | 59.2 | ||
| 12 | 68.0 | - | - | 68.3 | - | - | - | - | 61.1 | 61.8 | 61.3 | 56.3 | |
| 24 | 67.7 | - | - | 68.3 | - | - | - | - | 58.2 | 49.7 | 56.7 | - | |
Appendix B

References
- Konstantinidis, A.K.; Kuu, W.; Otten, L.; Nail, S.L.; Sever, R.R. Controlled nucleation in freeze-drying: Effects on pore size in the dried product layer, mass transfer resistance, and primary drying rate. J. Pharm. Sci. 2011, 100, 3453–3470. [Google Scholar] [CrossRef] [PubMed]
- Luoma, J.; Magill, G.; Kumar, L.; Yusoff, Z. Controlled Ice Nucleation Using ControLyo® Pressurization-Depressurization Method. In Lyophilization of Pharmaceuticals and Biologicals—New Technologies and Approaches, 1st ed.; Ward, K., Matejtschuk, P., Eds.; Humana Press: New York, NY, USA, 2019; pp. 57–77. [Google Scholar]
- Awotwe-Otoo, D.; Agarabi, C.; Read, E.K.; Lute, S.; Brorson, K.A.; Khan, M.A.; Shah, R.B. Impact of controlled ice nucleation on process performance and quality attributes of a lyophilized monoclonal antibody. Int. J. Pharm. 2013, 450, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Vollrath, I.; Friess, W.; Freitag, A.; Hawe, A.; Winter, G. Does controlled nucleation impact the properties and stability of lyophilized monoclonal antibody formulations? Eur. J. Pharm. Biopharm. 2018, 129, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Vollrath, I.; Friess, W.; Freitag, A.; Hawe, A.; Winter, G. Comparison of ice fog methods and monitoring of controlled nucleation success after freeze-drying. Int. J. Pharm. 2019, 558, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Scientific, S. ControLyo® Technology. Available online: https://www.spscientific.com/ControLyo/ (accessed on 20 August 2020).
- Allmendinger, A.; Schilder, G.; Mietzner, R.; Butt, Y.L.; Luemkemann, J.; Lema Martinez, C. Controlled Nucleation during Freeze Drying Using Vacuum-Induced Surface Freezing. Invention Disclosure rd633018. 2017. Available online: http://www.researchdisclosure.com (accessed on 20 August 2020).
- Millrock Technology: FreezeBooster® Controlled Nucleation Technology. Available online: https://www.millrocktech.com/freezebooster-controlled-nucleation-technology/ (accessed on 20 August 2020).
- Martin Christ: Controlled Nucleation LyoCoN. Available online: https://www.martinchrist.de/de/gefriertrocknung/controlled-nucleation-lyocon/ (accessed on 20 August 2020).
- IMA: VERISEQ® NUCLEATION. Available online: https://ima.it/pharma/lab4life/veriseq-nucleation/ (accessed on 20 August 2020).
