Crizotinib Resistance Mediated by Autophagy Is Higher in the Stem-Like Cell Subset in ALK-Positive Anaplastic Large Cell Lymphoma, and This Effect Is MYC-Dependent

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
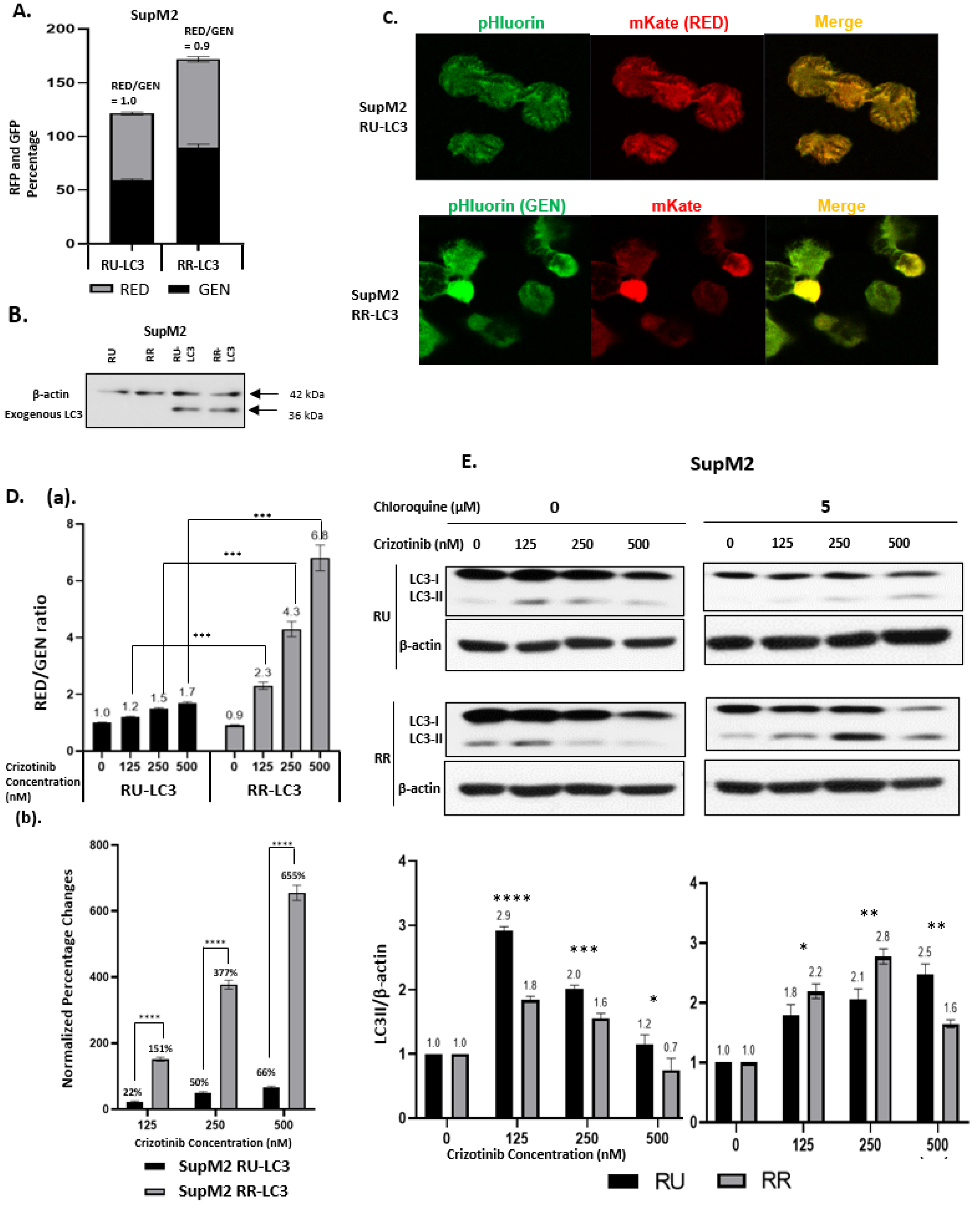
2.1. Generation of LC3-Transfected RR and RU Cells
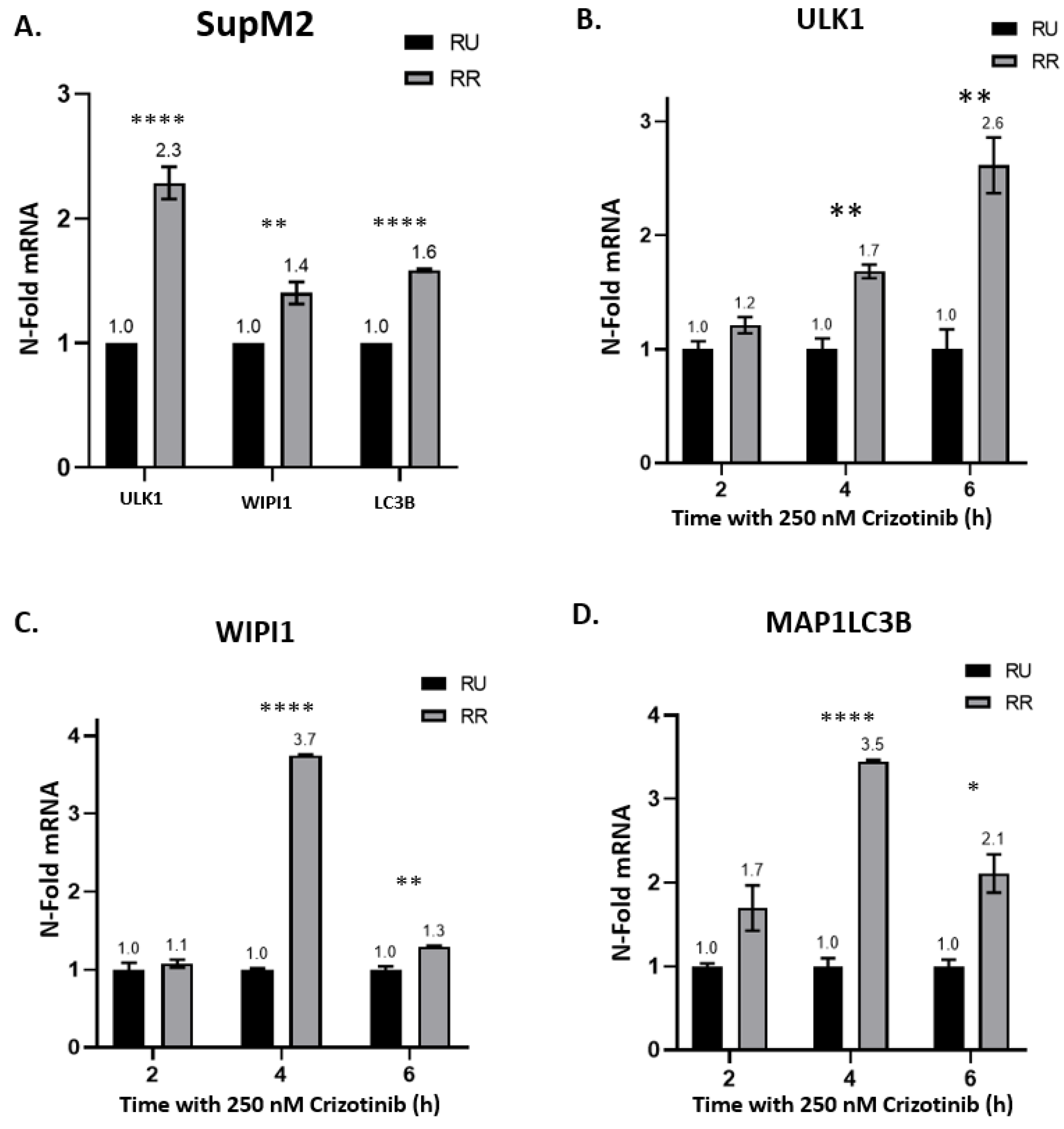
2.2. Crizotinib-Induced Autophagy Is Significantly Enhanced in RR Cells
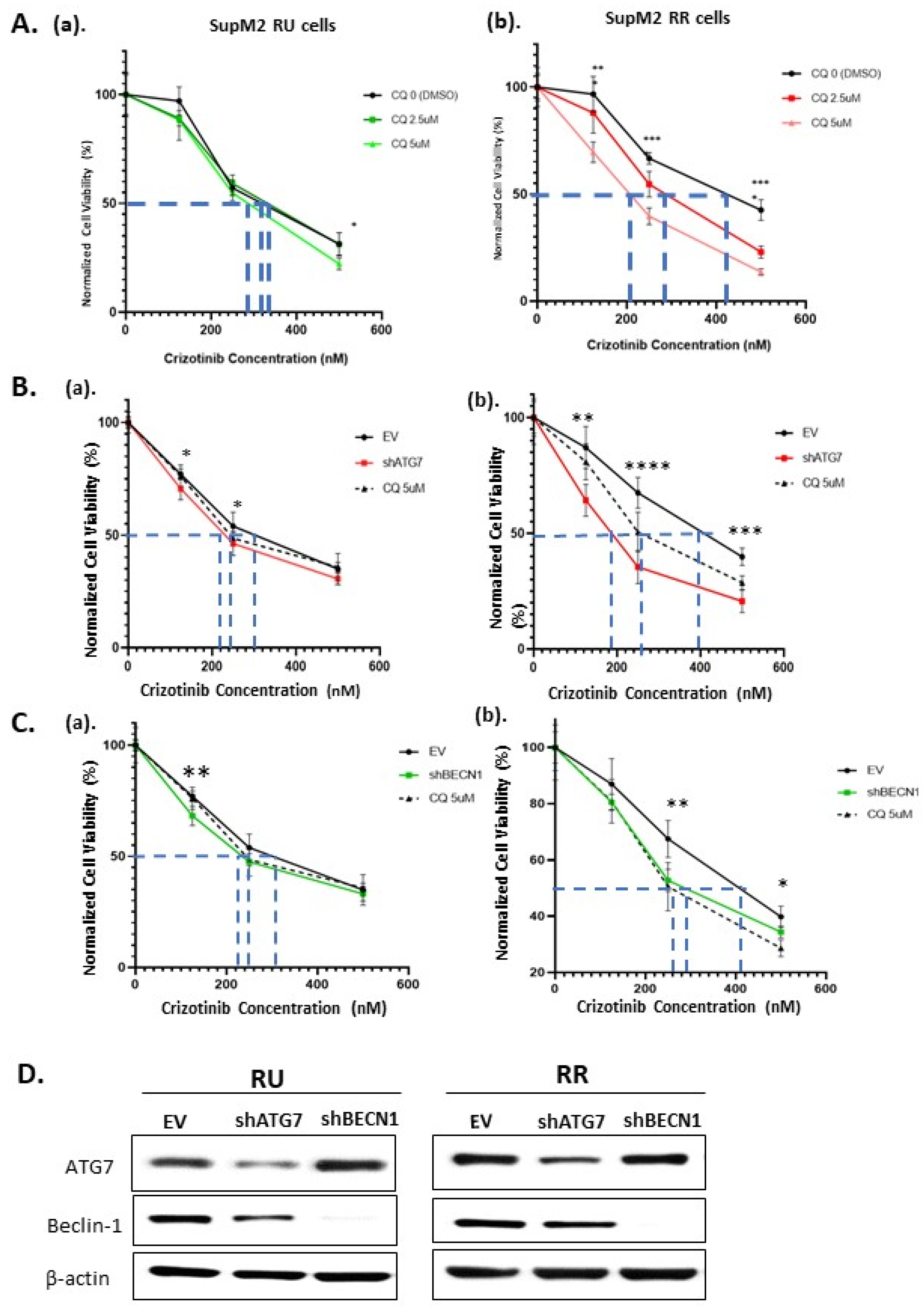
2.3. The Autophagy Response in RR Cells Contributes to Crizotinib Resistance
2.4. The Loss of Cell Viability in RR Cells Is Due to Autophagy Inhibition
2.5. Higher MYC Expression Enhances Crizotinib Chemoresistance via Autophagy in RR Cells
3. Discussion
4. Materials and Methods
4.1. Culture of Cell Lines
4.2. Cell Viability Assays
4.3. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.4. Vectors and Plasmids
4.5. Lentiviral Based Transduction
4.6. Antibodies and Chemicals
4.7. Western Blot
4.8. Flow Cytometric Quantification of Autophagic Flux
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 1–32. [Google Scholar] [PubMed] [Green Version]
- Bialik, S.; Dasari, S.K.; Kimchi, A. Autophagy-dependent cell death—Where, how and why a cell eats itself to death. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Zhou, J. Aberrant regulation of autophagy in mammalian diseases. Biol. Lett. 2018, 14, 20170540. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D.J.; Garcia-Arencibia, M.; Hochfeld, W.E.; Rubinsztein, D.C. Autophagy and misfolded proteins in neurodegeneration. Exp. Neurol. 2012, 238, 22–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014, 157, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Wileman, T. Autophagy as a defence against intracellular pathogens. Essays Biochem. 2013, 55, 153–163. [Google Scholar]
