Compounds from Natural Sources as Protein Kinase Inhibitors


Abstract
:1. Protein Kinases as Therapeutic Targets
2. Classes of Protein Kinase Inhibitors
3. Natural Compounds as Kinase Inhibitors
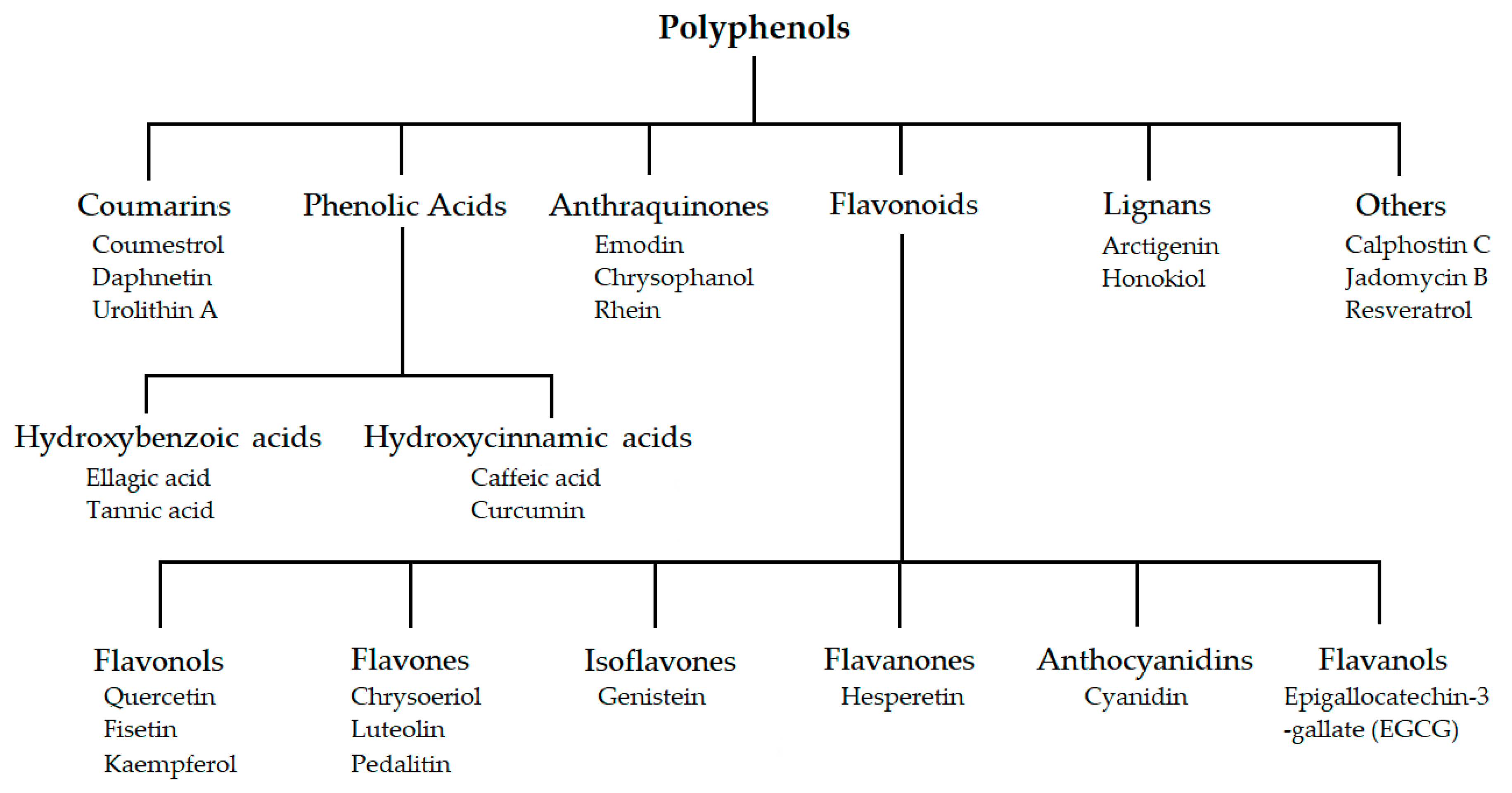
3.1. Polyphenol Analogues
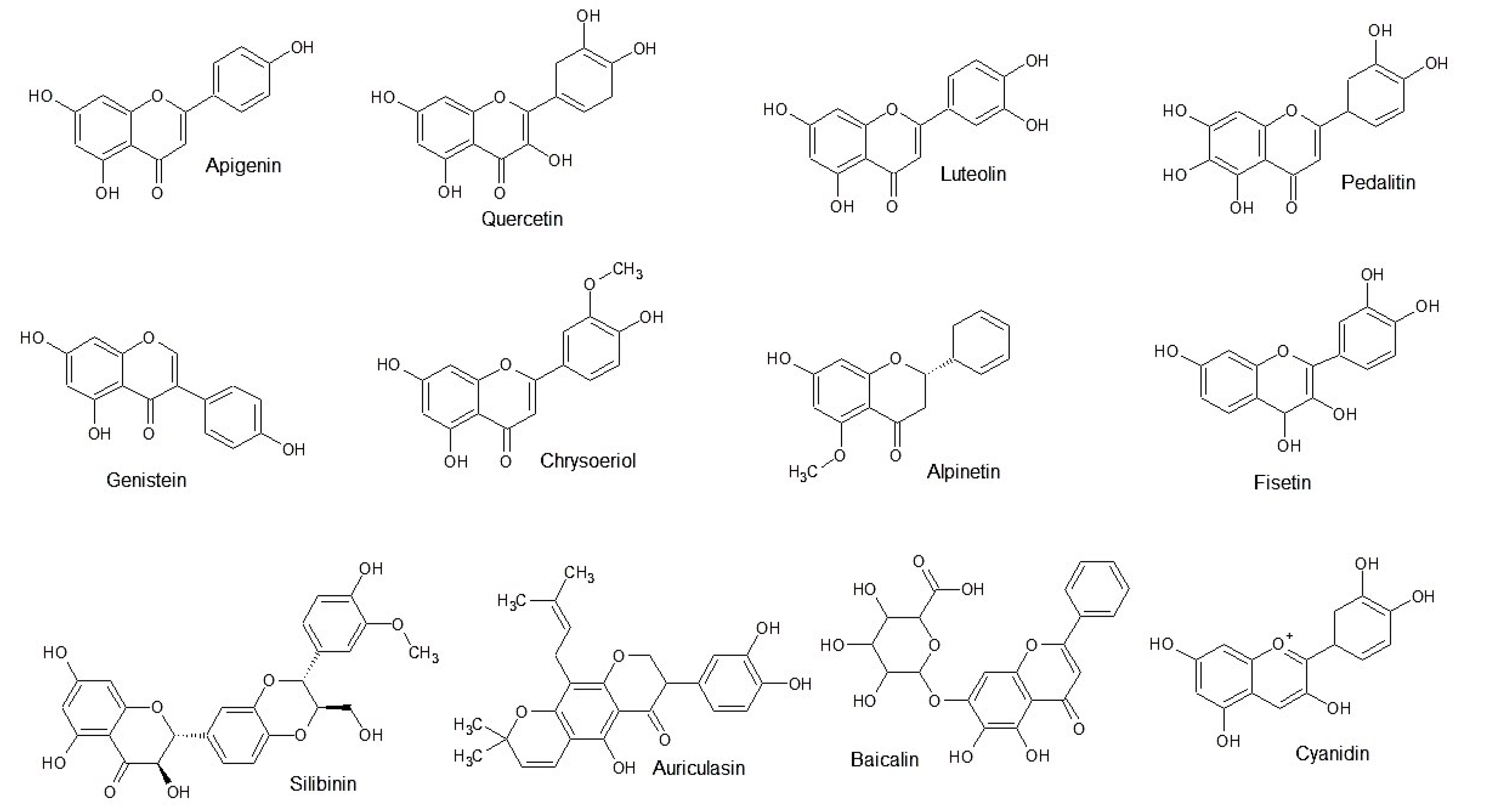
3.1.1. Flavonoid Analogues
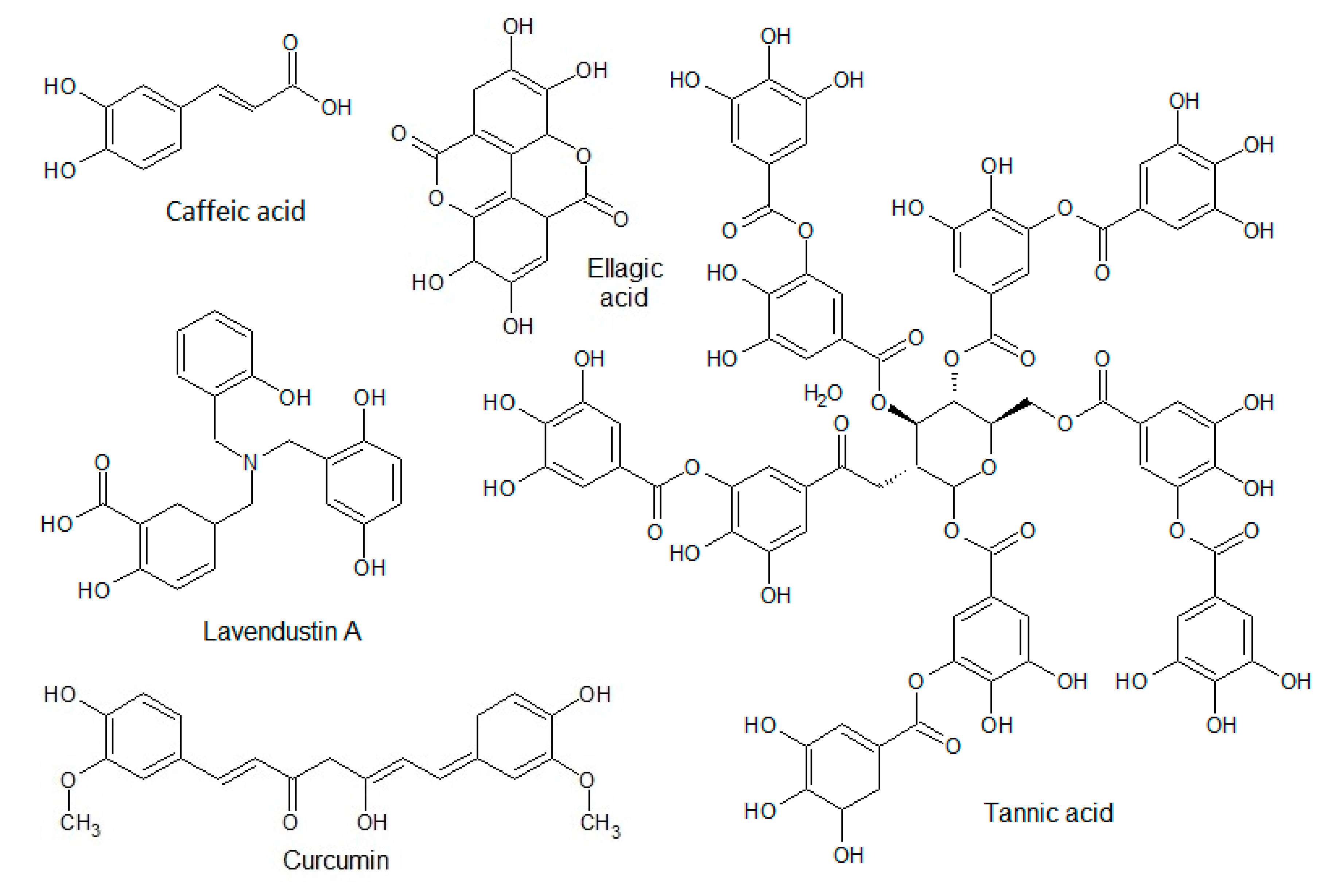
3.1.2. Phenolic Acids
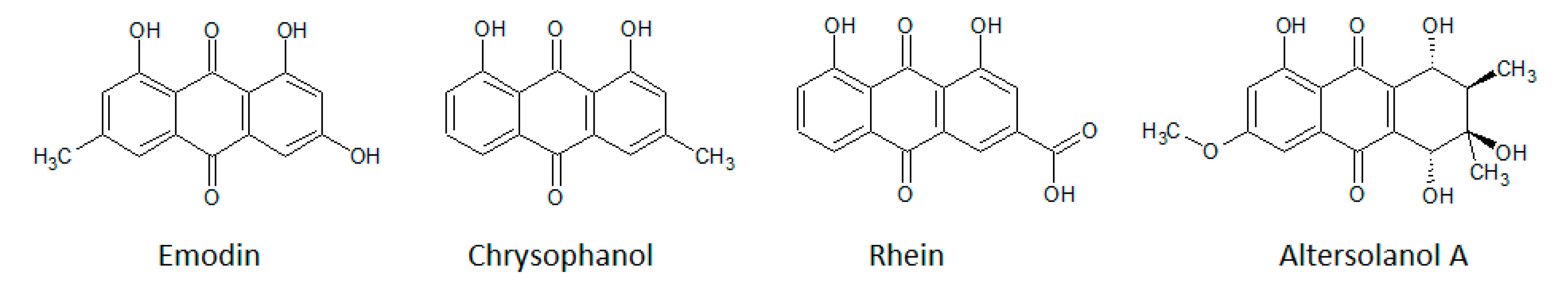
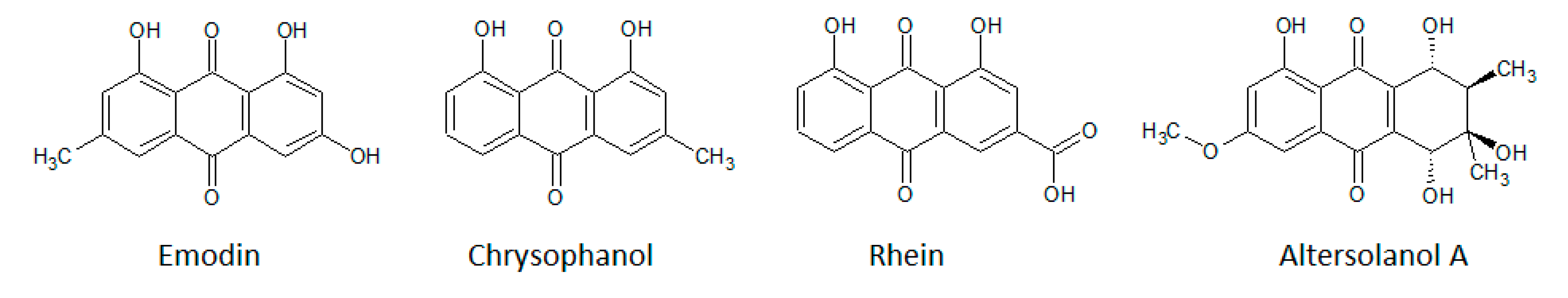
3.1.3. Anthraquinones
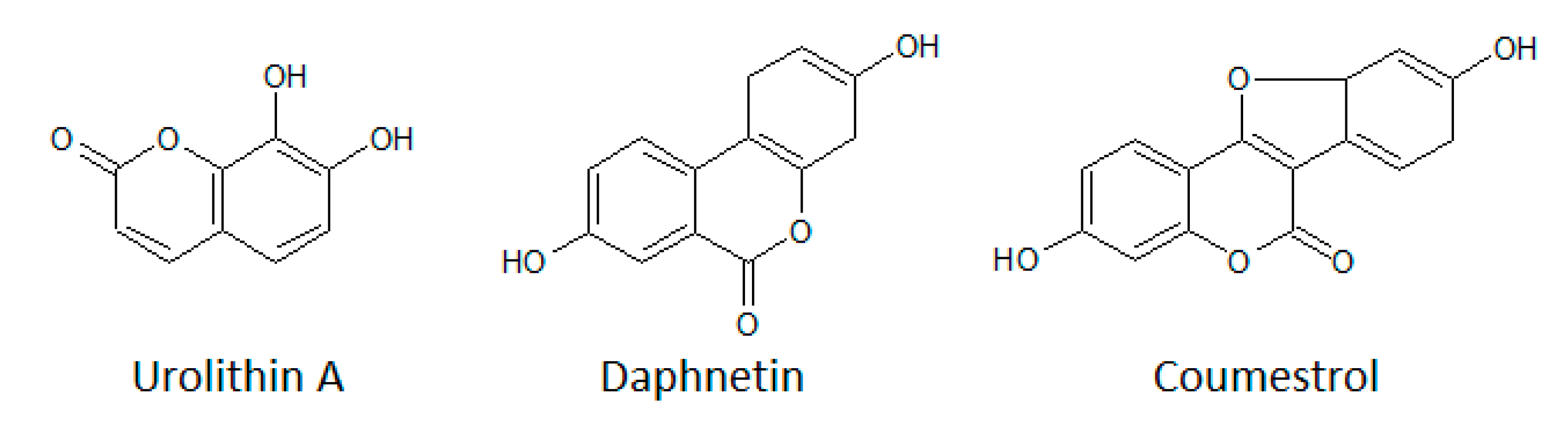
3.1.4. Coumarins
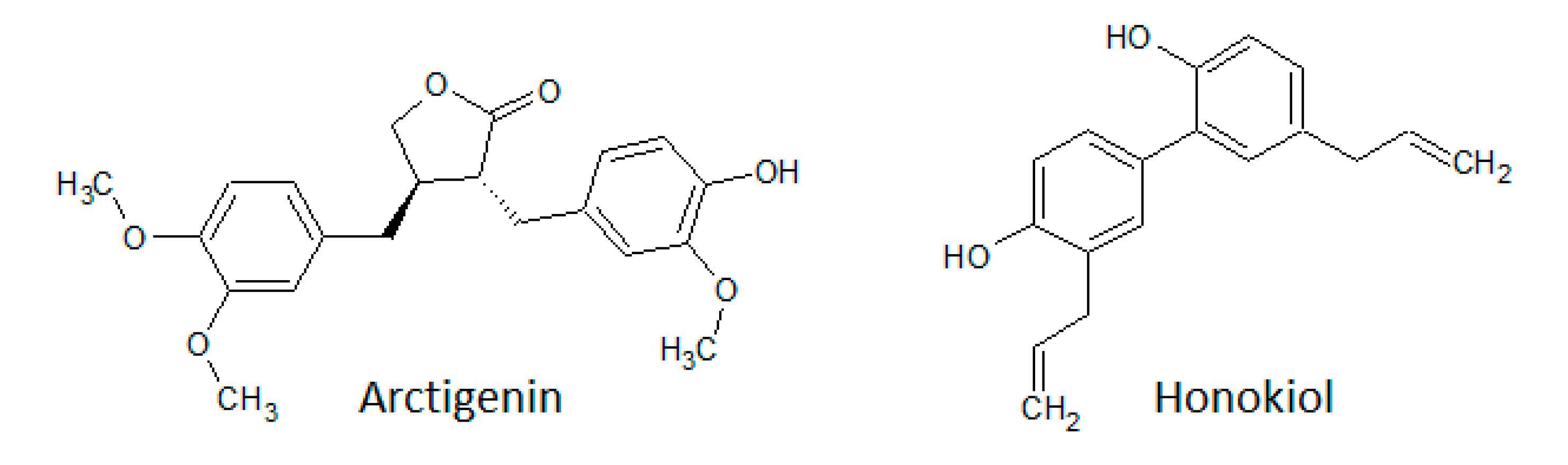
3.1.5. Lignans
3.1.6. Other Polyphenols
3.2. Indolocarbazole Analogues
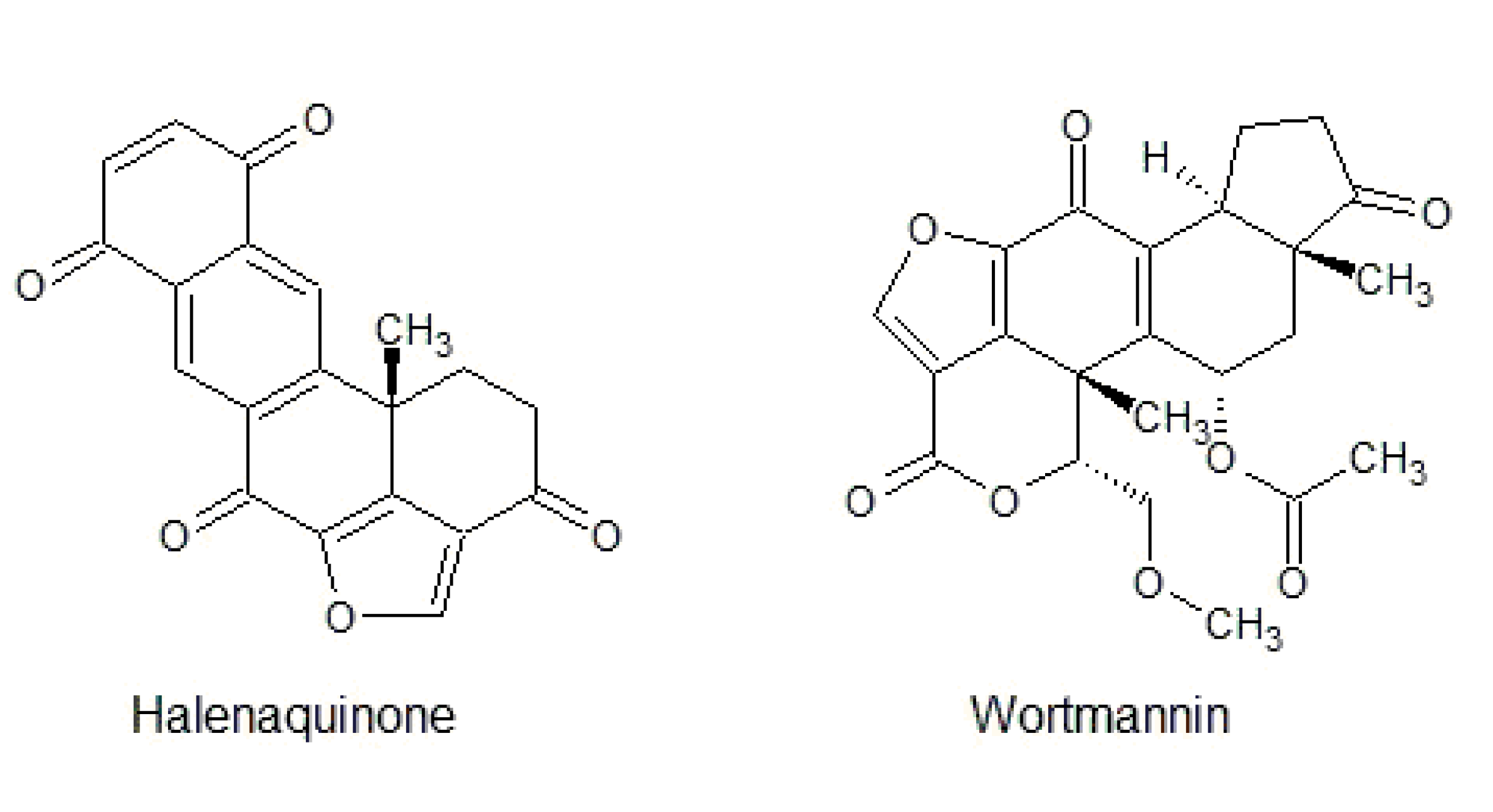
3.3. Furanosteroid Analogues
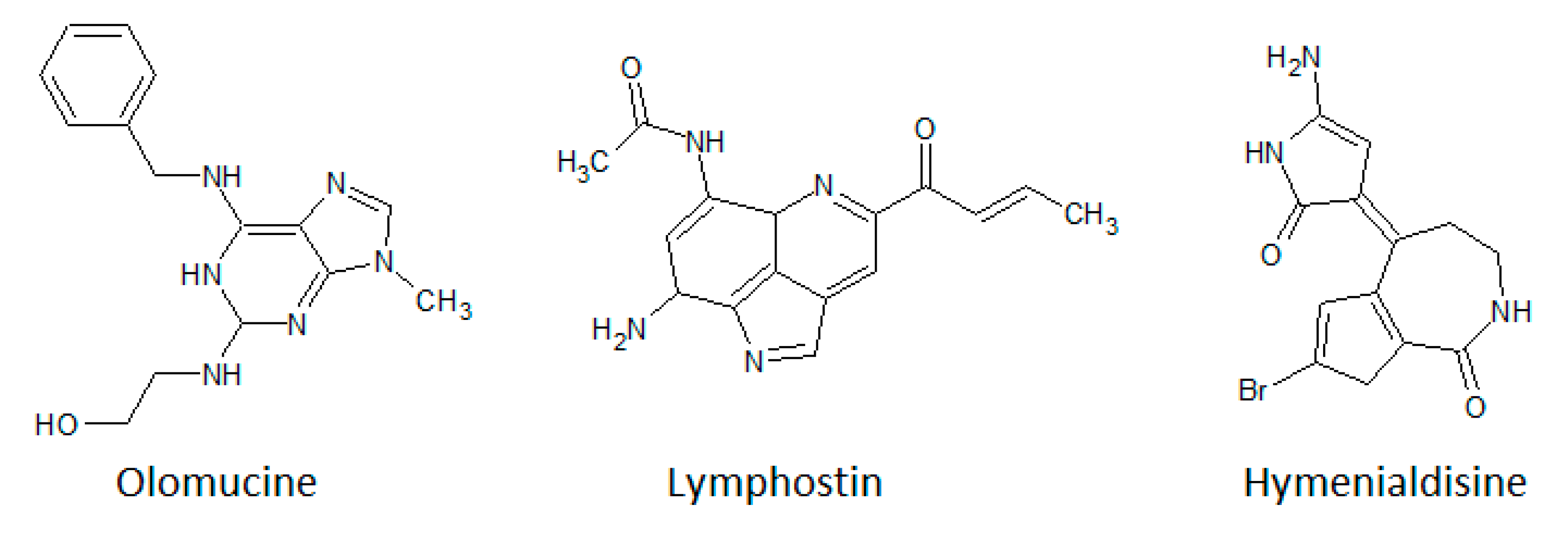
3.4. Purine Analogues
3.5. Other Natural Substances
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hunter, T. Protein kinases and phosphatases: The yin and yang of protein phosphorylation and signaling. Cell 1995, 80, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P. The origins of protein phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanks, S.K.; Hunter, T. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995, 9, 576–596. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Radzio-Andzelm, E. Three protein kinase structures define a common motif. Structure 1994, 2, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Briedis, K.M.; Starr, A.; Bourne, P.E. Analysis of the human kinome using methods including fold recognition reveals two novel kinases. PLoS ONE 2008, 3, e1597. [Google Scholar] [CrossRef]
- Schwarz, P.A.; Murray, B.W. Protein kinase chemistry and drug discovery. Bioorg. Chem. 2011, 39, 192–210. [Google Scholar] [CrossRef]
- Wilson, L.J.; Linley, A.; Hammond, D.E.; Hood, F.E.; Coulson, J.M.; MacEwan, D.J.; Ross, S.J.; Slupsky, J.R.; Smith, P.D.; Eyers, P.A.; et al. New perspectives, opportunities, and challenges in exploring the human protein kinome. Cancer Res. 2018, 78, 15–29. [Google Scholar] [CrossRef] [Green Version]
- Kanev, G.; de Graaf, C.; de Esch, I.J.P.; Leurs, R.; Würdinger, T.; Westerman, B.A.; Kooistra, A.J. The Landscape of Atypical and Eukaryotic Protein Kinases. Trends Pharmacol. Sci. 2019, 40, 818–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, A.H.; Khursigara, G.; Sun, X.; Franke, T.F.; Chao, M.V. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol. Cell. Biol. 2001, 21, 893–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, K.M.; Anderson, N.G. The protein kinase B/Akt signalling pathway in human malignancy. Cell. Signal. 2002, 14, 381–395. [Google Scholar] [CrossRef]
- Richter, E.A.; Ruderman, N.B. AMPK and the biochemistry of exercise: Implicationsfor human health and disease. Biochem. J. 2009, 418, 261–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaylova, M.M.; Shaw, R.J. The AMP-activated protein kinase (AMPK) signaling pathway coordinates cell growth, autophagy & metabolism. Nat. Cell. Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Obsilova, V.; Obsil, T. Structural Aspects of Protein Kinase ASK1 Regulation. Adv. Biol. Regul. 2017, 66, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Barnum, K.J.; O’Connell, M.J. Cell cycle regulation by checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Malumbers, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol. Res. 2019, 139, 471–488. [Google Scholar] [CrossRef]
- Ahmad, K.A.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2—A key suppressor of apoptosis. Adv. Enzym. Regul. 2008, 48, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Ortega, C.E.; Seidner, Y.; Dominguez, I. Mining CK2 in cancer. PLoS ONE 2014, 9, e115609. [Google Scholar] [CrossRef]
- Rabalski, A.; Gyenis, L.; Litchfield, D.W. Molecular Pathways: Emergence of Protein Kinase CK2 (CSNK2) as a Potential Target to Inhibit Survival and DNA Damage Response and Repair Pathways in Cancer Cells. Clin. Cancer Res. 2016, 22, 2840–2847. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Berezov, A.; Wang, Q.; Zhang, G.; Drebin, J.; Murali, R.; Greene, M.I. ErbB receptors: From oncogenes to targeted cancer therapies. J. Clin. Investig. 2007, 117, 2051–2058. [Google Scholar] [CrossRef] [Green Version]
- Mitri, Z.; Constantine, T.; O’Regan, R. The HER2 Receptor in Breast Cancer: Pathophysiology, Clinical Use, and New Advances in Therapy. Chemother. Res. Pract. 2012, 2012, 743193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, S.; Xia, Y.; Khoi, P.N.; Ung, T.T.; Yoon, H.J.; Kim, N.H.; Kim, K.K.; Jung, Y.D. Cadmium induces matrix metalloproteinase-9 expression via ROS-dependent EGFR, NF-κB, and AP-1 pathways in human endothelial cells. Toxicology 2015, 338, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Runkle, K.B.; Kharbanda, A.; Stypulkowski, E.; Cao, X.J.; Wang, W.; Garcia, B.A.; Witze, E.S. Inhibition of DHHC20-Mediated EGFR Palmitoylation Creates a Dependence on EGFR Signaling. Mol. Cell. 2016, 62, 385–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Seger, R. The ERK signaling cascade—Views rom different subcellular compartments. BioFactors 2009, 35, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Wortzel, I.; Seger, R. The ERK cascade: Distinct functions within various subcellular organelles. Genes Cancer 2011, 2, 195–209. [Google Scholar] [CrossRef]
- Zhou, J.; Yi, Q.; Tang, L. The roles of nuclear focal adhesion kinase (FAK) on Cancer: A focused review. J. Exp. Clin. Cancer Res. 2019, 38, 250. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.T. Nuclear FAK: A new mode of gene regulation from cellular adhesion. Mol. Cells 2013, 36, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Eswakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef]
- Belov, A.A.; Mohammadi, M. Molecular Mechanisms of Fibroblast Growth Factor Signaling in Physiology and Pathology. Cold Spring Harb. Perspect. Biol. 2013, 5, a015958. [Google Scholar] [CrossRef]
- Katoh, M. FGFR inhibitors: Effect on cancer cells, tumor micro environment and whole-body homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayasam, G.V.; Tulasi, V.K.; Sodhi, R.; Davis, J.A.; Ray, A. Glycogen synthase kinase 3: More than a namesake. Br. J. Pharmacol. 2009, 156, 885–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandini, G.; Frasca, F.; Mineo, R.; Sciacca, L.; Vignri, R.; Belfiore, A. Insulin/insulin-like Growth Factor I Hybrid Receptors Have Different Biological Characteristics Depending on the Insulin Receptor Isoform Involved. J. Biol. Chem. 2002, 277, 39684–39695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvan, V.; Logvinova, A.; Sperandio, S.; Ichijo, H.; Bredesen, D.E. Type 1 insulin-like growth factor receptor (IGF-IR) signaling inhibits apoptosis signal-regulating kinase 1(ASK1). J. Biol. Chem. 2003, 278, 13325–13332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandi, E.; Rothwarf, D.M.; Delhase, M.; Hayakawa, M.; Karin, M. The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell 1997, 91, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Hayden, M. New regulators of NF-κB in inflammation. Nat. Rev. Immunol. 2008, 8, 837–848. [Google Scholar] [CrossRef]
