The Aging Stress Response and Its Implication for AMD Pathogenesis

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Aging Stress Response
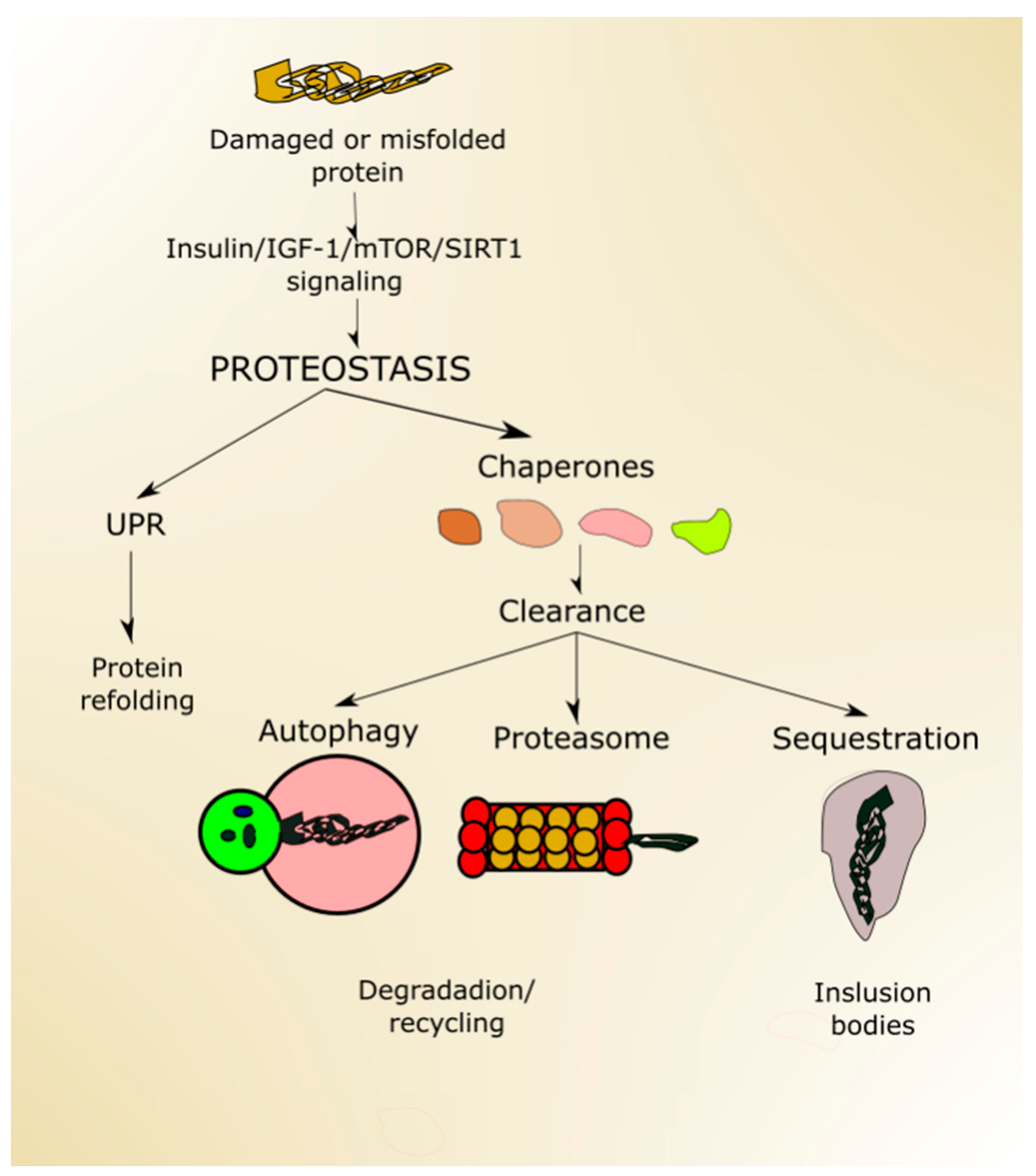
2.1. Nutrient-Signaling Pathways in Aging
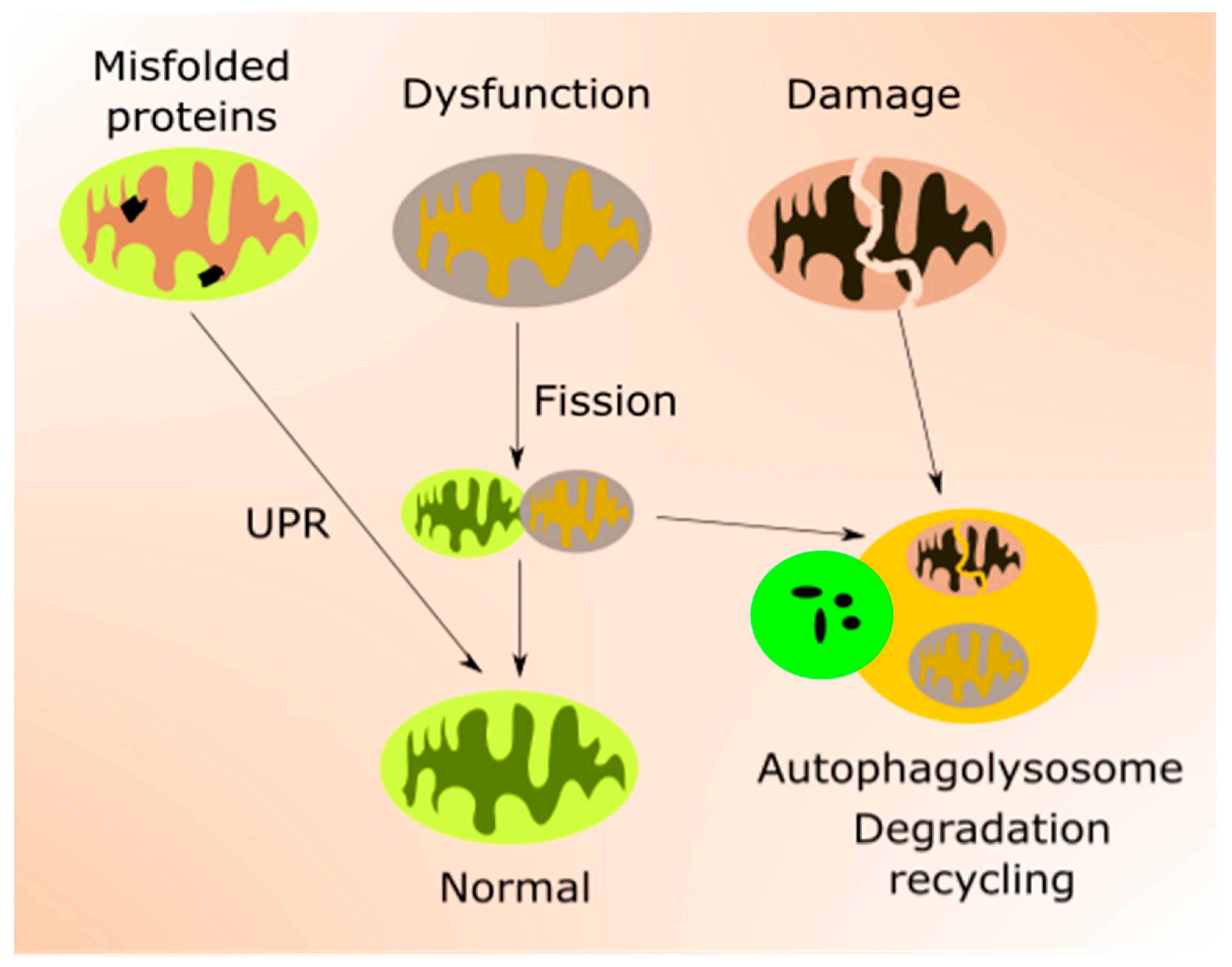
2.2. Mitochondria in Aging
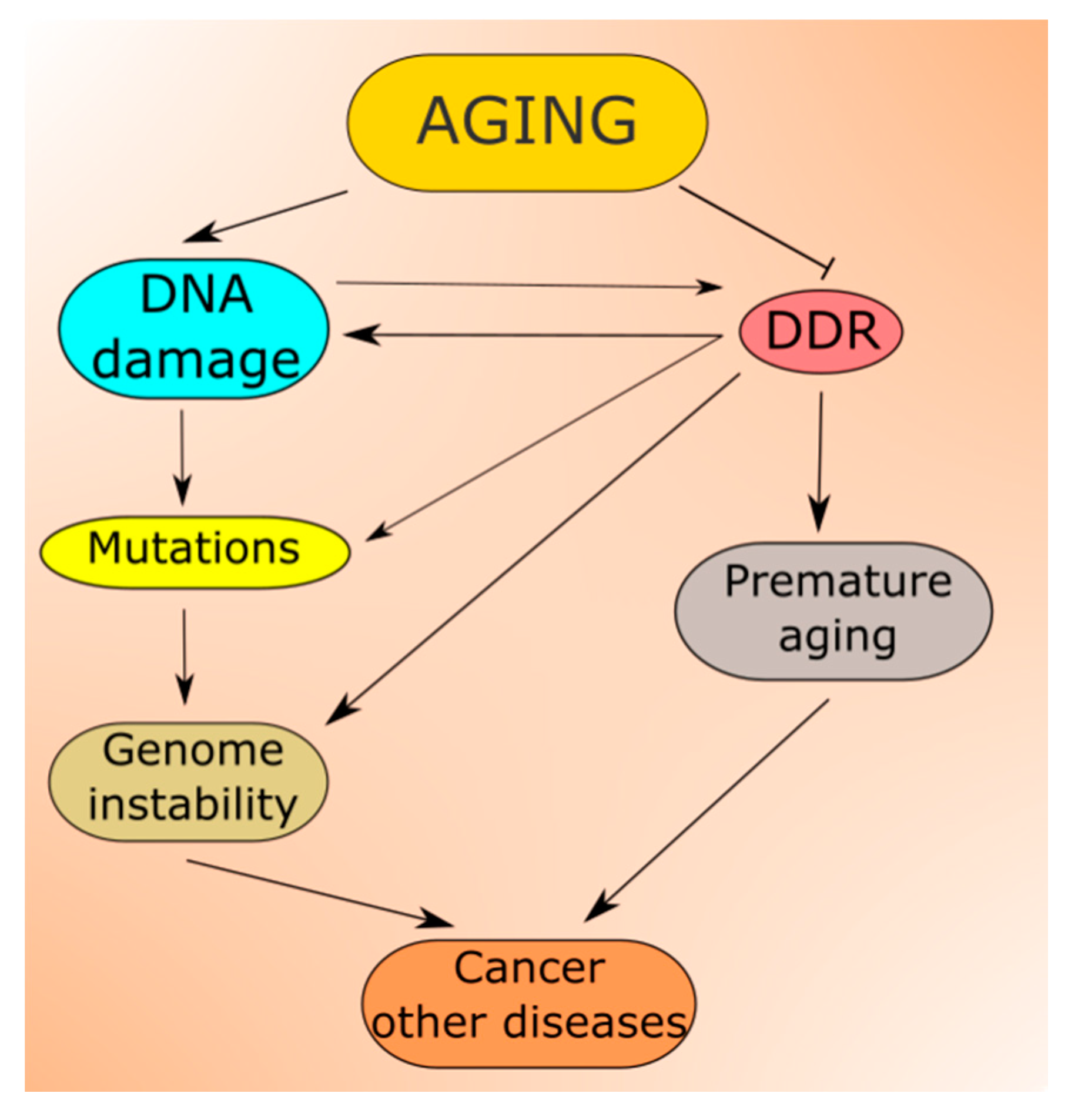
2.3. DDR in Aging
2.4. Autophagy and Aging
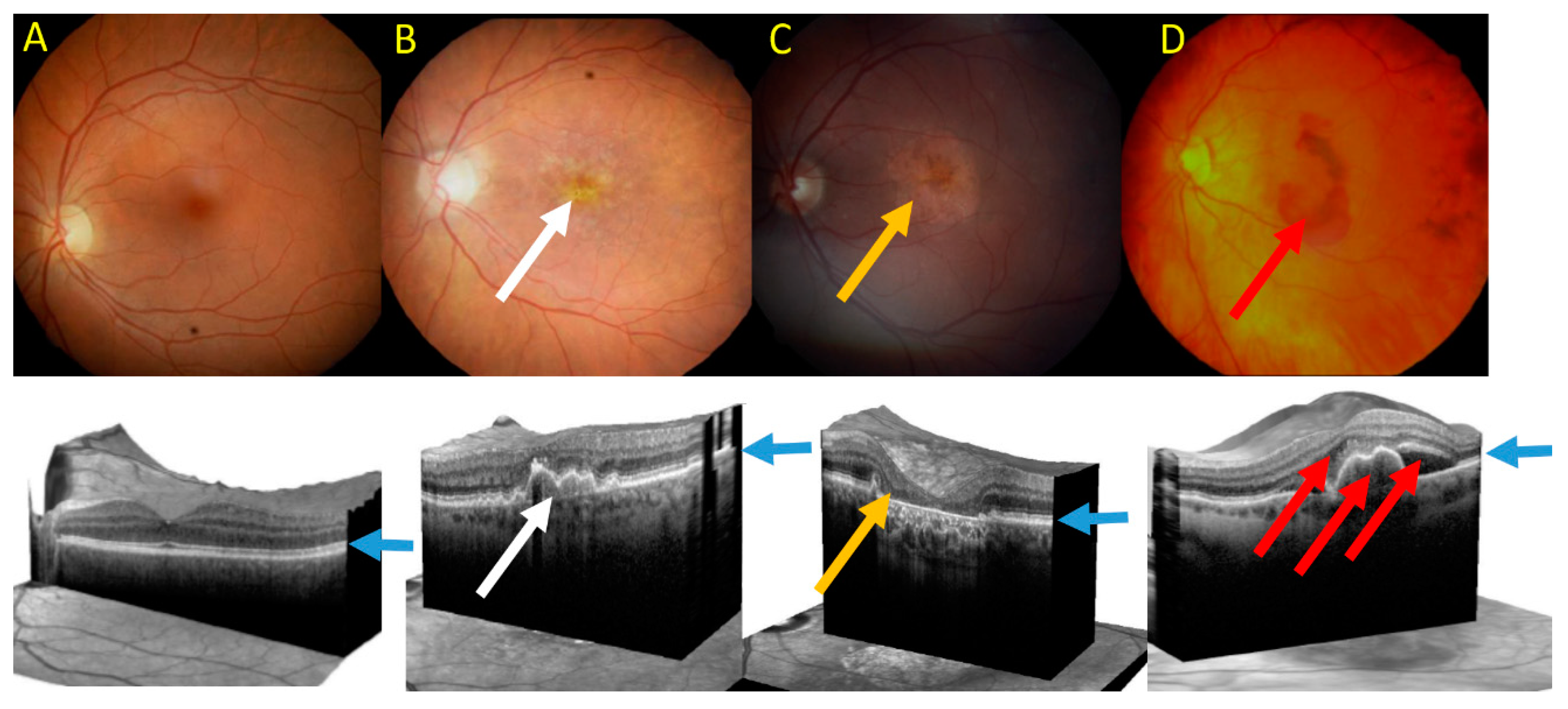
3. Age-Related Macular Degeneration—A Disease of Aging Stress Response
3.1. Nutrient Signaling in AMD
3.2. Autophagy in AMD
3.3. DDR in AMD
3.4. mtQC in AMD
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Carmona, J.J.; Michan, S. Biology of Healthy Aging and Longevity. Rev. Investig. Clin. Organo Hosp. Enferm. Nutr. 2016, 68, 7–16. [Google Scholar]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Yankner, B.A. The aging stress response. Mol. Cell 2010, 40, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.-Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Heal 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Beatty, S.; Koh, H.; Phil, M.; Henson, D.; Boulton, M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134. [Google Scholar] [CrossRef]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Pefani, D.-E.; Pateras, I.S.; Trougakos, I.P. Integrating the DNA damage and protein stress responses during cancer development and treatment. J. Pathol. 2018, 246, 12–40. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, S.G.; Boulton, M.E. Consequences of oxidative stress in age-related macular degeneration. Mol. Asp. Med. 2012, 33, 399–417. [Google Scholar] [CrossRef]
- Carneiro, Â.; Andrade, J.P. Nutritional and Lifestyle Interventions for Age-Related Macular Degeneration: A Review. Oxidative Med. Cell. Longev. 2017, 2017, 6469138. [Google Scholar] [CrossRef]
- Ferrucci, L.; Gonzalez-Freire, M.; Fabbri, E.; Simonsick, E.; Tanaka, T.; Moore, Z.; Salimi, S.; Sierra, F.; de Cabo, R. Measuring biological aging in humans: A quest. Aging Cell 2020, 19, e13080. [Google Scholar] [CrossRef]
- Pan, H.; Finkel, T. Key proteins and pathways that regulate lifespan. J. Biol. Chem. 2017, 292, 6452–6460. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Glowacki, S.; Kauppinen, A.; Kaarniranta, K. Mitochondrial and nuclear DNA damage and repair in age-related macular degeneration. Int. J. Mol. Sci. 2013, 14, 2996–3010. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Pawlowska, E.; Szczepanska, J.; Kaarniranta, K. Interplay between Autophagy and the Ubiquitin-Proteasome System and Its Role in the Pathogenesis of Age-Related Macular Degeneration. Int. J. Mol. Sci. 2019, 20, 210. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.T.; Błasiak, J.; Niittykoski, M.; Kinnunen, K.; Kauppinen, A.; Salminen, A.; Kaarniranta, K. DNA damage response and autophagy in the degeneration of retinal pigment epithelial cells-Implications for age-related macular degeneration (AMD). Ageing Res. Rev. 2017, 36, 64–77. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Kajdanek, J.; Morawiec, J.; Pawlowska, E.; Blasiak, J. PGC-1α Protects RPE Cells of the Aging Retina against Oxidative Stress-Induced Degeneration through the Regulation of Senescence and Mitochondrial Quality Control. The Significance for AMD Pathogenesis. Int. J. Mol. Sci. 2018, 19, 2317. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 100858. [Google Scholar] [CrossRef]
- Johnson, S.C. Nutrient Sensing, Signaling and Ageing: The Role of IGF-1 and mTOR in Ageing and Age-Related Disease. Sub-Cell. Biochem. 2018, 90, 49–97. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Calorie restriction: Decelerating mTOR-driven aging from cells to organisms (including humans). Cell Cycle 2010, 9, 683–688. [Google Scholar] [CrossRef]
- Vestergaard, P.F.; Hansen, M.; Frystyk, J.; Espelund, U.; Christiansen, J.S.; Jørgensen, J.O.; Fisker, S. Serum levels of bioactive IGF1 and physiological markers of ageing in healthy adults. Eur. J. Endocrinol. 2014, 170, 229–236. [Google Scholar] [CrossRef]