- Geidobler, R.; Winter, G. Controlled ice nucleation in the field of freeze-drying: Fundamentals and technology review. Eur. J. Pharm. Biopharm. 2013, 85, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Gitter, J.H.; Geidobler, R.; Presser, I.; Winter, G. A Comparison of Controlled Ice Nucleation Techniques for Freeze-Drying of a Therapeutic Antibody. J. Pharm. Sci. 2018, 107, 2748–2754. [Google Scholar] [CrossRef] [PubMed]
- Lam, P.; Patapoff, T.W. An improved method for visualizing the morphology of lyophilized product cakes. PDA J. Pharm. Sci. Technol 2011, 65, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Haeuser, C.; Goldbach, P.; Huwyler, J.; Friess, W.; Allmendinger, A. Imaging Techniques to Characterize Cake Appearance of Freeze-Dried Products. J. Pharm. Sci. 2018, 107, 2810–2822. [Google Scholar] [CrossRef] [PubMed]
- Haeuser, C.; Goldbach, P.; Huwyler, J.; Friess, W.; Allmendinger, A. Impact of dextran on thermal properties, product quality attributes, and monoclonal antibody stability in freeze-dried formulations. Eur. J. Pharm. Biopharm. 2020, 147, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Kasper, J.C.; Friess, W. The freezing step in lyophilization: Physico-chemical fundamentals, freezing methods and consequences on process performance and quality attributes of biopharmaceuticals. Eur. J. Pharm. Biopharm. 2011, 78, 248–263. [Google Scholar] [CrossRef] [PubMed]
- Searles, J.A.; Carpenter, J.F.; Randolph, T.W. The ice nucleation temperature determines the primary drying rate of lyophilization for samples frozen on a temperature-controlled shelf. J. Pharm. Sci. 2001, 90, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Geidobler, R.; Konrad, I.; Winter, G. Can controlled ice nucleation improve freeze-drying of highly-concentrated protein formulations? J. Pharm. Sci. 2013, 102, 3915–3919. [Google Scholar] [CrossRef] [PubMed]
- Arsiccio, A.; Barresi, A.; De Beer, T.; Oddone, I.; Van Bockstal, P.J.; Pisano, R. Vacuum Induced Surface Freezing as an effective method for improved inter- and intra-vial product homogeneity. Eur. J. Pharm. Biopharm. 2018, 128, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Oddone, I.; Van Bockstal, P.J.; De Beer, T.; Pisano, R. Impact of vacuum-induced surface freezing on inter- and intra-vial heterogeneity. Eur. J. Pharm. Biopharm. 2016, 103, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geidobler, R.; Mannschedel, S.; Winter, G. A new approach to achieve controlled ice nucleation of supercooled solutions during the freezing step in freeze-drying. J. Pharm. Sci. 2012, 101, 4409–4413. [Google Scholar] [CrossRef] [PubMed]
- Oddone, I.; Arsiccio, A.; Duru, C.; Malik, K.; Ferguson, J.; Pisano, R.; Matejtschuk, P. Vacuum-Induced Surface Freezing for the Freeze-Drying of the Human Growth Hormone: How Does Nucleation Control Affect Protein Stability? J. Pharm. Sci. 2020, 109, 254–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allmendinger, A.; Butt, Y.L.; Mietzner, R.; Schmidt, F.; Luemkemann, J.; Lema Martinez, C. Controlling ice nucleation during lyophilization: Process Optimization of vacuum-induced surface freezing. Processes 2020, 8, 1263. [Google Scholar] [CrossRef]
- Kramer, M.; Sennhenn, B.; Lee, G. Freeze-drying using vacuum-induced surface freezing. J. Pharm. Sci. 2002, 91, 433–443. [Google Scholar] [CrossRef] [PubMed]


| Type of Protein | Protein Conc. (mg/mL) | Nominal Fill Volume/ Vial Configuration | Additional Configuration |
|---|---|---|---|
| mAb1 | 10 | 1 mL/2 cc | - |