- Wang, W.; Guo, M.; Hu, L.; Cai, J.; Zeng, Y.; Luo, J.; Shu, Z.; Li, W.; Huang, Z. The zinc finger protein ZNF268 is overexpressed in human cervical cancer and contributes to tumorigenesis via enhancing NF-kappaB signaling. J. Biol. Chem. 2012, 287, 42856–42866. [Google Scholar] [CrossRef] [Green Version]
- Häcker, H.; Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, 357, re13. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; O’Shea, J.J. Janus kinases in immune cell signaling. Immunol. Rev. 2009, 228, 273–287. [Google Scholar] [CrossRef] [Green Version]
- Sharfe, N.; Dadi, H.K.; Roifman, C.M. JAK3 protein tyrosine kinase mediates interleukin-7-induced activation of phosphatidylinositol-3’ kinase. Blood 1995, 86, 2077–2085. [Google Scholar] [CrossRef] [Green Version]
- Zeke, A.; Misheva, M.; Reményi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, R.; Freitag, W.; He, B.; Zhang, Y.; Mivechi, N.F. c-Jun NH2-terminal kinase targeting and phosphorylation of heat shock factor-1 suppress its transcriptional activity. J. Biol. Chem. 2000, 275, 18210–18218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Singh, U.K.; Kini, S.G.; Garq, V.; Aqrawal, S.; Tomar, P.K.; Pathak, P.; Chaodhary, A.; Gupta, P.; Malik, A. JNK pathway signaling: A novel and smarter therapeutic targets for various biological diseases. Future Med. Chem. 2015, 7, 2065–2086. [Google Scholar] [CrossRef] [PubMed]
- Samraj, A.K.; Stroch, C.; Fischer, U.; Schulze-Osthoff, K. The tyrosine kinase Lck is a positive regulator of the apoptosis pathway controlling Bak expression. Oncogene 2006, 25, 186–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochl, R.; Thelen, F.; Vanes, L.; Brazao, T.F.; Fountain, K.; Xie, J.; Huang, C.L.; Lyck, R.; Stein, J.V.; Tybulewicz, V.L. WNK1 kinase balances T cell adhesion versus migration in vivo. Nat. Immunol. 2016, 17, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Vogel, L.B.; Fujita, D.J. p70 phosphorylation and binding to p56lck is an early event in interleukin-2-induced onset of cell cycle progression in T-lymphocytes. J. Biol. Chem. 1995, 270, 2506–2511. [Google Scholar] [CrossRef] [Green Version]
- Lipton, J.O.; Sahin, M. The neurology of mTOR. Neuron 2014, 84, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Troya, S.; Pérez-Pérez, M.E.; Florencio, F.J.; Crespo, J.L. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy 2008, 4, 851–865. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Hua, H.; Li, M.; Liu, S.; Kong, Q.; Shao, T.; Wang, J.; Luo, Y.; Wang, Q.; Luo, T.; et al. mTORC2 promotes type I insulin-like growth factor receptor and insulin receptor activation through the tyrosine kinase activity of mTOR. Cell Res. 2016, 26, 46–65. [Google Scholar] [CrossRef] [Green Version]
- Gagliardi, P.A.; Puliafito, A.; Primo, L. PDK1: At the crossroad of cancer signaling pathways. Sem. Cancer Biol. 2018, 48, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Torres-Quesada, O.; Mayrhofer, J.; Stefan, E. The many faces of compartmentalized PKA signalosomes. Cell. Signal. 2017, 37, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.G.; Lygren, B.; Dokurno, P.; Hoshi, N.; McConnachie, G.; Taskén, K.; Carlson, C.R.; Scott, J.D.; Barford, D. Molecular Basis of AKAP Specificity for PKA Regulatory Subunits. Mol. Cell. 2006, 24, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Kumar, S.; Gautam, P.K.; Tomar, M.S.; Verma, S.P.; Singh, S.P.; Kumar, S.; Acharya, A. Protein kinase C-α and the regulation of diverse cell responses. Biomol. Concepts 2017, 8, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Newton, A.C. Protein kinase C as a tumor suppressor. Semin. Cancer Biol. 2018, 48, 18–26. [Google Scholar] [CrossRef]
- Fiscus, R.R. Involvement of Cyclic GMP and Protein Kinase G in the Regulation of Apoptosis and Survival in Neural Cells. Neurosignals 2002, 11, 175–190. [Google Scholar] [CrossRef]
- Feil, R.; Lohman, S.M.; de Jonge, H.; Walter, U.; Hofman, F. Cyclic GMP-Dependent Protein Kinases and the Cardiovascular System. Circ. Res. 2003, 93, 907–916. [Google Scholar] [CrossRef] [Green Version]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Src protein tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef]
- Stacker, S.A.; Achen, M.G. The VEGF signaling pathway in cancer: The road ahead. Chin. J. Cancer 2013, 32, 297–302. [Google Scholar] [CrossRef]
- Lamalice, L.; Houle, F.; Huot, J. Phosphorylation of Tyr1214 within VEGFR-2 triggers the recruitment of Nck and activation of Fyn leading to SAPK2/p38 activation and endothelial cell migration in response to VEGF. J. Biol. Chem. 2006, 281, 34009–34020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, H.P.; McMurtrey, A.; Kowalski, J.; Yan, M.; Keyt, B.A.; Dixit, V.; Ferrara, N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 1998, 273, 30336–30343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, L.R.; Komander, D.; Alesi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, J.G.; Applebury, M.L. The photoreceptor guanylate cyclase is an autophosphorylating protein kinase. J. Biol. Chem. 1996, 271, 27083–27089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ségaliny, A.I.; Tellez-Gabriel, M.; Heymann, M.-F.; Heymann, D. Receptor tyrosine kinases: Characterization, mechanism of action and therapeutic interests for bone cancers. J. Bone Oncol. 2015, 4, 1–12. [Google Scholar] [CrossRef]
- Madhusudan, S.; Ganesan, T.S. Tyrosine kinase inhibitors in cancer therapy. Clin. Biochem. 2004, 37, 618–635. [Google Scholar] [CrossRef] [PubMed]
- Shchemelinin, I.; Šefc, L.; Nečas, E. Protein Kinases, Their function and Implication in Cancer and Other Diseases. Folia Biol. (Praha) 2006, 52, 81–100. [Google Scholar] [PubMed]
- Lipsick, J. A History of Cancer Research: Tyrosine Kinases. Cold Spring Harb. Perspect. Biol. 2019, 11, a035592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belfiore, A.; Frasca, A.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin Receptor Isoforms and Insulin Receptor/Insulin-Like Growth Factor Receptor Hybrids in Physiology and Disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, N.; Lennartsson, J. The PDGF/PDGFR pathway as a drug target. Mol. Asp. Med. 2018, 62, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araùjo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. FGFR a promising druggabe target in cancer: Molecular biology and new drugs. Crit. Rev. Oncol. Hematol. 2017, 113, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karaman, S.; Leppaänen, V.-M.; Alitalo, K. Vascular endothelial growth factor signaling in development and disease. Development 2018, 145, dev151019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skaper, S.D. Nerve growth factor: A neuroimmune crosstalk mediator for all seasons. Immunology 2017, 151, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gocek, E.; Moulas, A.N.; Studziński, G.P. Non-receptor protein tyrosine kinases signaling pathways in normal and cancer cells. Crit. Rev. Clin. Lab. Sci. 2014, 51, 125–137. [Google Scholar] [CrossRef]
- Azavedo, A.; Silva, S.; Rueff, J. Non-receptor Tyrosine Kinases Role and Significance in Hemetological Malignaces. In Tyrosine Kinases as Druggable Targets in Cancer; Ren, H., Ed.; IntechOpen: London, UK, 2019; pp. 1–33. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, Z.K.; Lewcook, J.W. ACS Chemical Neuroscience Spotlight on CEP-1347. ACS Chem. Neurosci. 2011, 2, 3–4. [Google Scholar] [CrossRef] [Green Version]