- Kenyon, C. The plasticity of aging: Insights from long-lived mutants. Cell 2005, 120, 449–460. [Google Scholar] [CrossRef]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cao, J.; Hu, K.; He, X.; Yun, D.; Tong, T.; Han, L. Sirtuins and their Biological Relevance in Aging and Age-Related Diseases. Aging Dis. 2020, 11, 927–945. [Google Scholar] [CrossRef]
- Anderson, K.A.; Madsen, A.S.; Olsen, C.A.; Hirschey, M.D. Metabolic control by sirtuins and other enzymes that sense NAD(+), NADH, or their ratio. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H. Mechanisms and disease implications of sirtuin-mediated autophagic regulation. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Poulose, N.; Raju, R. Sirtuin regulation in aging and injury. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2442–2455. [Google Scholar] [CrossRef] [PubMed]
- Fusco, S.; Maulucci, G.; Pani, G. Sirt1: Def-eating senescence? Cell Cycle 2012, 11, 4135–4146. [Google Scholar] [CrossRef]
- Gurd, B.J. Deacetylation of PGC-1α by SIRT1: Importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl. Physiol. Nutr. Metab. 2011, 36, 589–597. [Google Scholar] [CrossRef]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef]
- Kjøbsted, R.; Hingst, J.R.; Fentz, J.; Foretz, M.; Sanz, M.; Pehmøller, C.; Shum, M.; Marette, A.; Mounier, R.; Treebak, J.T.; et al. AMPK in skeletal muscle function and metabolism. FASEB J. 2018, 32, 1741–1777. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Age-related changes in AMPK activation: Role for AMPK phosphatases and inhibitory phosphorylation by upstream signaling pathways. Ageing Res. Rev. 2016, 28, 15–26. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Galluzzi, L.; Freije, J.M.P.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef] [PubMed]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef] [PubMed]
- Theurey, P.; Pizzo, P. The Aging Mitochondria. Genes 2018, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Simoes, I.C.M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jędrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar] [CrossRef]
- Dai, D.F.; Chiao, Y.A.; Martin, G.M.; Marcinek, D.J.; Basisty, N.; Quarles, E.K.; Rabinovitch, P.S. Mitochondrial-Targeted Catalase: Extended Longevity and the Roles in Various Disease Models. Prog. Mol. Biol. Transl. Sci. 2017, 146, 203–241. [Google Scholar] [CrossRef]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef]
- Lee, H.C.; Wei, Y.H. Oxidative stress, mitochondrial DNA mutation, and apoptosis in aging. Exp. Biol. Med. 2007, 232, 592–606. [Google Scholar]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.D.; Quain, D.E.; Smart, K.A. The impact of media composition and petite mutation on the longevity of a polyploid brewing yeast strain. Lett. Appl. Microbiol. 2000, 31, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Woo, D.K.; Poyton, R.O. The absence of a mitochondrial genome in rho0 yeast cells extends lifespan independently of retrograde regulation. Exp. Gerontol. 2009, 44, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.-S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef]
- Rambold, A.S.; Kostelecky, B.; Lippincott-Schwartz, J. Together we are stronger: Fusion protects mitochondria from autophagosomal degradation. Autophagy 2011, 7, 1568–1569. [Google Scholar] [CrossRef]
- Zuo, Z.; Jing, K.; Wu, H.; Wang, S.; Ye, L.; Li, Z.; Yang, C.; Pan, Q.; Liu, W.J.; Liu, H.F. Mechanisms and Functions of Mitophagy and Potential Roles in Renal Disease. Front. Physiol. 2020, 11, 935. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef]
- Purushotham, A.; Schug, T.T.; Li, X. SIRT1 performs a balancing act on the tight-rope toward longevity. Aging 2009, 1, 669–673. [Google Scholar] [CrossRef]
- Valero, T. Mitochondrial biogenesis: Pharmacological approaches. Curr. Pharm. Des. 2014, 20, 5507–5509. [Google Scholar] [CrossRef]