- He, L.; Zhang, J.; Zhao, J.; Ma, N.; Kim, S.W.; Qiao, S.; Ma, X. Autophagy: The Last Defense against Cellular Nutritional Stress. Adv. Nutr. 2018, 9, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.H.; Young, L.N. Mechanisms of Autophagy Initiation. Annu. Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Ho, C.J.; Gorski, S.M. Molecular Mechanisms Underlying Autophagy-Mediated Treatment Resistance in Cancer. Cancers 2019, 11, 1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maycotte, P.; Aryal, S.; Cummings, C.T.; Thorburn, J.; Morgan, M.J.; Thorburn, A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 2012, 8, 200–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.G.; Macleod, K.F. Autophagy, cancer stem cells and drug resistance. J. Pathol. 2019, 247, 708–718. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Quintanilla-Martinez, L. The 2016 updated WHO classification of lymphoid neoplasias. Hematol. Oncol. 2017, 1, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Montes-Mojarro, I.A.; Steinhilber, J.; Bonzheim, I.; Quintanilla-Martinez, L.; Fend, F. The Pathological Spectrum of Systemic Anaplastic Large Cell Lymphoma (ALCL). Cancers 2018, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Lamant, L.; Meggetto, F.; al Saati, T.; Brugieres, L.; de Paillerets, B.B.; Dastugue, N.; Bernheim, A.; Rubie, H.; Terrier-Lacombe, M.J.; Robert, A.; et al. High incidence of the t(2;5)(p23;q35) translocation in anaplastic large cell lymphoma and its lack of detection in Hodgkin’s disease. Comparison of cytogenetic analysis, reverse transcriptase-polymerase chain reaction, and P-80 immunostaining. Blood 1996, 87, 284–291. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L. Look AT Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef]
- Werner, M.T.; Zhao, C.; Zhang, Q.; Wasik, M.A. Nucleophosmin-anaplastic lymphoma kinase: The ultimate oncogene and therapeutic target. Blood 2017, 129, 823–831. [Google Scholar] [CrossRef] [Green Version]
- Bonzheim, I.; Steinhilber, J.; Fend, F.; Lamant, L.; Quintanilla-Martinez, L. ALK-positive anaplastic large cell lymphoma: An evolving story. Front. Biosci. 2015, 7, 248–259. [Google Scholar]