- Scharpfenecker, M.; van Dinther, M.; Liu, Z.; van Bezooijen, R.L.; Zhao, Q.; Pukac, L.; Lö, C.W.; ten Dijke, P. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J. Cell. Sci. 2007, 120, 964–972. [Google Scholar] [CrossRef] [Green Version]
- Fabbro, D.; Cowan-Jacob, S.; Moebits, H. Ten things you should know about protein kinases: IUPHAR Review 14. Br. J. Pharmacol. 2015, 172, 2675–2700. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Rommel, C. PI3K and Cancer: Lessons, Challenges and Opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted terapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Lahiry, P.; Torkamani, A.; Schork, N.J.; Hegele, R. Kinase mutations in human disease: Interpreting genotype-phenotype relationships. Nat. Rev. Genet. 2010, 11, 60–74. [Google Scholar] [CrossRef]
- Montor, W.R.; Salas, A.R.O.S.E.; de Melo, F.H.M. Receptor tyrosine kinases and downstream pathways as druggable targets for cancer treatment: The current arsenal of inhibitors. Mol. Cancer 2018, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.C.; Shokat, K.M. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu. Rev. Biochem. 2011, 80, 769–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Gray, N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer. 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Kufareva, I.; Abagyan, R. Type-II kinase inhibitor docking, screening, and profiling using modified structures of active kinase states. J. Med. Chem. 2008, 51, 7921–7932. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Davies, S.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef]
- Roskoski, R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Zuccotto, F.; Ardini, E.; Casale, E.; Angiolini, M. Through the “gatekeeper door”: Exploiting the active kinase conformation. J. Med. Chem. 2009, 53, 2681–2694. [Google Scholar] [CrossRef] [PubMed]
- Gavrin, L.K.; Saiah, E. Approaches to discover non-ATP site kinase inhibitors. Med. Chem. Comm. 2013, 4, 41–51. [Google Scholar] [CrossRef]
- Blanc, J.; Geney, R.; Menet, C. Type II kinase inhibitors: An opportunity in cancer for rational design. Anti-Cancer Agents Med. Chem. 2013, 13, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Lamba, V.; Ghosh, I. New directions in targeting protein kinases: Focusing upon true allosteric and bivalent inhibitors. Curr. Pharm. Des. 2012, 18, 2936–2945. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.S.; Zhang, C.; Shokat, K.M.; Taunton, J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 2005, 308, 1318–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, E.L.; Sordella, R.; Bell, D.W.; Godin-Heymann, N.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Driscoll, D.R.; Fidias, P.; Lynch, T.J. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. USA 2005, 102, 7665–7670. [Google Scholar] [CrossRef] [Green Version]
- Leproult, E.; Barluenga, S.; Moras, D.; Wurtz, J.-M.; Winssinger, N. Cysteine mapping in conformationally distinct kinase nucleotide binding sites: Application to the design of selective covalent inhibitors. J. Med. Chem. 2011, 54, 1347–1355. [Google Scholar] [CrossRef]
- Liu, Q.; Sabnis, Y.; Zhao, Z.; Zhang, T.; Buhrlage, S.J.; Jones, L.H.; Gray, N.S. Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. 2013, 20, 146–159. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef]
- Zhao, X.; Guo, X.; Shen, J.; Hua, D. Alpinetin inhibits proliferation and migration of ovarian cancer cells via suppression of STAT3 signaling. Mol. Med. Rep. 2018, 18, 4030–4036. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.-D.; Moon, K.-D.; Park, K.-H.; Lee, Y.-S.; Seo, K.-I. Effects of auriculasin on vascular endothelial growth factor (VEGF)-induced angiogenesis via regulation of VEGF receptor 2 signaling pathways in vitro and in vivo. Food Chem. Toxicol. 2018, 121, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, J.; Xiao, X.Q. Cantharidin inhibits angiogenesis by suppressing VEGF-induced JAK1/STAT3, ERK and AKT signaling pathways. Arch. Pharm. Res. 2015, 38, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Park, J.; Tak, K.H.; Bu, S.Y.; Kim, E. Chemopreventive effects of curcumin on chemically induced mouse skin carcinogenesis in BK5.insulin-like growth factor-1 transgenic mice. In Vitro Cell Dev. Biol. Anim. 2014, 50, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Golonko, A.; Lewandowska, H.; Świsłocka, R.; Jasińska, U.T.; Priebe, W.; Lewandowski, W. Curcumin as tyrosine kinase inhibitor in cancer treatment. Eur. J. Med. Chem. 2019, 181, 111512. [Google Scholar] [CrossRef]
- Leu, T.H.; Su, S.L.; Chuang, Y.C.; Maa, M.C. Direct inhibitory effect of curcumin on Src and focal adhesion kinase activity. Biochem. Pharmacol. 2003, 66, 2323–2331. [Google Scholar] [CrossRef]
- Muto, A.; Hori, M.; Sasaki, A.; Saitoh, I.; Yasuda, T.; Maekawa, T.; Uchida, T.; Asakura, K.; Nakazato, T.; Kaneda, T.; et al. Emodin has a cytotoxic activity against human multiple mylenoma as a Janus-activated kinase 2 inhibitor. Mol. Cancer Ther. 2007, 6, 987–994. [Google Scholar] [CrossRef] [Green Version]
- Ueno, N.; Kiyokawa, N.; Hung, M. Growth suppression of low Her-2/neu-expressing expressing breast cancer cell line MDA-MB-435 by tyrosine kinase inhibitor emodin. Oncol. Rep. 1996, 3, 509–511. [Google Scholar] [CrossRef]
- Fan, Y.; Xue, W.; Schachner, M.; Zhao, W. Honokiol Eliminates Glioma/Glioblastoma Stem Cell-Like Cells Via JAK-STAT3 Signaling and Inhibits Tumor Progression by Targeting Epidermal Growth Factor Receptor. Cancers 2018, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Cook, M.T.; Liang, Y.; Besch-Williford, C.; Hyder, S.M. Luteolin inhibits lung metastasis, cell migration, and viability of triple-negative breast cancer cells. Breast Cancer 2017, 9, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Cook, M.T. Mechanism of metastasis suppression by luteolin in breast cancer. Breast Cancer 2018, 10, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Sui, J.; Xie, K.; Xie, M. Inhibitory effect of luteolin on the proliferation of human breast cancer cell lines induced by epidermal growth factor. Acta Physiol. Sin. 2016, 68, 27–34. [Google Scholar] [CrossRef]
- Nagata, H.; Yano, H.; Sasaki, K.; Sato, S.; Nakanishi, S.; Takahashi, I.; Tamaoki, T. Inhibition of lymphocyte kinase Lck and phosphatidylinositol 3-kinase by a novel immunosuppressant, lymphostin. Biosci. Biotechnol. Biochem. 2002, 66, 501–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Li, J.; Liu, T.; Li, S.; Feng, J.; Yu, Q.; Zhang, J.; Chen, J.; Zhou, Y.; Ji, J.; et al. Quercetin shows anti-tumor effect in hepatocellular carcinoma LM3 cells by abrogating JAK2/STAT3 signaling pathway. Cancer Med. 2019, 8, 4806–4820. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Joung, P.; Kanq, D.Y.; Sp, N.; Hwang, T.S.; Sasidharakurp, H.; Lee, C.H.; Cho, K.H.; Park, K.D.; Lee, H.K.; et al. Tannic acid inhibits EGFR/STAT1/3 and enhances p38/STAT1 signalling axis in breast cancer cells. J. Cell. Mol. Med. 2017, 21, 720–734. [Google Scholar] [CrossRef]
- Lee, R.X.; Qingdi Quentin, L.I.; Reed, E. β-elemene effectively suppresses the growth and survival of both platinum-sensitive and -resistant ovarian tumor cells. Anticancer Res. 2012, 32, 3103–3113. [Google Scholar]