- Xiong, S.; Salazar, G.; Patrushev, N.; Ma, M.; Forouzandeh, F.; Hilenski, L.; Alexander, R.W. Peroxisome Proliferator-Activated Receptor γ Coactivator-1α Is a Central Negative Regulator of Vascular Senescence. Arter. Thromb. Vasc. Biol. 2013, 33, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.; Fang, E.F.; Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. DNA Damage, DNA Repair, Aging, and Neurodegeneration. Cold Spring Harb. Perspect. Med. 2015, 5, a025130. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; Vijg, J. Does Damage to DNA and Other Macromolecules Play a Role in Aging? If So, How? J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2009, 64A, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Beroukhim, R.; Golub, T.R. Genomic evolution of cancer models: Perils and opportunities. Nature reviews. Cancer 2019, 19, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Boulianne, B.; Feldhahn, N. Transcribing malignancy: Transcription-associated genomic instability in cancer. Oncogene 2018, 37, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Duijf, P.H.G.; Nanayakkara, D.; Nones, K.; Srihari, S.; Kalimutho, M.; Khanna, K.K. Mechanisms of Genomic Instability in Breast Cancer. Trends Mol. Med. 2019, 25, 595–611. [Google Scholar] [CrossRef]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef]
- Evangelakou, Z.; Manola, M.; Gumeni, S.; Trougakos, I.P. Nutrigenomics as a tool to study the impact of diet on aging and age-related diseases: The Drosophila approach. Genes Nutr. 2019, 14, 12. [Google Scholar] [CrossRef]
- Gorbunova, V.; Seluanov, A.; Mao, Z.; Hine, C. Changes in DNA repair during aging. Nucleic Acids Res. 2007, 35, 7466–7474. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Gurkar, A.U.; Wang, Y.; Vijg, J.; Hoeijmakers, J.H.J.; Robbins, P.D. Nuclear Genomic Instability and Aging. Annu. Rev. Biochem. 2018, 87, 295–322. [Google Scholar] [CrossRef]
- Vijg, J.; Suh, Y. Genome instability and aging. Annu. Rev. Physiol. 2013, 75, 645–668. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, I.P.; Golubov, A.; Koturbash, I.V.; Kutanzi, K.; Martin, O.A.; Kovalchuk, O. Age-dependent changes in DNA repair in radiation-exposed mice. Radiat. Res. 2014, 182, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.S.; Ikram, S.; Bibi, N.; Mir, A. Hutchinson-Gilford Progeria Syndrome: A Premature Aging Disease. Mol. Neurobiol. 2018, 55, 4417–4427. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F.; Migliore, L. DNA repair in premature aging disorders and neurodegeneration. Curr. Aging Sci. 2010, 3, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Kubben, N.; Misteli, T. Shared molecular and cellular mechanisms of premature ageing and ageing-associated diseases. Nature reviews. Mol. Cell Biol. 2017, 18, 595–609. [Google Scholar] [CrossRef]
- Oshima, J.; Sidorova, J.M.; Monnat, R.J., Jr. Werner syndrome: Clinical features, pathogenesis and potential therapeutic interventions. Ageing Res. Rev. 2017, 33, 105–114. [Google Scholar] [CrossRef]
- Tiwari, V.; Wilson, D.M., 3rd. DNA Damage and Associated DNA Repair Defects in Disease and Premature Aging. Am. J. Hum. Genet. 2019, 105, 237–257. [Google Scholar] [CrossRef]
- Andressoo, J.O.; Hoeijmakers, J.H.; Mitchell, J.R. Nucleotide excision repair disorders and the balance between cancer and aging. Cell Cycle 2006, 5, 2886–2888. [Google Scholar] [CrossRef]
- de Boer, J.; Andressoo, J.O.; de Wit, J.; Huijmans, J.; Beems, R.B.; van Steeg, H.; Weeda, G.; van der Horst, G.T.; van Leeuwen, W.; Themmen, A.P.; et al. Premature aging in mice deficient in DNA repair and transcription. Science 2002, 296, 1276–1279. [Google Scholar] [CrossRef]
- Garinis, G.A.; van der Horst, G.T.; Vijg, J.; Hoeijmakers, J.H. DNA damage and ageing: New-age ideas for an age-old problem. Nat. Cell Biol. 2008, 10, 1241–1247. [Google Scholar] [CrossRef]