- Aubry, A.; Galiacy, S.; Allouche, M. Targeting ALK in Cancer: Therapeutic Potential of Proapoptotic Peptides. Cancers 2019, 11, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, R. Drug resistance in anaplastic lymphoma kinase-rearranged lung cancer. Cancer Sci. 2018, 109, 572–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccon, M.; Mologni, L.; Bisson, W.; Scapozza, L.; Gambacorti-Passerini, C. Crizotinib-resistant NPM-ALK mutants confer differential sensitivity to unrelated Alk inhibitors. Mol. Cancer Res. 2013, 11, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, R.; Lovly, C.M.; Shaw, A.T. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer: A paradigm for precision cancer medicine. Clin. Cancer Res. 2015, 21, 2227–2235. [Google Scholar] [CrossRef] [Green Version]
- Strullu, M.; Thomas, C.; Le Deley, M.C.; Chevance, A.; Kanold, J.; Bertrand, Y.; Jubert, C.; Dalle, J.H.; Paillard, C.; Baruchel, A.; et al. Hematopoietic stem cell transplantation in relapsed ALK+ anaplastic large cell lymphoma in children and adolescents: A study on behalf of the SFCE and SFGM-TC. Bone Marrow Transpl. 2015, 50, 795–801. [Google Scholar] [CrossRef]
- Torossian, A.; Broin, N.; Frentzel, J.; Daugrois, C.; Gandarillas, S.; Saati, T.A.; Lamant, L.; Brousset, P.; Giuriato, S.; Espinos, E. Blockade of crizotinib-induced BCL2 elevation in ALK-positive anaplastic large cell lymphoma triggers autophagy associated with cell death. Haematologica 2019, 104, 1428–1439. [Google Scholar] [CrossRef]
- Gelebart, P.; Hegazy, S.A.; Wang, P.; Bone, K.M.; Anand, M.; Sharon, D.; Hitt, M.; Pearson, J.D.; Ingham, R.J.; Ma, Y.; et al. Aberrant expression and biological significance of Sox2, an embryonic stem cell transcriptional factor, in ALK-positive anaplastic large cell lymphoma. Blood Cancer J. 2012, 2, e82. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Zhang, H.F.; Gupta, N.; Alshareef, A.; Wang, Q.; Huang, Y.H.; Lewis, J.T.; Douglas, D.N.; Kneteman, N.M.; Lai, R. A positive feedback loop involving the Wnt/beta-catenin/MYC/Sox2 axis defines a highly tumorigenic cell subpopulation in ALK-positive anaplastic large cell lymphoma. J. Hematol. Oncol. 2016, 9, 120. [Google Scholar] [CrossRef] [Green Version]
- Mitou, G.; Frentzel, J.; Desquesnes, A.; Le Gonidec, S.; AlSaati, T.; Beau, I.; Lamant, L.; Meggetto, F.; Espinos, E.; Codogno, P.; et al. Targeting autophagy enhances the anti-tumoral action of crizotinib in ALK-positive anaplastic large cell lymphoma. Oncotarget 2015, 6, 30149–30164. [Google Scholar] [CrossRef]
- Gupta, N.; Jung, K.; Wu, C.; Alshareef, A.; Alqahtani, H.; Damaraju, S.; Mackey, J.R.; Ghosh, S.; Sabri, S.; Abdulkarim, B.S.; et al. High Myc expression and transcription activity underlies intra-tumoral heterogeneity in triple-negative breast cancer. Oncotarget 2017, 8, 28101–28115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Gupta, N.; Huang, Y.H.; Zhang, H.F.; Alshareef, A.; Chow, A.; Lai, R. Oxidative stress enhances tumorigenicity and stem-like features via the activation of the Wnt/beta-catenin/MYC/Sox2 axis in ALK-positive anaplastic large-cell lymphoma. BMC Cancer 2018, 18, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanida, I.; Ueno, T.; Uchiyama, Y. A super-ecliptic, pHluorin-mKate2, tandem fluorescent protein-tagged human LC3 for the monitoring of mammalian autophagy. PLoS ONE 2014, 9, e110600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.; Gupta, N.; Wang, P.; Lewis, J.T.; Gopal, K.; Wu, F.; Ye, X.; Alshareef, A.; Abdulkarim, B.S.; Douglas, D.N.; et al. Triple negative breast cancers comprise a highly tumorigenic cell subpopulation detectable by its high responsiveness to a Sox2 regulatory region 2 (SRR2) reporter. Oncotarget 2015, 6, 10366–10373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [PubMed] [Green Version]