- Zhang, H.-W.; Hu, J.-J.; Fu, R.-Q.; Liu, X.; Zhang, Y.-H.; Li, J.; Liu, L.; Li, Y.-N.; Deng, Q.; Luo, Q.-S.; et al. Flavonoids inhibit cell proliferation and induce apoptosis and autophagy through downregulation of PI3Kg mediated PI3K/AKT/mTor/p70S6K/ULK signaling pathway in human brest cancer cells. Sci. Rep. 2018, 8, 11255. [Google Scholar] [CrossRef]
- Huo, M.; Chen, N.; Chi, G.; Yuan, X.; Guan, S.; Li, H.; Zhong, W.; Guo, W.; Soromou, L.W.; Gao, R.; et al. Traditional medicine alpinetin inhibits the inflammatory response in Raw 264.7 cells and mouse models. Int. Immunopharmacol. 2012, 12, 241–248. [Google Scholar] [CrossRef]
- Liu, J.; Cao, X.-C.; Xiao, Q.; Quan, M.-F. Apigenin inhibits HeLa sphere-forming cells through inactivation of casein kinase 2α. Mol. Med. Rep. 2015, 11, 665–669. [Google Scholar] [CrossRef]
- Shukla, S.; Kanwal, R.; Shankar, E.; Datt, M.; Chance, M.R.; Fu, P.; MacLennan, G.T.; Gupta, S. Apigenin blocks IKKα activation and suppresses prostate cancer progression. Oncotarget 2015, 6, 31216–31232. [Google Scholar] [CrossRef]
- Wang, K.S.; Li, J.; Wang, Z.; Mi, C.; Ma, J.; Piao, L.X.; Xu, G.H.; Li, X.; Jin, X. Artemisinin inhibits inflammatory response via regulating NF-κB and MAPK signaling pathways. Immunopharmacol. Immunotoxicol. 2017, 39, 28–36. [Google Scholar] [CrossRef]
- Awasthee, N.; Rai, V.; Chava, S.; Nallasamy, P.; Kunnumakkara, A.B.; Bishayee, A.; Chauhan, S.C.; Challagundla, K.B.; Gupta, S.C. Targeting IκappaB kinases for cancer therapy. Semin. Cancer Biol. 2019, 56, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Li, R.; Xu, L.; Qiu, Y.; Fu, S.; Liu, Y.; Wu, Z.; Hou, Y.; Hu, C.A. Effects of Baicalin on piglet monocytes involving PKC-MAPK signaling pathways induced by Haemophilus parasuis. BMC Vet. Res. 2019, 15, 98. [Google Scholar] [CrossRef] [PubMed]
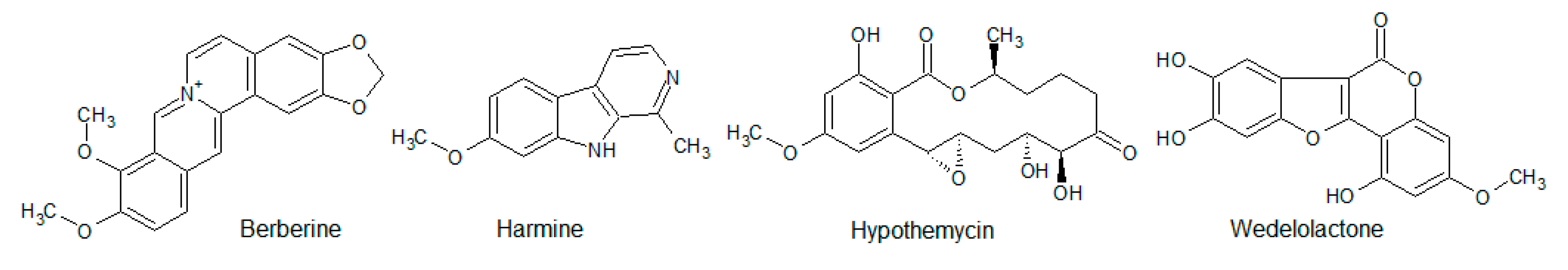
- Li, W.; Hua, B.; Saud, S.M.; Lin, H.; Hou, W.; Matter, M.S.; Jia, L.; Colburn, N.H.; Young, M.R. Berberine regulates AMP-activated protein kinase signaling pathways and inhibits colon tumorigenesis in mice. Mol. Carcinog. 2015, 54, 1096–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Zhang, M.; Dou, D.; Tao, X.; Kang, T. Berberine depresses contraction of smooth muscle via inhibiting myosin light-chain kinase. Pharm. Mag. 2017, 13, 454–458. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Liu, Z.; Li, J.; Zhang, Q.; Zhong, P.; Teng, T.; Chen, M.; Xie, Z.; Ji, A.; Li, Y. Epigallocatechin-3-gallate inhibits the growth and increases the apoptosis of human thyroid carcinoma cells through suppression of EGFR/RAS/RAF/MEK/ERK signaling pathway. Cancer Cell Int. 2019, 19, 43. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.R.; Kim, K.; Lee, K.H.; Kim, S.J.; Kim, J. Inhibition of casein kinase 2 enhances the death ligand- and natural killer cell-induced hepatocellular carcinoma cell death. Clin. Exp. Immunol. 2008, 152, 336–344. [Google Scholar] [CrossRef]
- Ismail, I.A.; Kang, K.S.; Lee, H.A.; Kim, J.W.; Sohn, Y.K. Genistein-induced neuronal apoptosis and G2/M cell cycle arrest is associated with MDC1 up-regulation and PLK1 down-regulation. Eur. J. Pharmacol. 2007, 575, 12–20. [Google Scholar] [CrossRef]
- Besley, C.; Rhinehart, D.P.; Ammons, T.; Goess, B.C.; Rawlings, J.S. Inhibition of phosphatidylinositol-3-kinase by the furanosesquiterpenoid hibiscone C. Bioorg. Med. Chem. Lett. 2017, 27, 3087–3091. [Google Scholar] [CrossRef]
- Lim, S.H.; Jung, S.K.; Byun, S.; Lee, E.J.; Hwang, J.A.; Seo, S.G.; Kim, Y.A.; Yu, J.G.; Lee, K.W.; Lee, H.J. Luteolin suppresses UVB-induced photoageing by targeting JNK1 and p90 RSK2. J. Cell Mol. Med. 2013, 17, 672–680. [Google Scholar] [CrossRef]
- Holder, S.; Zemskova, M.; Zhang, C.; Tabrizizad, M.; Bremer, R.; Neidigh, J.W.; Lilly, M.B. Characterization of a potent and selective small-molecule inhibitor of the PIM1 kinase. Mol. Cancer Ther. 2007, 6, 163–172. [Google Scholar] [CrossRef] [Green Version]
- Spencer, J.P.; Rice-Evans, C.; Williams, R.J. Modulation of pro-survival Akt/protein kinase B and ERK1/2 signaling cascades by quercetin and its in vivo metabolites underlie their action on neuronal viability. J. Biol. Chem. 2003, 278, 34783–34793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, M.; Milito, A.; Spagnuolo, C.; Carbone, V.; Rosén, A.; Minasi, P.; Lauria, F.; Russo, G.L. CK2 and PI3K are direct molecular targets of quercetin in chronic lymphocytic leukaemia. Oncotarget 2017, 8, 42571–42587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
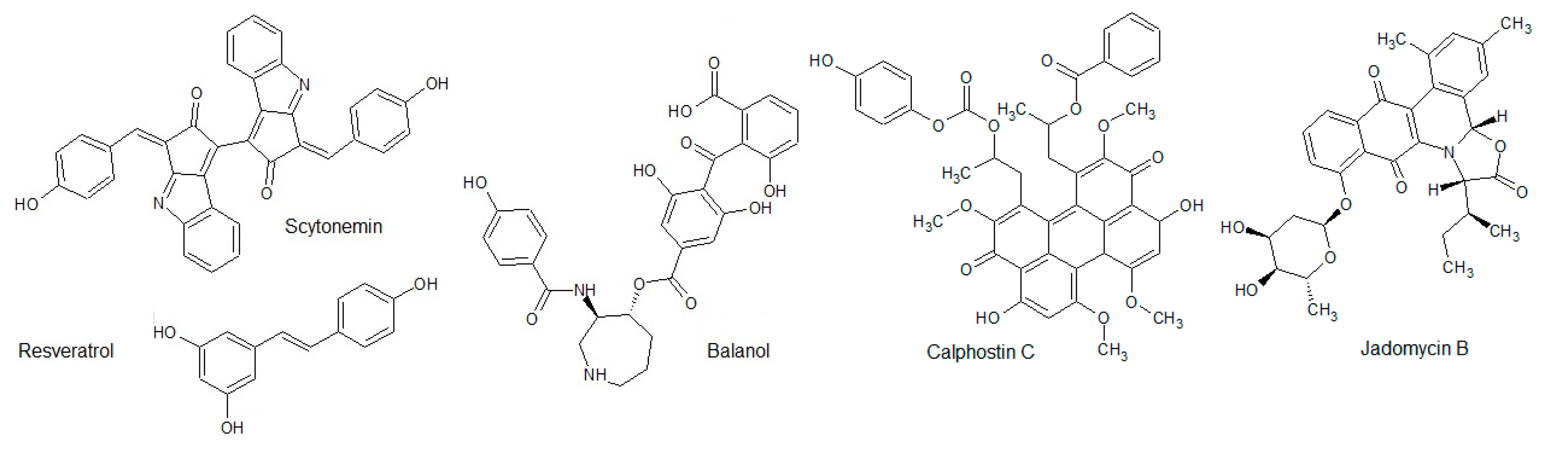
- Fang, J.Y.; Li, Z.H.; Li, Q.; Huang, W.S.; Kang, L.; Wang, J.P. Resveratrol affects protein kinase C activity and promotes apoptosis in human colon carcinoma cells. Asian Pac. J. Cancer Prev. 2012, 13, 6017–6022. [Google Scholar] [CrossRef]
- Kundu, J.K.; Surh, Y.J. Cancer chemopreventive and therapeutic potential of resveratrol: Mechanistic perspectives. Cancer Lett. 2008, 269, 243–261. [Google Scholar] [CrossRef] [PubMed]
- Subbaramaiah, K.; Chung, W.J.; Michaluart, P.; Telang, N.; Tanabe, T.; Inoue, H.; Jang, M.; Pezzuto, J.M.; Dannenberg, A.J. Resveratrol inhibits cyclooxygenase-2 transcription and activity in phorbol ester-treated human mammary epithelial cells. J. Biol. Chem. 1998, 273, 21875–21882. [Google Scholar] [CrossRef] [Green Version]
- Woo, J.H.; Lim, J.H.; Kim, Y.H.; Suh, S.I.; Min, D.S.; Chang, J.S.; Lee, Y.H.; Park, J.W.; Kwon, T.K. Resveratrol inhibits phorbol myristate acetate-induced matrix metalloproteinase-9 expression by inhibiting JNK and PKC delta signal transduction. Oncogene 2004, 23, 1845–1853. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Chang, L.; Ma, G.; Wang, T.; Ly, M.; Wang, Z.; Chen, L.; Wang, Y.; Gao, X.; Zhu, X. Delineation of Platelet Activation Pathway of Scutellarein Revealed Its Intracellular Target as Protein Kinase C. Biol. Pharm. Bul. 2016, 39, 181–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Liu, W.; Ma, L.; Liu, X.; Liu, Z.; Zhu, B. Scutellarin inhibits translocation of protein kinase C in diabetic thoracic aorta of the rat. Clin. Exp. Pharmacol. Physiol. 2012, 39, 136–140. [Google Scholar] [CrossRef]
- Zhang, H.; Dong, R.; Zhang, P.; Wang, Y. Songorine suppresses cell growth and metastasis in epithelial ovarian cancer via the Bcl-2/Bax and GSK3β/β-catenin signaling pathways. Oncol. Rep. 2019, 41, 3069–3079. [Google Scholar] [CrossRef]
- Middletom, E., Jr.; Kandaswami, C.; Theoharides, T.C. The Effects of Plant Flavonoids on Mammalian Cells: Implications for Inflammation, Heart Disease, and Cancer. Pharmacol. Rev. 2000, 52, 673–751. [Google Scholar]
- Hou, D.X.; Kumamoto, T. Flavonoids as protein kinase inhibitors for cancer chemoprevention: Direct binding and molecular modeling. Antioxid. Redox Signal. 2010, 13, 691–719. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 2000, 6, 909–919. [Google Scholar] [CrossRef]
- Li, C.; Liu, X.; Lin, X.; Chen, X. Structure-activity relationship of 7 flavonoids on recombinant human protein kinase CK2 holoenzyme. J. Cent. South Univ. Med. Sci. 2009, 34, 20–26. [Google Scholar]
- Lolli, G.; Cozza, G.; Mazzorana, M.; Tibaldi, E.; Cesaro, L.; Donella-Deana, A.; Meggio, F.; Venerando, A.; Franchin, C.; Sarno, S.; et al. Inhibition of protein kinase CK2 by flavonoids and tyrphostins. A structural insight. Biochemistry 2012, 51, 6097–6107. [Google Scholar] [CrossRef]