- Gredilla, R.; Garm, C.; Stevnsner, T. Nuclear and mitochondrial DNA repair in selected eukaryotic aging model systems. Oxidative Med. Cell. Longev. 2012, 2012, 282438. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, B.; van der Pluijm, I.; Moorhouse, M.J.; Kosteas, T.; Robinson, A.R.; Suh, Y.; Breit, T.M.; van Steeg, H.; Niedernhofer, L.J.; van IJcken, W.; et al. Delayed and Accelerated Aging Share Common Longevity Assurance Mechanisms. PLoS Genet. 2008, 4, e1000161. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.L.; Ghosh, A.K.; Bohr, V.A. Roles of Werner syndrome protein in protection of genome integrity. Dna Repair 2010, 9, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Maier, B.; Gluba, W.; Bernier, B.; Turner, T.; Mohammad, K.; Guise, T.; Sutherland, A.; Thorner, M.; Scrable, H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004, 18, 306–319. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Garinis, G.A.; Raams, A.; Lalai, A.S.; Robinson, A.R.; Appeldoorn, E.; Odijk, H.; Oostendorp, R.; Ahmad, A.; van Leeuwen, W.; et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 2006, 444, 1038–1043. [Google Scholar] [CrossRef]
- Palazzo, R.P.; Jardim, L.B.; Bacellar, A.; de Oliveira, F.R.; Maraslis, F.T.; Pereira, C.H.J.; da Silva, J.; Maluf, S.W. DNA damage and repair in individuals with ataxia-telangiectasia and their parents. Mutat. Res. Genet. Toxicol. Environ. Mutagenesis 2018, 836, 122–126. [Google Scholar] [CrossRef]
- Marechal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Zhong, H.-H.; Hu, S.-J.; Yu, B.; Jiang, S.-S.; Zhang, J.; Luo, D.; Yang, M.-W.; Su, W.-Y.; Shao, Y.-L.; Deng, H.-L.; et al. Apoptosis in the aging liver. Oncotarget 2017, 8, 102640–102652. [Google Scholar] [CrossRef]
- Choy, K.R.; Watters, D.J. Neurodegeneration in ataxia-telangiectasia: Multiple roles of ATM kinase in cellular homeostasis. Dev. Dyn. 2018, 247, 33–46. [Google Scholar] [CrossRef]
- Pizzamiglio, L.; Focchi, E.; Antonucci, F. ATM Protein Kinase: Old and New Implications in Neuronal Pathways and Brain Circuitry. Cells 2020, 9, 1969. [Google Scholar] [CrossRef] [PubMed]
- Moreno, D.F.; Jenkins, K.; Morlot, S.; Charvin, G.; Csikasz-Nagy, A.; Aldea, M. Proteostasis collapse, a hallmark of aging, hinders the chaperone-Start network and arrests cells in G1. eLife 2019, 8, e48240. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.C.; Grosso, R.A.; Fader, C.M. Hallmarks of Aging: An Autophagic Perspective. Front. Endocrinol. 2019, 9, 790. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nature reviews. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshimori, T. Autophagy and Longevity. Mol. Cells 2018, 41, 65–72. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy participates in, well, just about everything. Cell Death Differ. 2020, 27, 831–832. [Google Scholar] [CrossRef]
- Metaxakis, A.; Ploumi, C.; Tavernarakis, N. Autophagy in Age-Associated Neurodegeneration. Cells 2018, 7, 37. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Shane Anderson, A.; Loeser, R.F. Why is osteoarthritis an age-related disease? Best Pr. Res. Clin. Rheumatol. 2010, 24, 15–26. [Google Scholar] [CrossRef]
- Sacitharan, P.K.; Bou-Gharios, G.; Edwards, J.R. SIRT1 directly activates autophagy in human chondrocytes. Cell Death Discov. 2020, 6, 41. [Google Scholar] [CrossRef]
- Diot, A.; Morten, K.; Poulton, J. Mitophagy plays a central role in mitochondrial ageing. Mamm. Genome 2016, 27, 381–395. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, T.J.; Hearon, C.M., Jr.; Henson, G.D.; Seals, D.R. Mitochondrial quality control and age-associated arterial stiffening. Exp. Gerontol. 2014, 58, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Bhutto, I.; Lutty, G. Understanding age-related macular degeneration (AMD): Relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol. Asp. Med. 2012, 33, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.R.; Ferrington, D.A. Perspective on AMD Pathobiology: A Bioenergetic Crisis in the RPE. Investig. Opthalmology Vis. Sci. 2018, 59, AMD41. [Google Scholar] [CrossRef]
- Handa, J.T.; Bowes Rickman, C.; Dick, A.D.; Gorin, M.B.; Miller, J.W.; Toth, C.A.; Ueffing, M.; Zarbin, M.; Farrer, L.A. A systems biology approach towards understanding and treating non-neovascular age-related macular degeneration. Nat. Commun. 2019, 10, 3347. [Google Scholar] [CrossRef]