- Turco, E.; Fracchiolla, D.; Martens, S. Recruitment and Activation of the ULK1/Atg1 Kinase Complex in Selective Autophagy. J. Mol. Biol. 2020, 432, 123–134. [Google Scholar] [CrossRef]
- Mehrpour, M.; Botti, J.; Codogno, P. Mechanisms and regulation of autophagy in mammalian cells. Atlas Genet. Cytogenet. Oncol. Haematol. 2012. [Google Scholar] [CrossRef] [Green Version]
- Tsuyuki, S.; Takabayashi, M.; Kawazu, M.; Kudo, K.; Watanabe, A.; Nagata, Y.; Kusama, Y.; Yoshida, K. Detection of WIPI1 mRNA as an indicator of autophagosome formation. Autophagy 2014, 10, 497–513. [Google Scholar] [CrossRef] [Green Version]
- Noda, T.; Fujita, N.; Yoshimori, T. The late stages of autophagy: How does the end begin? Cell Death Differ. 2009, 16, 984–990. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, S.R.; Simonsen, A. Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, K.; Suzuki, N.N.; Fujioka, Y.; Mizushima, N.; Ohsumi, Y.; Inagaki, F. The crystal structure of microtubule-associated protein light chain 3, a mammalian homologue of Saccharomyces cerevisiae Atg8. Genes Cells 2004, 9, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef] [PubMed]
- Tsuneoka, M.; Umata, T.; Kimura, H.; Koda, Y.; Nakajima, M.; Kosai, K.; Takahashi, T.; Takahashi, Y.; Yamamoto, A. c-myc induces autophagy in rat 3Y1 fibroblast cells. Cell Struct. Funct. 2003, 28, 195–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, H.; He, J.; Yuan, Z.; Wu, Z.; Liu, B.; Lin, X.; Guan, J. PLK1 contributes to autophagy by regulating MYC stabilization in osteosarcoma cells. Onco Targets Ther. 2019, 12, 7527–7536. [Google Scholar] [CrossRef] [Green Version]
- Toh, P.P.; Luo, S.; Menzies, F.M.; Rasko, T.; Wanker, E.E.; Rubinsztein, D.C. Myc inhibition impairs autophagosome formation. Hum. Mol. Genet. 2013, 22, 5237–5248. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Yuwen, D.; Chen, J.; Zheng, B.; Gao, J.; Fan, M.; Xue, W.; Wang, Y.; Li, W.; Shu, Y.; et al. Exosomal Transfer Of Cisplatin-Induced miR-425-3p Confers Cisplatin Resistance In NSCLC Through Activating Autophagy. Int. J. Nanomed. 