- Baier, A.; Galicka, A.; Nazaruk, J.; Szyszka, R. Selected flavonoid compounds as promising inhibitors of protein kinase CK2α and CK2α’, the catalytic subunits of CK2. Phytochemistry 2017, 136, 39–45. [Google Scholar] [CrossRef]
- Baier, A.; Nazaruk, J.; Galicka, A.; Szyszka, R. Inhibitory influence of natural flavonoids on human protein kinase CK2 isoforms—Effect of the regulatory subunit. Mol. Cell. Biochem. 2018, 444, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Sicheri, F.; Moarefi, I.; Kuriyan, J. Crystal structure of the Src family tyrosine kinase Hck. Nature 1997, 385, 602–609. [Google Scholar] [CrossRef]
- Johnson, J.L.; Rupasinghe, S.G.; Stefani, F.; Schuler, M.A.; Gonzalez de Mejia, E. Citrus flavonoids luteolin, apigenin, and quercetin inhibit glycogen synthase kinase-3beta enzymatic activity by lowering the interaction energy within the binding cavity. J. Med. Food. 2011, 14, 325–333. [Google Scholar] [CrossRef]
- Lee, K.W.; Kang, N.J.; Heo, Y.S.; Rogozin, E.A.; Pugliese, A.; Hwang, M.K.; Bowden, G.T.; Bode, A.M.; Lee, H.J.; Dong, Z. Raf and MEK protein kinases are direct molecular targets for the chemopreventive effect of quercetin, a major flavonol in red wine. Cancer Res. 2008, 68, 946–955. [Google Scholar] [CrossRef] [Green Version]
- Peet, G.W.; Li, J. IkappaB kinases alpha and beta show a random sequential kinetic mechanism and are inhibited by staurosporine and quercetin. J. Biol. Chem. 1999, 274, 32655–32661. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Núñez, L.; Rivera, J.; Guerrero, J.A.; Martinez, C.; Vicente, V.; Lozano, M.L. Differential effects of quercetin, apigenin and genistein on signaling pathways of protease-activated receptors PAR1 and PAR4 in platelets. Br. J. Pharmacol. 2009, 158, 1548–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawara, H.; Akiyama, T.; Ishida, J.; Watanabe, S.; Suzuki, K. A specific inhibitor for tyrosine protein kinase from Pseudomonas. J. Antibiot. 1986, 39, 606–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, T.; Ishida, J.; Nakagawa, S.; Ogawara, H.; Watanabe, S.; Itoh, N.; Shibuya, M.; Fukami, Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J. Biol. Chem. 1987, 262, 5592–5595. [Google Scholar] [PubMed]
- Xu, L.; Ding, Y.; Catalona, W.J.; Yang, X.J.; Anderson, W.F.; Jovanovic, B.; Wellman, K.; Killmer, J.; Huang, K.; Scheidt, A.; et al. MEK4 function, genistein treatment, and invasion of human prostate cancer cells. J. Natl. Cancer Inst. 2009, 101, 1141–1155. [Google Scholar] [CrossRef] [Green Version]
- Agullo, G.; Gamet-Payrastre, L.; Manenti, S.; Viala, C.; Rémésy, C.; Chap, H.; Payrastre, B. Relationship between flavonoid Structure and inhibition of phosphatitylinosotol 3-Kinase: A comparison with tyrosine kinase and protein kinase C inhibition. Biochem. Pharmacol. 1997, 53, 1649–1657. [Google Scholar] [CrossRef]
- Polier, G.; Ding, J.; Konkimalla, B.V.; Eick, D.; Ribeiro, N.; Köhler, R.; Giaisi, M.; Efferth, T.; Desaubry, L.; Krammer, P.H.; et al. Wogonin and related natural flavones are inhibitors of CDK9 that induce apoptosis in cancer cells by transcriptional suppression of Mcl-1. Cell Death Dis. 2011, 2, e182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.K.; Bharate, S.B.; Vishwakarma, R.A. Cyclin-Dependent Kinase Inhibition by Flavoalkaloids. Mini Rev. Med. Chem. 2012, 12, 632–649. [Google Scholar] [CrossRef] [PubMed]
- Szaefer, H.; Kaczmarek, J.; Rybczynska, M.; Baer-Dubowska, W. The effect of plant phenols on the expression and activity of phorbol ester-induced PKC in mouse epidermis. Toxicology 2007, 230, 1–10. [Google Scholar] [CrossRef]
- Kashiwada, Y.; Nonaka, G.-I.; Nishioka, I.; Ballas, L.M.; Jiang, J.B.; Janzen, W.P.; Lee, H.-H. Tannins as selective inhibitors of protein kinase C. Bioorg. Med. Chem. Lett. 1992, 2, 239–244. [Google Scholar] [CrossRef]
- Yang, E.B.; Wei, L.; Zhang, K.; Chen, Y.Z.; Chen, W.N. Tannic acid, a potent inhibitor of epidermal growth factor receptor tyrosine kinase. J. Biochem. 2006, 139, 495–502. [Google Scholar] [CrossRef]
- Cozza, G.; Bonvini, P.; Zorzi, E.; Poletto, G.; Pagano, M.A.; Sarno, S.; Donella-Deana, A.; Zagotti, G.; Rosolen, A.; Pinna, L.A.; et al. Identification of ellagic acid as potent inhibitor of protein kinase CK2: A successful example of a virtual screening application. J. Med. Chem. 2006, 49, 2363–2366. [Google Scholar] [CrossRef]
- Cozza, G. The Development of CK2 Inhibitors: From Traditional Pharmacology to in Silico Rational Drug Design. Pharmaceuticals 2017, 10, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onoda, T.; Iinuma, H.; Sasaki, Y.; Hamada, M.; Isshiki, K.; Naganawa, H.; Takeuchi, T.; Tatsuta, K.; Umezawa, K. Isolation of a novel tyrosine kinase inhibitor, lavendustin A, from Streptomyces griseolavendus. J. Nat. Prod. 1989, 52, 1252–1257. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-Y.; Persons, P.E.; Spada, A.P.; Bednarq, R.A.; Levitzki, A.; Zilberstein, A. Kinetic Analysis of the Inhibition of the Epidermal Growth Factor Receptor Tyrosine Kinase by Lavendustin-A and Its Analogue. J. Biol. Chem. 1991, 66, 21105–21112. [Google Scholar]
- O’Dell, T.J.; Kandel, E.R.; Grant, S.G. Long-term potentiation in the hippocampus is blocked by tyrosine kinase inhibitors. Nature 1991, 353, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Nardini, M.; Scaccini, C.; Packer, L.; Virgili, F. In vitro inhibition of the activity of phosphorylase kinase, protein kinase C and protein kinase A by caffeic acid and a procyanidin-rich pine bark (Pinus marittima) extract. Biochim. Biophys. Acta 2000, 1474, 219–225. [Google Scholar] [CrossRef]
- Kang, N.J.; Lee, K.W.; Shin, B.J.; Jung, S.K.; Hwang, M.K.; Bode, A.M.; Heo, Y.-S.; Lee, H.J.; Dong, Z. Caffeic acid, a phenolic phytochemical in coffee, directly inhibits Fyn kinase activity and UVB-induced COX-2 expression. Carcinogenesis 2009, 30, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmmoud, Y.A. Modulation of protein kinase C by curcumin; inhibition and activation switched by calcium ions. Br. J. Pharmacol. 2007, 150, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Pany, S.; You, Y.; Das, J. Curcumin inhibits PKCα activity by binding to its C1 domain. Biochemistry 2017, 55, 6327–6336. [Google Scholar] [CrossRef] [Green Version]
- Banerjeea, S.; Jib, C.; Mayfielda, J.E.; Goelc, A.; Xiaob, J.; Dixona, J.E.; Guo, X. Ancient drug curcumin impedes 26S proteasome activity by direct inhibition of dual-specificity tyrosine-regulated kinase 2. Proc. Natl. Acad. Sci. USA 2018, 115, 8155–8160. [Google Scholar] [CrossRef] [Green Version]
- Jayasuriya, H.; Koonchanok, N.M.; Grahlen, R.L.; McLaughlin, J.L.; Chang, C.-J. Emodin, a protein tyrosine kinase inhibitor from Polyonum cuspidatum. J. Nat. Prod. 1992, 55, 696–698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hung, M.C. Sensitization of HER-2/neu-overexpressing non-small cell lung cancer cells to chemotherapeutic drugs by tyrosine kinase inhibitor emodin. Oncogene 1996, 12, 571–576. [Google Scholar]