- Ao, J.; Wood, J.P.; Chidlow, G.; Gillies, M.C.; Casson, R.J. Retinal pigment epithelium in the pathogenesis of age-related macular degeneration and photobiomodulation as a potential therapy? Clin. Exp. Ophthalmol. 2018, 46, 670–686. [Google Scholar] [CrossRef]
- Desmettre, T.J. Epigenetics in Age-related Macular Degeneration (AMD). J. Fr. Ophtalmol. 2018, 41, e407–e415. [Google Scholar] [CrossRef]
- Mansoor, N.; Wahid, F.; Azam, M.; Shah, K.; den Hollander, A.I.; Qamar, R.; Ayub, H. Molecular Mechanisms of Complement System Proteins and Matrix Metalloproteinases in the Pathogenesis of Age-Related Macular Degeneration. Curr. Mol. Med. 2019, 19, 705–718. [Google Scholar] [CrossRef]
- Wang, S.; Wang, X.; Cheng, Y.; Ouyang, W.; Sang, X.; Liu, J.; Su, Y.; Liu, Y.; Li, C.; Yang, L.; et al. Autophagy Dysfunction, Cellular Senescence, and Abnormal Immune-Inflammatory Responses in AMD: From Mechanisms to Therapeutic Potential. Oxidative Med. Cell. Longev. 2019, 2019, 3632169. [Google Scholar] [CrossRef]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Salminen, A.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Age-Related Macular Degeneration (AMD): Alzheimer’s Disease in the Eye? J. Alzheimer’s Dis. 2011, 24, 615–631. [Google Scholar] [CrossRef] [PubMed]
- Ratnayaka, J.A.; Serpell, L.C.; Lotery, A.J. Dementia of the eye: The role of amyloid beta in retinal degeneration. Eye 2015, 29, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Morgese, M.G.; Schiavone, S.; Trabace, L. Emerging role of amyloid beta in stress response: Implication for depression and diabetes. Eur. J. Pharmacol. 2017, 817, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative Stress, Amyloid-β Peptide, and Altered Key Molecular Pathways in the Pathogenesis and Progression of Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1345–1367. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef]
- Park, D.H.; Connor, K.M.; Lambris, J.D. The Challenges and Promise of Complement Therapeutics for Ocular Diseases. Front. Immunol. 2019, 10, 1007. [Google Scholar] [CrossRef]
- Grassmann, F.; Heid, I.M.; Weber, B.H.F.; International, A.M.D.G.C. Recombinant Haplotypes Narrow the ARMS2/HTRA1 Association Signal for Age-Related Macular Degeneration. Genetics 2017, 205, 919–924. [Google Scholar] [CrossRef]
- van Asten, F.; Chiu, C.Y.; Agrón, E.; Clemons, T.E.; Ratnapriya, R.; Swaroop, A.; Klein, M.L.; Fan, R.; Chew, E.Y. No CFH or ARMS2 Interaction with Omega-3 Fatty Acids, Low versus High Zinc, or β-Carotene versus Lutein and Zeaxanthin on Progression of Age-Related Macular Degeneration in the Age-Related Eye Disease Study 2: Age-Related Eye Disease Study 2 Report No. 18. Ophthalmology 2019, 126, 1541–1548. [Google Scholar] [CrossRef]
- Blasiak, J.; Pawlowska, E.; Chojnacki, J.; Szczepanska, J.; Chojnacki, C.; Kaarniranta, K. Zinc and Autophagy in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2020, 21, 4994. [Google Scholar] [CrossRef]
- Merle, B.M.; Silver, R.E.; Rosner, B.; Seddon, J.M. Adherence to a Mediterranean diet, genetic susceptibility, and progression to advanced macular degeneration: A prospective cohort study. Am. J. Clin. Nutr. 2015, 102, 1196–1206. [Google Scholar] [CrossRef]
- Keenan, T.D.; Agrón, E.; Mares, J.; Clemons, T.E.; van Asten, F.; Swaroop, A.; Chew, E.Y. Adherence to the Mediterranean Diet and Progression to Late Age-Related Macular Degeneration in the Age-Related Eye Disease Studies 1 and 2. Ophthalmology 2020, 127, 1515–1528. [Google Scholar] [CrossRef] [PubMed]
- Raimundo, M.; Mira, F.; Cachulo, M.D.L.; Barreto, P.; Ribeiro, L.; Farinha, C.; Laíns, I.; Nunes, S.; Alves, D.; Figueira, J.; et al. Adherence to a Mediterranean diet, lifestyle and age-related macular degeneration: The Coimbra Eye Study—report 3. Acta Ophthalmol 2018, 96, e926–e932. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, E.M.; Emri, E.; Merle, B.M.J.; Colijn, J.M.; Kersten, E.; Cougnard-Gregoire, A.; Dammeier, S.; Meester-Smoor, M.; Pool, F.M.; de Jong, E.K.; et al. A new perspective on lipid research in age-related macular degeneration. Prog. Retin. Eye Res. 2018, 67, 56–86. [Google Scholar] [CrossRef] [PubMed]