2019, 14, 8121–8132. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Li, J.; Zhou, L.; Han, J.; Liu, R.; Zhang, H.; Ning, T.; Gao, Z.; Liu, B.; Chen, X.; et al. The c-Myc/miR-27b-3p/ATG10 regulatory axis regulates chemoresistance in colorectal cancer. Theranostics 2020, 10, 1981–1996. [Google Scholar] [CrossRef]
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and cancer stem cells: Molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.J.; Glanville, J.; Knight, S.; Jacobsen, S.E.; et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.L.; Simon, A.K.; Prescott, M.; Menendez, J.A.; Liu, F.; Wang, F.; Wang, C.; Wolvetang, E.; Vazquez-Martin, A.; Zhang, J. Autophagy in stem cells. Autophagy 2013, 9, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R.; et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J. Clin. Investig. 2009, 119, 1109–1123. [Google Scholar] [CrossRef]
- Calabretta, B.; Salomoni, P. Inhibition of autophagy: A new strategy to enhance sensitivity of chronic myeloid leukemia stem cells to tyrosine kinase inhibitors. Leuk Lymphoma 2011, 52, 54–59. [Google Scholar] [CrossRef]
- Baquero, P.; Dawson, A.; Mukhopadhyay, A.; Kuntz, E.M.; Mitchell, R.; Olivares, O.; Ianniciello, A.; Scott, M.T.; Dunn, K.; Nicastri, M.C.; et al. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia 2019, 33, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Delomenie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013, 32, 2261–2272. [Google Scholar] [CrossRef]
- Espina, V.; Mariani, B.D.; Gallagher, R.I.; Tran, K.; Banks, S.; Wiedemann, J.; Huryk, H.; Mueller, C.; Adamo, L.; Deng, J.; et al. Malignant precursor cells pre-exist in human breast DCIS and require autophagy for survival. PLoS ONE 2010, 5, e10240. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, D.; Liu, Y.; Su, Z.; Zhang, L.; Chen, F.; Zhou, Y.; Wu, Y.; Yu, M.; Zhang, Z.; et al. Role of the Hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells. Cancer Cell Int. 2013, 13, 119. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Shang, Z.; Zhou, Y.; Hu, X.; Chen, Y.; Fan, Y.; Wei, X.; Wu, L.; Liang, Q.; Zhang, J.; et al. Autophagy mediates glucose starvation-induced glioblastoma cell quiescence and chemoresistance through coordinating cell metabolism, cell cycle, and survival. Cell Death Dis. 2018, 9, 213. [Google Scholar] [CrossRef] [Green Version]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J. 2018, 285, 1751–1766. [Google Scholar] [CrossRef] [Green Version]
- Schlafli, A.M.; Berezowska, S.; Adams, O.; Langer, R.; Tschan, M.P. Reliable LC3 and p62 autophagy marker detection in formalin fixed paraffin embedded human tissue by immunohistochemistry. Eur. J. Histochem. 2015, 59, 2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, C.; Hassan, B.; Haque, M.; Song, Y.; Li, J.; Liu, D.; Lipke, E.; Chen, W.; Giuriato, S.; Lai, R. Crizotinib Resistance Mediated by Autophagy Is Higher in the Stem-Like Cell Subset in ALK-Positive Anaplastic Large Cell Lymphoma, and This Effect Is MYC-Dependent. Cancers 2021, 13, 181. https://doi.org/10.3390/cancers13020181
Shang C, Hassan B, Haque M, Song Y, Li J, Liu D, Lipke E, Chen W, Giuriato S, Lai R. Crizotinib Resistance Mediated by Autophagy Is Higher in the Stem-Like Cell Subset in ALK-Positive Anaplastic Large Cell Lymphoma, and This Effect Is MYC-Dependent. Cancers. 2021; 13(2):181. https://doi.org/10.3390/cancers13020181
Chicago/Turabian StyleShang, Chuquan, Bardes Hassan, Moinul Haque, Yuqi Song, Jing Li, Dongzhe Liu, Eva Lipke, Will Chen, Sylvie Giuriato, and Raymond Lai. 2021. "Crizotinib Resistance Mediated by Autophagy Is Higher in the Stem-Like Cell Subset in ALK-Positive Anaplastic Large Cell Lymphoma, and This Effect Is MYC-Dependent" Cancers 13, no. 2: 181. https://doi.org/10.3390/cancers13020181