- Yim, H.; Lee, Y.H.; Lee, C.H.; Lee, S.K. Emodin, an Anthraquinone Derivative Isolated from the Rhizomes of Rheum palmatum, Selectively Inhibits the Activity of Casein Kinase II as a Competitive Inhibitor. Planta Med. 1999, 65, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.C.; Harris, D.R.; Kirkman, L.M.D.; Perez, A.M.; Qian, Y.; Schermerhorn, J.T.; Hong, M.Y.; Winston, D.S.; Xu, L.; Brandt, G.S. FIKK Kinase, a Ser/Thr Kinase Important to Malaria Parasites, Is Inhibited by Tyrosine Kinase Inhibitors. ACS Omega 2017, 2, 6605–6612. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Cha, E.Y.; Sul, J.Y.; Song, I.S.; Kim, J.Y. Chrysophanic Acid Blocks Proliferation of Colon Cancer Cells by Inhibiting EGFR/mTOR Pathway. Phytother. Res. 2011, 25, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wu, J.; Gao, Y.; Luo, Y.; Yang, D.; Wang, P. The pharmacological properties of chrysophanol, the recent advances. Biomed. Pharmacother. 2020, 125, 110002. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, X.; Fang, L.; Liu, F.; Cai, R.; Peng, C.; Qi, Y. Rhein exerts pro- and anti-inflammatory actions by targeting IKKβ inhibition in LPS-activated macrophages. Free. Radic. Biol. Med. 2014, 72, 104–112. [Google Scholar] [CrossRef]
- Henamayee, S.; Banik, K.; Sailo, B.L.; Shabnam, B.; Harsha, C.; Srilakshmi, S.; Vgm, N.; Baek, S.H.; Ahn, K.S.; Kunnumakkara, A.B. Therapeutic Emergence of Rhein as a Potential Anticancer Drug: A Review of Its Molecular Targets and Anticancer Properties. Molecules 2020, 25, 10. [Google Scholar] [CrossRef]
- Aly, A.H.; Debbab, A.; Edrada-Ebel, R.A.; Müller, W.E.G.; Kubbutat, M.H.G.; Wray, V.; Ebel, R.; Proksch, P. Protein kinase inhibitors and other cytotoxic metabolites from the fungal endophyte Stemphylium botryosum isolated from Chenopodium album. Mycosphere 2010, 1, 153–162. [Google Scholar]
- Yang, E.B.; Zhao, Y.N.; Zhang, K.; Mack, P. Daphnetin, One of Coumarin Derivatives, Is a Protein Kinase Inhibitor. Biochem. Biophys. Res. Commun. 1999, 260, 682–685. [Google Scholar] [CrossRef]
- Cozza, G.; Gianoncelli, A.; Bonvini, P.; Zorzi, E.; Pasquale, R.; Rosolen, A.; Pinna, L.A.; Meggio, F.; Zagotto, G.; Moro, S. Urolithin as a converging scaffold linking ellagic acid and coumarin analogues: Design of potent protein kinase CK2 inhibitors. ChemMedChem 2011, 6, 2273–2286. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Hsieh, D.; Yang, Y.-L.; Xu, Z.; Peto, C.; Jablons, D.M.; You, L. Coumestrol from the national cancer Institute’snatural product library is a novel inhibitor ofprotein kinase CK2. BMC Pharmacol. Toxicol. 2013, 14, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banik, K.; Ranaware, A.M.; Deshpande, V.; Nalawade, S.P.; Padmavathi, G.; Bordoloi, D.; Sailo, B.L.; Shanmugam, M.K.; Fan, L.; Arfuso, F.; et al. Honokiol for cancer therapeutics: A traditional medicine that can modulate multiple oncogenic targets. Pharmacol. Res. 2019, 144, 192–209. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.P.; Lee, W.L.; Tang, Y.Q.; Yap, W.H. Honokiol: A Review of Its Anticancer Potential and Mechanisms. Cancers 2020, 12, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, M.K.; Jang, Y.P.; Kim, Y.C.; Kim, A.G. Arctigenin, a phenylpropanoid dibenzylbutyrolactone lignin, inhibits MAP kinases and AP-1 activation via potent MKK inhibition: Role in TNF-α inhibition. Int. Immunopharmacol. 2004, 4, 1419–1429. [Google Scholar] [CrossRef]
- Tang, S.; Zhou, W.; Zhong, X.; Xu, J.; Huang, H.; Zheng, X.; Zhang, J.; Yang, S.; Shang, P.; Tang, Q.; et al. Arctigenin prevents the progression of osteoarthritis by targeting PI3K/Akt/NF-κB axis: In vitro and in vivo studies. J. Cell. Mol. Med. 2020, 24, 4183–4193. [Google Scholar] [CrossRef]
- Kulanthaivel, P.; Hallock, Y.F.; Boros, C.; Hamilton, S.M.; Janzen, W.P.; Ballas, L.M.; Loomis, C.R.; Jiang, J.B.; Katz, B.; Steiner, J.R.; et al. Balanol: A novel and potent inhibitor of protein kinase C from the fungus Verticillium balanoides. J. Am. Chem. Soc. 1993, 115, 6452–6453. [Google Scholar] [CrossRef]
- Setyawan, J.; Koide, K.; Diller, T.C.; Bunnage, M.E.; Taylor, S.S.; Nicolaou, K.C.; Brunton, L.L. Inhibition of Protein Kinases by Balanol: Specificity within the Serine/Threonine Protein Kinase Subfamily. Mol. Pharmacol. 1999, 56, 370–376. [Google Scholar] [CrossRef]
- Tesmer, J.J.; Tesmer, V.M.; Lodowski, D.T.; Steinhagen, H.; Huber, J. Structure of Human G Protein-Coupled Receptor Kinase 2 in Complex with the Kinase Inhibitor Balanol. J. Med. Chem. 2010, 53, 1867–1870. [Google Scholar] [CrossRef] [Green Version]
- Koidel, K.; Bunnage, M.E.; Paloma, L.G.; Kanter, J.R.; Taylor, S.S.; Brunton, L.L.; Nicolaou, K.C. Molecular design and biological activity of potent and selective protein kinase inhibitors related to balanol. Chem. Biol. 1995, 2, 601–608. [Google Scholar] [CrossRef] [Green Version]
- Proteau, P.J.; Gerwick, W.H.; Garcia-Pichel, F.; Castenholz, R. The structure of scytonemin, an ultraviolet sunscreen pigment from the sheaths of cyanobacteria. Experientia 1993, 49, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Pirola, L.; Fröjdö, M. Resveratrol: One Molecule, Many Targets. IUBMB Life 2008, 60, 323–332. [Google Scholar] [CrossRef]
- Zang, M.; Xu, S.; Maitland-Toolan, K.A.; Zuccollo, A.; Hou, X.; Jiang, B.; Wierzbicki, M.; Verbeuren, T.J.; Cohen, R.A. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes 2006, 55, 2180–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundu, J.K.; Shin, Y.K.; Kim, S.H.; Surh, Y.J. Resveratrol inhibits phorbol ester-induced expression of COX-2 and activation of NF-kappaB in mouse skin by blocking IkappaB kinase activity. Carcinogenesis 2006, 27, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Slater, S.J.; Seiz, J.L.; Cook, A.C.; Stagliano, B.A.; Buzas, C.J. Inhibition of protein kinase C by resveratrol. Biochim. Biophys. Acta 2003, 1637, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.; Ando, K.; Nakano, H.; Iida, T.; Ohno, H.; Morimoto, M.; Tamaoki, T. Calphostins (UCN-1028), novel and specific inhibitors of protein kinase C. I. Fermentation, isolation, physico-chemical properties and biological activities. J. Antibiot. 1989, 42, 1470–1474. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.; Nakano, H.; Morimoto, M.; Tamaoki, T. Calphostin C (UCN-1028C), a novel microbial compound, is a highly potent and specific inhibitor of protein kinase C. Biochem. Biophys. Res. Commun. 1989, 159, 548–553. [Google Scholar] [CrossRef]
- Pollack, I.F.; Kawecki, S. The effect of calphostin C, a potent photodependent protein kinase C inhibitor, on the proliferation of glioma cells in vitro. J. Neuro-Oncol. 1997, 31, 255–266. [Google Scholar] [CrossRef]
- Doull, J.L.; Singh, A.K.; Hoare, M.; Ayer, S.W. Conditions for the production of jadomycin B by Streptomyces venezuelae ISP5230: Effects of heat shock, ethanol treatment and phage infection. J. Ind. Microbiol. 1994, 13, 120–125. [Google Scholar] [CrossRef]