- Skowronska-Krawczyk, D.; Chao, D.L. Long-Chain Polyunsaturated Fatty Acids and Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2019, 1185, 39–43. [Google Scholar] [CrossRef]
- Hogg, R.E.; Woodside, J.V.; McGrath, A.; Young, I.S.; Vioque, J.L.; Chakravarthy, U.; de Jong, P.T.; Rahu, M.; Seland, J.; Soubrane, G.; et al. Mediterranean Diet Score and Its Association with Age-Related Macular Degeneration: The European Eye Study. Ophthalmology 2017, 124, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Jiang, N.; Chu, Y.; Postnikova, O.; Varghese, R.; Horvath, A.; Cheema, A.K.; Golestaneh, N. Dysregulated metabolic pathways in age-related macular degeneration. Sci. Rep. 2020, 10, 2464. [Google Scholar] [CrossRef]
- Bonelli, R.; Woods, S.M.; Ansell, B.R.E.; Heeren, T.F.C.; Egan, C.A.; Khan, K.N.; Guymer, R.; Trombley, J.; Friedlander, M.; Bahlo, M.; et al. Systemic lipid dysregulation is a risk factor for macular neurodegenerative disease. Sci. Rep. 2020, 10, 12165. [Google Scholar] [CrossRef]
- Golestaneh, N.; Chu, Y.; Cheng, S.K.; Cao, H.; Poliakov, E.; Berinstein, D.M. Repressed SIRT1/PGC-1α pathway and mitochondrial disintegration in iPSC-derived RPE disease model of age-related macular degeneration. J. Transl. Med. 2016, 14, 344. [Google Scholar] [CrossRef]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell. Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef]
- Hernández-Sánchez, C.; López-Carranza, A.; Alarcón, C.; de La Rosa, E.J.; de Pablo, F. Autocrine/paracrine role of insulin-related growth factors in neurogenesis: Local expression and effects on cell proliferation and differentiation in retina. Proc. Natl. Acad. Sci. USA 1995, 92, 9834–9838. [Google Scholar] [CrossRef]
- Lambooij, A.C.; van Wely, K.H.; Lindenbergh-Kortleve, D.J.; Kuijpers, R.W.; Kliffen, M.; Mooy, C.M. Insulin-like growth factor-I and its receptor in neovascular age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2192–2198. [Google Scholar] [CrossRef] [PubMed]
- Arroba, A.I.; Wallace, D.; Mackey, A.; de la Rosa, E.J.; Cotter, T.G. IGF-I maintains calpastatin expression and attenuates apoptosis in several models of photoreceptor cell death. Eur. J. Neurosci. 2009, 30, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Arroba, A.I.; Campos-Caro, A.; Aguilar-Diosdado, M.; Valverde, Á.M. IGF-1, Inflammation and Retinal Degeneration: A Close Network. Front. Aging Neurosci. 2018, 10, 203. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.J.; Conley, Y.P.; Gorin, M.B.; Gensler, G.; Lai, C.Q.; Shang, F.; Taylor, A. Associations between genetic polymorphisms of insulin-like growth factor axis genes and risk for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9099–9107. [Google Scholar] [CrossRef] [PubMed]
- Arroba, A.I.; Rodríguez-de la Rosa, L.; Murillo-Cuesta, S.; Vaquero-Villanueva, L.; Hurlé, J.M.; Varela-Nieto, I.; Valverde, Á.M. Autophagy resolves early retinal inflammation in Igf1-deficient mice. Dis. Models Mech. 2016, 9, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Castellino, N.; Longo, A.; Avitabile, T.; Russo, A.; Fallico, M.; Bonfiglio, V.; Toro, M.D.; Rejdak, R.; Nowomiejska, K.; Murabito, P.; et al. Circulating insulin-like growth factor-1: A new clue in the pathogenesis of age-related macular degeneration. Aging 2018, 10, 4241–4247. [Google Scholar] [CrossRef]
- Narayan, D.S.; Chidlow, G.; Wood, J.P.; Casson, R.J. Glucose metabolism in mammalian photoreceptor inner and outer segments. Clin. Exp. Ophthalmol. 2017, 45, 730–741. [Google Scholar] [CrossRef]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef]
- Samant, R.S.; Livingston, C.M.; Sontag, E.M.; Frydman, J. Distinct proteostasis circuits cooperate in nuclear and cytoplasmic protein quality control. Nature 2018, 563, 407–411. [Google Scholar] [CrossRef]