| 10 mL/20 cc 1 | - | ||
| Stochastically nucleated samples | |||
| 20 mL/50 cc | - | ||
| 100 | 1 mL/2 cc | - | |
| 10 mL/20 cc 2 | - | ||
| Stochastically nucleated samples spiked to moisture levels between 1–4% | |||
| Samples nucleated at −15 °C (“Ice fog” method) | |||
| 20 mL/50 cc | - | ||
| enzyme | 2.5 | 0.9 mL/6 cc 1 | - |
| 10 mL/20 cc | - | ||
| 20 mL/50 cc 2 | - | ||
| Samples nucleated at −15 °C (“Ice fog” method) | |||
| Samples dried conservatively (“Ice fog” and “partial vacuum” method) |
| (a) Warmest Achieved Nucleation Temperatures | |||||||
| Type of Protein | Protein Conc. | Total Solid Content | Vial Format (cc) | Nominal Fill (mL) | Highest Controlled Nucleation Temperature (°C) | ||
| Depressuri-zation | Partial Vacuum | Ice Fog | |||||
| mAb1 | 10 mg/mL | 9% | 2 | 1 | STN | −5 | −5 |
| 20 | 10 | −5 | −5 | −5 | |||
| 50 | 20 | −5 | −5 | −5 | |||
| 100 mg/mL | 18% | 2 | 1 | STN | −15 | −5 | |
| 20 | 10 | −5 | −15 | −5 | |||
| 50 | 20 | −5 | −15 | −5 | |||
| enzyme | 2.5 mg/mL | 11% | 6 | 0.9 | −10 | −5 | −5 |
| 20 | 10 | −5 | −5 | −5 | |||
| 50 | 20 | −5 | −15 | −5 | |||
| (b) Nucleation Temperatures Used in this Study | |||||||
| Type of Protein | Protein Conc. | Total Solid Content | Vial Format (cc) | Nominal Fill (mL) | Highest Controlled Nucleation Temperature (°C) | ||
| Depressuri-zation | Partial Vacuum | Ice Fog | |||||
| mAb1 | 10 mg/mL | 9% | 2 | 1 | STN | −5 | −5 |
| 20 | 10 | −5 | −5 | −5 | |||
| 50 | 20 | −5 | −5 | −5 | |||
| 100 mg/mL | 18% | 2 | 1 | STN | −15 | n.p. | |
| 20 | 10 | −5 | −15 | −5, −15 | |||
| 50 | 20 | −5 | −15 | n.p. | |||
| enzyme | 2.5 mg/mL | 11% | 6 | 0.9 | −10 | −5 | n.p. |
| 20 | 10 | −5 | −5 | n.p. | |||
| 50 | 20 | −10 | −15 | −10, −15 | |||
| Nucleation Technique | Partial Vacuum | Depressurization | Ice Fog | Stochastical Nucleation |
|---|---|---|---|---|
| mAb1 10 mg/mL 20 cc vial | −5 °C | −5 °C | −5 °C | - |
| mAb1 100 mg/mL 20 cc vial | −15 °C | −5 °C | −5 °C | - |
| enzyme 2.5 mg/mL 50 cc vial | −15 °C | −15 °C | −15 °C | - |
| Nucleation Technique | Partial Vacuum | Depressurization | Ice Fog | Stochastical Nucleation |
|---|---|---|---|---|
| Nucleation Temperature (°C) | −15 | −5 | −5 | - |
| (a) |  | |||
| (b) |  | |||
| (a) PDMS Cake Embedding | ||
|---|---|---|
| Nucleation Technique/ Temperature | Nucleation Temperature −10 °C | Nucleation Temperature −15 °C |
| Depressur. |  |  |
| Ice fog |  |  |
| Partial vacuum | Nucleation temperature of −10 °C was not achieved |  |
| (b) µCT | ||
| Nucleation Technique/ Temperature | Conservative Cycle −15 °C | Aggressive Cycle −15 °C |
| Ice fog |  |  |
| Partial vacuum |  |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luoma, J.; Ingham, E.; Lema Martinez, C.; Allmendinger, A. Comparison of Techniques to Control Ice Nucleation during Lyophilization. Processes 2020, 8, 1439. https://doi.org/10.3390/pr8111439
Luoma J, Ingham E, Lema Martinez C, Allmendinger A. Comparison of Techniques to Control Ice Nucleation during Lyophilization. Processes. 2020; 8(11):1439. https://doi.org/10.3390/pr8111439
Chicago/Turabian StyleLuoma, Jacob, Erika Ingham, Carmen Lema Martinez, and Andrea Allmendinger. 2020. "Comparison of Techniques to Control Ice Nucleation during Lyophilization" Processes 8, no. 11: 1439. https://doi.org/10.3390/pr8111439
APA StyleLuoma, J., Ingham, E., Lema Martinez, C., & Allmendinger, A. (2020). Comparison of Techniques to Control Ice Nucleation during Lyophilization. Processes, 8(11), 1439. https://doi.org/10.3390/pr8111439