- Fu, D.-H.; Jiang, W.; Zheng, J.T.; Zhao, G.Y.; Li, Y.; Yi, H.; Li, Z.R.; Jiang, J.D.; Yang, K.Q.; Wang, Y.; et al. Jadomycin B, an Aurora-B kinase inhibitor discovered through virtual screening. Mol. Cancer Ther. 2008, 7, 2386–2393. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Wang, N.; Zhang, T.; Zhang, B.; Sajeevan, T.P.; Joseph, V.; Armstrong, L.; He, S.; Yan, X.; Naman, C.B. A Systematic Review of Recently Reported Marine Derived Natural Product Kinase Inhibitors. Mar. Drugs 2019, 17, 493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omura, S.; Iwai, Y.; Hirano, A.; Nakagawa, A.; Awaya, J.; Tsuchya, H.; Takahashi, Y.; Masuma, R. A new alkaloid AM-2282 of Streptomyces origin. Taxonomy, fermentation, isolation and preliminary characterization. J. Antibiot. 1977, 30, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Tamaoki, T.; Nomoto, H.; Takahashi, I.; Kato, Y.; Morimoto, M.; Tomita, F. Staurosporine, a potent inhibitor of phospholipid/Ca++dependent protein kinase. Biochem. Biophys. Res. Commun. 1986, 135, 397–402. [Google Scholar] [CrossRef]
- Lamers, M.B.; Antson, A.A.; Hubbard, R.E.; Scott, R.K.; Williams, D.H. Structure of the protein tyrosine kinase domain of C-terminal Src kinase (CSK) in complex with staurosporine. J. Mol. Biol. 1999, 285, 713–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meggio, F.; Donella-Deana, A.; Ruzzene, M.; Brunati, A.M.; Cesaro, L.; Guerra, B.; Meyer, T.; Mett, H.; Fabbro, D.; Furet, P.; et al. Different susceptibility of protein kinases to staurosporine inhibition. Kinetic studies and molecular bases for the resistance of protein kinase CK2. Eur. J. Biochem. 1995, 234, 317–322. [Google Scholar] [CrossRef]
- Nakanishi, S.; Kakita, S.; Takahashi, I.; Kawahara, K.; Tsukuda, E.; Sano, T.; Yamada, K.; Yoshida, M.; Kase, H.; Matsuda, Y.; et al. Wortmannin, a microbial product inhibitor of myosin light chain kinase. J. Biol. Chem. 1992, 267, 2157–2163. [Google Scholar]
- Bain, J.; Plater, L.; Elliot, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.C.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A futher update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Shreder, K.R.; Gai, W.; Corral, S.; Ferris, D.K.; Rosenblum, J.S. Wortmannin, a widely used phosphoinositide 3-kinase inhibitor, also potently inhibits mammalian polo-like kinase. Chem. Biol. 2005, 12, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Roll, D.M.; Scheuer, P.J.; Matsumoto, G.K.; Clardy, J. Halenaquinone, a pentacyclic polyketide from a marine sponge. J. Am. Chem. Soc. 1983, 105, 6177–6178. [Google Scholar] [CrossRef]
- Kobayashi, M.; Shimizu, N.; Kyogoku, Y.; Kitagawa, I. Halenaquinol and halenaquinol sulfate, pentacyclic hydroquinones from the Okinawa marine sponge Xestospongia sapra. Chem. Pharm. Bull. 1985, 33, 1305–1308. [Google Scholar] [CrossRef]
- Skropeta, D.; Pastro, N.; Zivanovic, A. Kinase Inhibitors from Marine Sponges. Mar. Drugs 2011, 9, 2131–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, H.; Matsunaga, K.; Saito, M.; Hagiya, S.; Furukawa, K.; Nakamura, H.; Ohizumi, Y. Halenaquinone, a novel phosphatidylinositol 3-kinase inhibitor from a marine sponge, induces apoptosis in PC12 cells. Eur. J. Pharmacol. 2001, 413, 37–45. [Google Scholar] [CrossRef]
- Vesely, J.; Havlicek, L.; Strnad, M.; Blow, J.J.; Donella-Deana, A.; Pinna, L.; Letham, D.S.; Kato, J.; Detivaud, L.; Leclerc, S.; et al. Inhibition of cyclin-dependent kinases by purine analogues. Eur. J. Biochem. 1994, 224, 771–786. [Google Scholar] [CrossRef]
- Meijer, L.; Borgne, A.; Mulner, O.; Chong, J.P.J.; Blow, J.J.; Inagaki, N.; Inagaki, M.; Delcros, J.-G.; Moulinoux, J.-P. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 1997, 243, 527–536. [Google Scholar] [CrossRef]
- Nagata, H.; Ochiai, K.; Aotani, Y.; Ando, K.; Yoshida, M.; Takahashi, I.; Tamaoki, T. Lymphostin (LK6-A), a Novel Immunosuppressant from Streptomyces sp. KYI1783: Taxonomyof the Producing Organism, Fermentation, Isolation and Biological Activities. J. Antibiot. 1997, 50, 537–542. [Google Scholar] [CrossRef] [Green Version]
- Aotani, Y.; Nagata, H.; Yoshida, M. Lymphostin (LK6-A), a novel immunosuppressant from Streptomyces sp. KY11783: Structural elucidation. J. Antibiot. 1997, 50, 543–545. [Google Scholar] [CrossRef] [Green Version]
- Miyanaga, A.; Janso, J.E.; McDonald, L.; He, M.; Liu, H.B.; Barbieri, L.; Eustaquio, A.S.; Fielding, E.N.; Carter, G.T.; Jensen, P.R.; et al. Discovery and Assembly-Line Biosynthesis of the Lymphostin Pyrroloquinoline Alkaloid Family of mTOR Inhibitors in Salinispora Bacteria. J. Am. Chem. Soc. 2011, 133, 13311–13313. [Google Scholar] [CrossRef] [Green Version]
- Nair, M.S.R.; Carey, S.T. Metabolites of pyrenomycetes XIII: Structure of (+) hypothemycin, an antibiotic macrolide from Hypomyces trichothecoides. Tetrahedron Lett. 1980, 21, 2011–2012. [Google Scholar] [CrossRef]
- Isaka, M.; Suyarnsestakorn, C.; Tanticharoen, M.; Kongsaeree, P.; Thebtaranonth, Y. Aigialomycins A-E, new resorcylic macrolides from the marine mangrove fungus Aigialus parvus. J. Org. Chem. 2002, 67, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, A.; Kennedy, J.; Murli, S.; Reid, R.; Santi, D.V. Targeted covalent inactivation of protein kinases by resorcylic acid lactone polyketides. Proc. Natl. Acad. Sci. USA 2006, 103, 4234–4239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göckler, N.; Jofre, G.; Papadopulos, C.; Soppa, U.; Tejedor, F.J.; Becker, W. Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. FEBS J. 2009, 276, 6324–6337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rüben, K.; Wurzlbauer, A.; Walte, A.; Sippl, W.; Bracher, F.; Becker, W. Selectivity Profiling and Biological Activity of Novel β-Carbolines as Potent and Selective DYRK1 Kinase Inhibitors. PLoS ONE 2015, 10, e0132453. [Google Scholar] [CrossRef]
- Kobori, M.; Yang, Z.; Gong, D.; Heissmeyer, V.; Zhu, H.; Jung, Y.-K.; Gagidis, M.A.M.; Rao, A.; Sekine, T.; Ikegami, F.; et al. Wedelolactone suppresses LPS-induced caspase-11 expression by directly inhibiting the IKK Complex. Cell Death Differ. 2004, 11, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
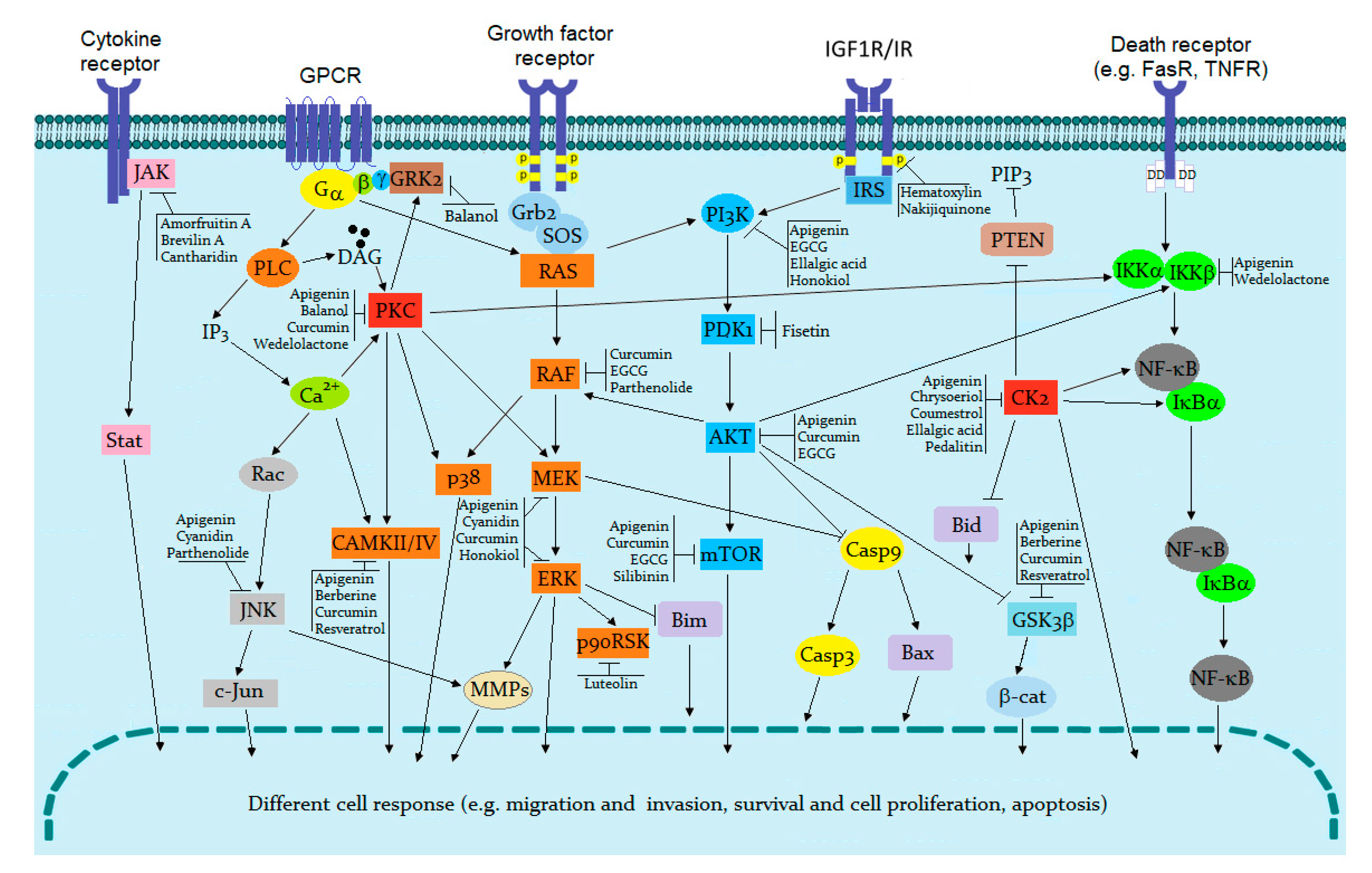










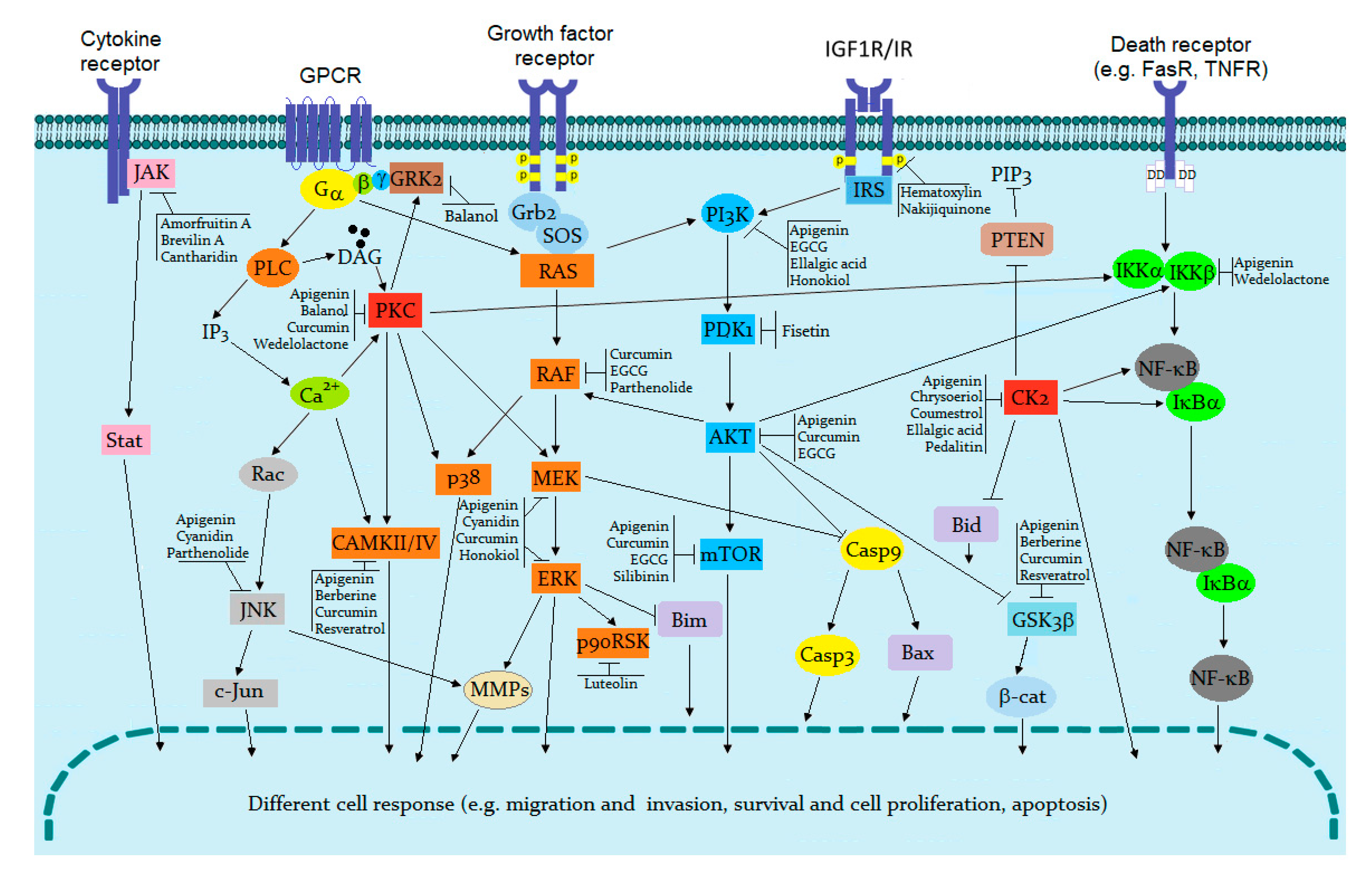{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ePK | Function in the Cell | Ref. |
|---|---|---|
| Akt/PKB |
| [10,11] |
| AMPK |
| [12,13] |
| ASK1 |
| [14] |
| CDKs |
| [15,16,17] |
| CK2, CK2α |
| [18,19,20] |
| EGFR |
| [21,22,23,24] |
| ERK1/2 |
| [25,26] |
| FAK1 |
| [27,28] |
| FGFR |
| [29,30,31] |
| GSK3β |
| [32] |
| IGF1-R |
| [33,34] |
| IKKα |
| [35,36,37,38] |
| JAK1-3 |
| [39,40] |
| JNK |
| [41,42,43] |
| Lck |
| [44,45,46] |
| mTOR |
| [47,48,49,50] |
| PDK1 |
| [51] |
| ||
| PKA |
| [52,53] |
| PKC |
| [54,55] |
| PKG |
| [56,57] |
| Src |
| [58,59] |
| VEGFR1/3 |
| [60,61,62] |
| Compound | Target | Biological Effect | Ref. |
|---|---|---|---|
| Tyr Protein Kinases | |||
| Alpinetin | Stat3↓ | Altered protein expression levels of cyclin-D1, c-Myc, survivin, Bcl-2, Bax, TIMP-1, TIMP-2, MMP-2, MMP-9, as well as cleaved caspase-3 and PARP in SKOV3 cells. | [101] |
| Auriculasin | VEGFR2↓ | Inhibition of angiogenesis by modulating VEGFR2-related signaling pathways. Inhibition of VEGFR2 activation, as well as phosphorylation of intracellular downstream protein kinases AKT, mTOR, PI3K, p38, ERK, and Src. | [102] |
| Cantharidin | JAK1/ Stat3↓ | Suppression of VEGF-induced activation of Stat3 and inhibition of JAK1 and ERK phosphorylation. | [103] |
| Curcumin | IGF-1R↓ | Inhibition of phosphorylation: IGF1R, IRS1, AKT, S6K, and 4EBP1 in the mouse keratinocyte cell line. | [104,105] |
| Src↓, PTK2↓ | Inhibition activity of v-Src led to a reduction of Src-Tyr phosphorylation, decreased Src-mediated Shc phosphorylation, ERK activation, and cell proliferation in v-Src transformed cells. | [106] | |