- Shin, W.H.; Park, J.H.; Chung, K.C. The central regulator p62 between ubiquitin proteasome system and autophagy and its role in the mitophagy and Parkinson’s disease. BMB Rep. 2020, 53, 56–63. [Google Scholar] [CrossRef]
- Felszeghy, S.; Viiri, J.; Paterno, J.J.; Hyttinen, J.M.T.; Koskela, A.; Chen, M.; Leinonen, H.; Tanila, H.; Kivinen, N.; Koistinen, A.; et al. Loss of NRF-2 and PGC-1α genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.T.M.; Kannan, R.; Felszeghy, S.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. The Regulation of NFE2L2 (NRF2) Signalling and Epithelial-to-Mesenchymal Transition in Age-Related Macular Degeneration Pathology. Int. J. Mol. Sci. 2019, 20, 5800. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Sinha, D.; Blasiak, J.; Kauppinen, A.; Vereb, Z.; Salminen, A.; Boulton, M.E.; Petrovski, G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013, 9, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Jia, L.; Khan, N.; Lin, C.; Mitter, S.K.; Boulton, M.E.; Dunaief, J.L.; Klionsky, D.J.; Guan, J.L.; Thompson, D.A.; et al. Deletion of autophagy inducer RB1CC1 results in degeneration of the retinal pigment epithelium. Autophagy 2015, 11, 939–953. [Google Scholar] [CrossRef]
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef]
- Kosmidou, C.; Efstathiou, N.E.; Hoang, M.V.; Notomi, S.; Konstantinou, E.K.; Hirano, M.; Takahashi, K.; Maidana, D.E.; Tsoka, P.; Young, L.; et al. Issues with the Specificity of Immunological Reagents for NLRP3: Implications for Age-related Macular Degeneration. Sci. Rep. 2018, 8, 461. [Google Scholar] [CrossRef]
- Denton, D.; Xu, T.; Kumar, S. Autophagy as a pro-death pathway. Immunol. Cell Biol. 2015, 93, 35–42. [Google Scholar] [CrossRef]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef]
- Jang, K.H.; Hwang, Y.; Kim, E. PARP1 Impedes SIRT1-Mediated Autophagy during Degeneration of the Retinal Pigment Epithelium under Oxidative Stress. Mol. Cells 2020, 43, 632–644. [Google Scholar] [CrossRef]
- Cui, R.; Tian, L.; Lu, D.; Li, H.; Cui, J. Exendin-4 Protects Human Retinal Pigment Epithelial Cells from H2O2-Induced Oxidative Damage via Activation of NRF2 Signaling. Ophthalmic Res. 2020, 63, 404–412. [Google Scholar] [CrossRef]
- Gong, C.; Qiao, L.; Feng, R.; Xu, Q.; Zhang, Y.; Fang, Z.; Shen, J.; Li, S. IL-6-induced acetylation of E2F1 aggravates oxidative damage of retinal pigment epithelial cell line. Exp. Eye Res. 2020, 200, 108219. [Google Scholar] [CrossRef] [PubMed]
- Mahendra, C.K.; Tan, L.T.H.; Pusparajah, P.; Htar, T.T.; Chuah, L.H.; Lee, V.S.; Low, L.E.; Tang, S.Y.; Chan, K.G.; Goh, B.H. Detrimental Effects of UVB on Retinal Pigment Epithelial Cells and Its Role in Age-Related Macular Degeneration. Oxidative Med. Cell. Longev. 2020, 2020, 1904178. [Google Scholar] [CrossRef] [PubMed]
- Muangnoi, C.; Sharif, U.; Ratnatilaka Na Bhuket, P.; Rojsitthisak, P.; Paraoan, L. Protective Effects of Curcumin Ester Prodrug, Curcumin Diethyl Disuccinate against H2O2-Induced Oxidative Stress in Human Retinal Pigment Epithelial Cells: Potential Therapeutic Avenues for Age-Related Macular Degeneration. Int. J. Mol. Sci. 2019, 20, 3367. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wei, Q.; Yu, J. The cGAS/STING pathway: A sensor of senescence-associated DNA damage and trigger of inflammation in early age-related macular degeneration. Clin. Interv. Aging 2019, 14, 1277–1283. [Google Scholar] [CrossRef]
- Murphy, M.P. Modulating mitochondrial intracellular location as a redox signal. Sci. Signal. 2012, 5, pe39. [Google Scholar] [CrossRef]
- Pinto, M.; Moraes, C.T. Mechanisms linking mtDNA damage and aging. Free Radic. Biol. Med. 2015, 85, 250–258. [Google Scholar] [CrossRef]
- Hiona, A.; Leeuwenburgh, C. The role of mitochondrial DNA mutations in aging and sarcopenia: Implications for the mitochondrial vicious cycle theory of aging. Exp. Gerontol. 2008, 43, 24–33. [Google Scholar] [CrossRef]
- Lin, H.; Xu, H.; Liang, F.Q.; Liang, H.; Gupta, P.; Havey, A.N.; Boulton, M.E.; Godley, B.F. Mitochondrial DNA damage and repair in RPE associated with aging and age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3521–3529. [Google Scholar] [CrossRef]
- Brown, E.E.; Lewin, A.S.; Ash, J.D. Mitochondria: Potential Targets for Protection in Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2018, 1074, 11–17. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Fisher, C.R.; Kowluru, R.A. Mitochondrial Defects Drive Degenerative Retinal Diseases. Trends Mol. Med. 2020, 26, 105–118. [Google Scholar] [CrossRef]
- Hyttinen, J.M.T.; Viiri, J.; Kaarniranta, K.; Błasiak, J. Mitochondrial quality control in AMD: Does mitophagy play a pivotal role? Cell. Mol. Life Sci. 2018, 75, 2991–3008. [Google Scholar] [CrossRef] [PubMed]
- Somasundaran, S.; Constable, I.J.; Mellough, C.B.; Carvalho, L.S. Retinal pigment epithelium and age-related macular degeneration: A review of major disease mechanisms. Clin. Exp. Ophthalmol. 2020, ceo.13834. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Ebeling, M.C.; Kapphahn, R.J.; Terluk, M.R.; Fisher, C.R.; Polanco, J.R.; Roehrich, H.; Leary, M.M.; Geng, Z.; Dutton, J.R.; et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 2017, 13, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Golestaneh, N.; Chu, Y.; Xiao, Y.Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2017, 8, e2537. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, H.; Hoek, J.B. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 2017, 16, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L. Mitochondrial DNA degradation: A quality control measure for mitochondrial genome maintenance and stress response. Enzymes 2019, 45, 311–341. [Google Scholar] [CrossRef]
- Brown, E.E.; DeWeerd, A.J.; Ildefonso, C.J.; Lewin, A.S.; Ash, J.D. Mitochondrial oxidative stress in the retinal pigment epithelium (RPE) led to metabolic dysfunction in both the RPE and retinal photoreceptors. Redox Biol. 2019, 24, 101201. [Google Scholar] [CrossRef]
- Egger, A.; Samardzija, M.; Sothilingam, V.; Tanimoto, N.; Lange, C.; Salatino, S.; Fang, L.; Garcia-Garrido, M.; Beck, S.; Okoniewski, M.J.; et al. PGC-1α determines light damage susceptibility of the murine retina. PLoS ONE 2012, 7, e31272. [Google Scholar] [CrossRef]
- Rosales, M.A.B.; Shu, D.Y.; Iacovelli, J.; Saint-Geniez, M. Loss of PGC-1α in RPE induces mesenchymal transition and promotes retinal degeneration. Life Sci. Alliance 2019, 2, e201800212. [Google Scholar] [CrossRef]
- Satish, S.; Philipose, H.; Rosales, M.A.B.; Saint-Geniez, M. Pharmaceutical Induction of PGC-1α Promotes Retinal Pigment Epithelial Cell Metabolism and Protects against Oxidative Damage. Oxidative Med. Cell. Longev. 2018. [Google Scholar] [CrossRef]
- Sridevi Gurubaran, I.; Viiri, J.; Koskela, A.; Hyttinen, J.M.T.; Paterno, J.J.; Kis, G.; Antal, M.; Urtti, A.; Kauppinen, A.; Felszeghy, S.; et al. Mitophagy in the Retinal Pigment Epithelium of Dry Age-Related Macular Degeneration Investigated in the NFE2L2/PGC-1α(-/-) Mouse Model. Int. J. Mol. Sci. 2020, 21, 1976. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blasiak, J.; Pawlowska, E.; Sobczuk, A.; Szczepanska, J.; Kaarniranta, K. The Aging Stress Response and Its Implication for AMD Pathogenesis. Int. J. Mol. Sci. 2020, 21, 8840. https://doi.org/10.3390/ijms21228840
Blasiak J, Pawlowska E, Sobczuk A, Szczepanska J, Kaarniranta K. The Aging Stress Response and Its Implication for AMD Pathogenesis. International Journal of Molecular Sciences. 2020; 21(22):8840. https://doi.org/10.3390/ijms21228840
Chicago/Turabian StyleBlasiak, Janusz, Elzbieta Pawlowska, Anna Sobczuk, Joanna Szczepanska, and Kai Kaarniranta. 2020. "The Aging Stress Response and Its Implication for AMD Pathogenesis" International Journal of Molecular Sciences 21, no. 22: 8840. https://doi.org/10.3390/ijms21228840
APA StyleBlasiak, J., Pawlowska, E., Sobczuk, A., Szczepanska, J., & Kaarniranta, K. (2020). The Aging Stress Response and Its Implication for AMD Pathogenesis. International Journal of Molecular Sciences, 21(22), 8840. https://doi.org/10.3390/ijms21228840