| Emodin | JAK2↓ | Inhibition of IL-6-induced JAK2/Stat3 pathway induced apoptosis. | [107] |
| Her2/neu↓ | Suppression of Her2/neu PTK activity and proliferation; repression of transformation and metastasis. | [108] | |
| Honokiol | EGFR↓ | Inhibition of U251 and U-87 MG human glioma/glioblastoma cell viability, colony formation, and promoted apoptosis. Inhibition of cell migration/proliferation and invasion. Induction of apoptosis and reduction of Bcl-2 expression, accompanied by an increase in Bax expression. Reduced expression of EGFR, CD133, and nestin. Suppression of AKT and ERK signaling pathway activation. | [109] |
| Luteolin | VEGF/ VEGFR2↓ | Decreased VEGF, cell migration, and viability of triple-negative breast cancer cell lines MDA-MB-435. | [110,111] |
| EGFR↓ | Inhibition of EGF-induced activities of EGFR signaling pathway in human breast cancer cell lines and PI3K/AKT, MAPK/ERK1/2, Stat3 signal pathways. | [112] | |
| Lymphostin | Lck↓ | Inhibition of Src family kinase Lck activity in Jurkat T cells. | [113] |
| Quercetin | JAK2/Stat3↓ | Inhibition of hepatocellular carcinoma progression by modulating cell apoptosis, migration, invasion, and autophagy. Effects partly related to the JAK2/Stat3 signaling pathway. | [114] |
| Tannic acid | EGFR↓ Stat1/3↓ | Tannic acid binding to EGFR inhibited the tyrosine kinase activity, modulated the EGFR/JAK2/Stat1/3 and p38/ Stat1/ p21WAF1/CIP1 pathways, and induced G1-arrest and intrinsic apoptosis in breast carcinomas. | [115] |
| Ser/Thr Protein Kinases | |||
| β-elemene | Cdc2↓ | Cell cycle G2/M phase was arrested in A2780 and A2780/CP human ovarian carcinoma cells in vitro, mediated by alterations in cyclin and CDK expression, the down-regulation of Cdc2, cyclin A, and cyclin B1, and the upregulation of p21WAF1/CIP1 and p53 proteins. | [116] |
| Acacetin Genkwanin Isorhamnetin | Akt/PKB↓ | Cell cycle arrested at G2/M as a result of PI3Kγ inhibition and inactivation of PI3K, AKT, mTOR, p70RSK, and ULK, resulting in apoptosis in human breast cancer cells. | [117] |
| Alpinetin | ERK↓ | Phosphorylation of IκBα protein, p65, p38, and ERK inhibited in LPS stimulated RAW 264.7 cells. | [118] |
| Apigenin | CK2α↓ | Inactivation of CK2α resulted in inhibition of the sphere-forming cell capacity of HeLa. | [119] |
| IKKα↓ | Direct binding with IKKα attenuated kinase activity and suppressed NF-ĸB/p65 activation in human prostate cancer PC-3 and 22Rv1 cells. | [120] | |
| Artemisinin | IKKα↓ | Exhibited anti-inflammatory activities in TPA-induced skin inflammation in mice; inhibited the expression of TRAF2 and RIP1; inhibited TNFα induced NF-κB reporter gene expression, phosphorylation, and degradation of IκBα, and p65 nuclear translocation. | [121,122] |
| Baicalin | PKC↓, MAPK↓ | ROS production reduced, suppressed Casp3 cleavage for inducing apoptosis. Inhibited activation of PKC/MAPK signaling pathway for down regulating JNK, p38, ERK, PKCα, and PKCδ in piglet monocytes stimulated by Haemophilus parasuis. | [123] |
| Berberine | AMPK↑, mTOR↓ | AMPK activated, as a major regulator of metabolic pathways, mTOR inhibited. mTOR targets: 4EBP1 and p70RSK down-regulated. | [124] |
| MLCK↓ | Reduced amplitude of contraction in isolated duodenum and gastric strips in rats by inhibition of MLCK and down-regulation of MLC20 and Mg2+-ATPase activity. | [125] | |
| EGCG | RAF↓, MEK1/2↓, ERK1/2↓ | Reduced protein levels of pEGFR, H-RAS, p-RAF, p-MEK1/2, and pERK1/2 in human thyroid carcinoma cells. Inhibition of the growth by induced apoptosis and down-regulated angiogenesis. | [126] |
| Emodin | CK2↓ | CK2 inhibition cancer cells to Fas and TRAIL ligand-induced apoptosis. CK2 inhibition enhanced the cytotoxicity of natural killer cells HepG2 and Hep3B in vivo. | [127] |
| Genistein | PLK1↓ | In human neuroblastoma SK-N-MC cells, the protein level of MDC1, p53, p21WAF/CIP1, and Bax increased in a dose-dependent manner. Phosphorylation of Chk2 and Cdc25C increased. In addition, consistent with PLK1 down-regulation, Cdc25C phosphorylation inhibited at Ser-198. Down-regulation of proteins Chk2, Cdc25C, Cyclin B1, and Cdc2 as well as Bcl-2 resulted in neuronal apoptosis and G2/M cell cycle arrest. | [128] |
| Hibiscone C | PI3K↓ | Hibiscone C competitively inhibited PI3K activity in intact cells, slowed proliferation, and induced cell death. | [129] |
| Luteolin | p90RSK↓, JNK1↓, | Luteolin exhibited anti-photoaging effects in vitro and in vivo by suppression of JNK1 and p90RSK activity and may have potential as a treatment for the prevention of skin aging. | [130] |
| Quercetagetin | PIM1↓ | PIM1 activity in intact RWPE2 prostate cancer cells inhibited in a dose-dependent manner. RWPE2 cells showed pronounced growth inhibition at inhibitor concentrations that blocked PIM1 kinase activity. The ability of quercetagetin to inhibit the growth of other prostate epithelial cell lines varied in proportion to their levels of PIM1 protein. | [131] |
| Quercetin | Akt/PKB↓, ERK↓ | Akt/PKB and ERK inhibited, resulting in reduced phosphorylation of BAD and a strong activation of caspase-3. | [132] |
| CK2↓, Akt/PKB↓ | CK2 and PI3K/Akt pathways inhibited in chronic lymphocytic leukaemia HG3 cells. | [133] | |
| Resveratrol | PKC↓, MAPK↓, IKKβ↓ | TPA-induced expression of PKC inhibited in human mammary and oral epithelial cells PKCδ in human cervical cancer and affected PKC activity, inducing apoptosis in human colon carcinoma cells. Activity of kinases: PKC, MAPK, IKKβ, and transcription factors: Stat3, HIFα, NF-κB, AP-1, were repressed, resulting in various responses to oncogenic stimuli. | [134,135,136,137] |
| Scutellarein | PKC↓ | Platelet adhesion and aggregation induced by multiple G protein coupled receptor agonists, such as thrombin, was inhibited in a concentration-dependent manner. Scutellarein had a mild effect on intracellular Ca2+ mobilization and cAMP levels. The role of scutellarein as PKC inhibitor was confirmed by PKC activity analysis and molecular docking with PKCα and β. | [138] |
| PKC βI↓, PKC βII↓, PKCδ↓ | PKC activity in the membrane fraction of thoracic aorta smooth muscle cells of diabetic rats inhibited. The translocation inhibition of PKC in vivo and in vitro in diabetic rats may have value as a drug in the treatment of diabetic complications via its inhibition of PKC βI, βII, and δ. | [139] | |
| Songorine | GSK3β↓ | Cell growth and metastasis in epithelial ovarian cancer suppressed via the GSK3β/β-catenin and Bcl2/Bax signaling pathways. | [140] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baier, A.; Szyszka, R. Compounds from Natural Sources as Protein Kinase Inhibitors. Biomolecules 2020, 10, 1546. https://doi.org/10.3390/biom10111546
Baier A, Szyszka R. Compounds from Natural Sources as Protein Kinase Inhibitors. Biomolecules. 2020; 10(11):1546. https://doi.org/10.3390/biom10111546
Chicago/Turabian StyleBaier, Andrea, and Ryszard Szyszka. 2020. "Compounds from Natural Sources as Protein Kinase Inhibitors" Biomolecules 10, no. 11: 1546. https://doi.org/10.3390/biom10111546
APA StyleBaier, A., & Szyszka, R. (2020). Compounds from Natural Sources as Protein Kinase Inhibitors. Biomolecules, 10(11), 1546. https://doi.org/10.3390/biom10111546