SIRT1 Activation Attenuates the Cardiac Dysfunction Induced by Endothelial Cell-Specific Deletion of CRIF1

 ,
, 

Abstract
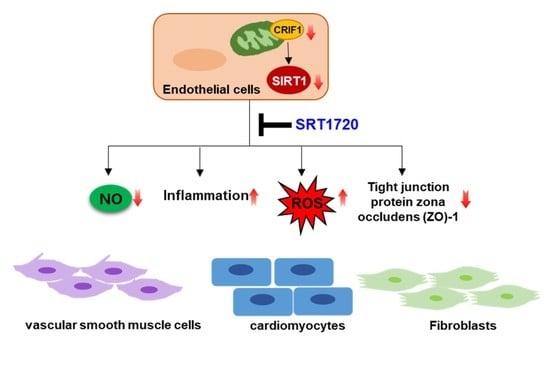
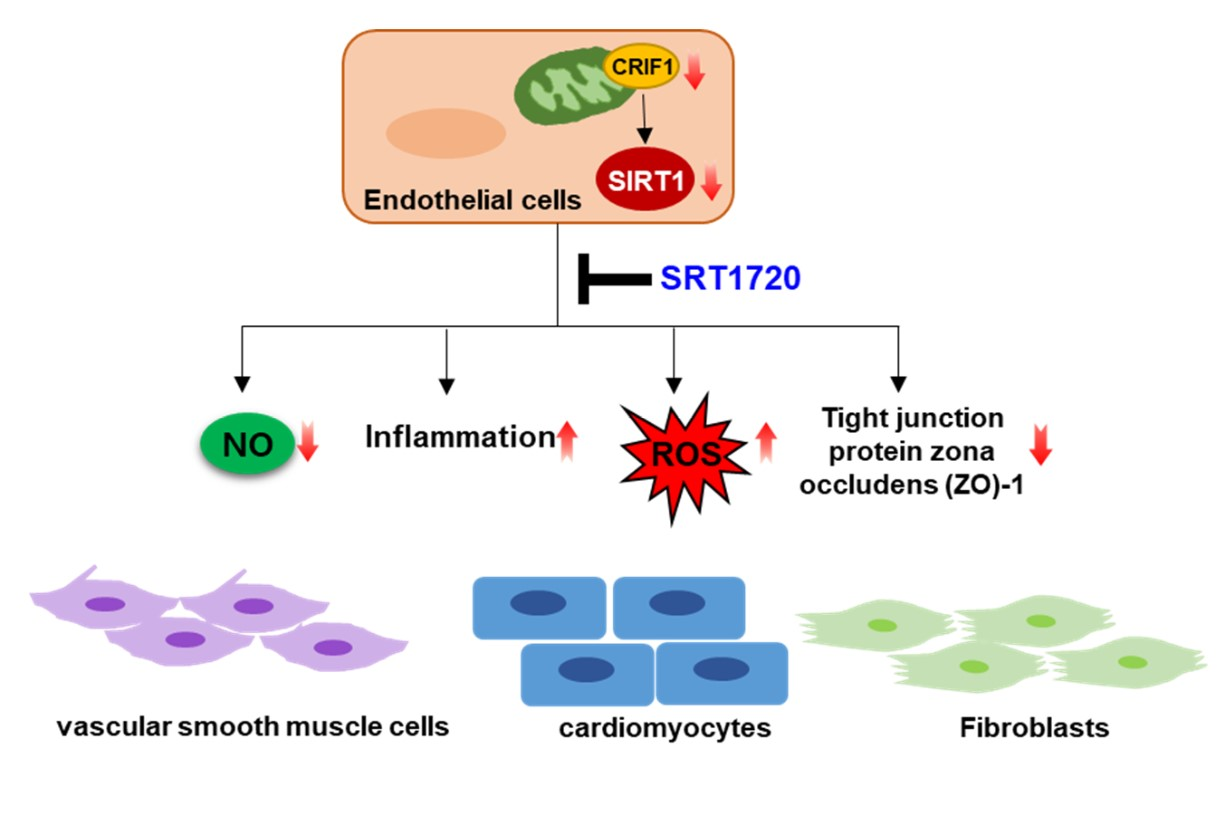
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
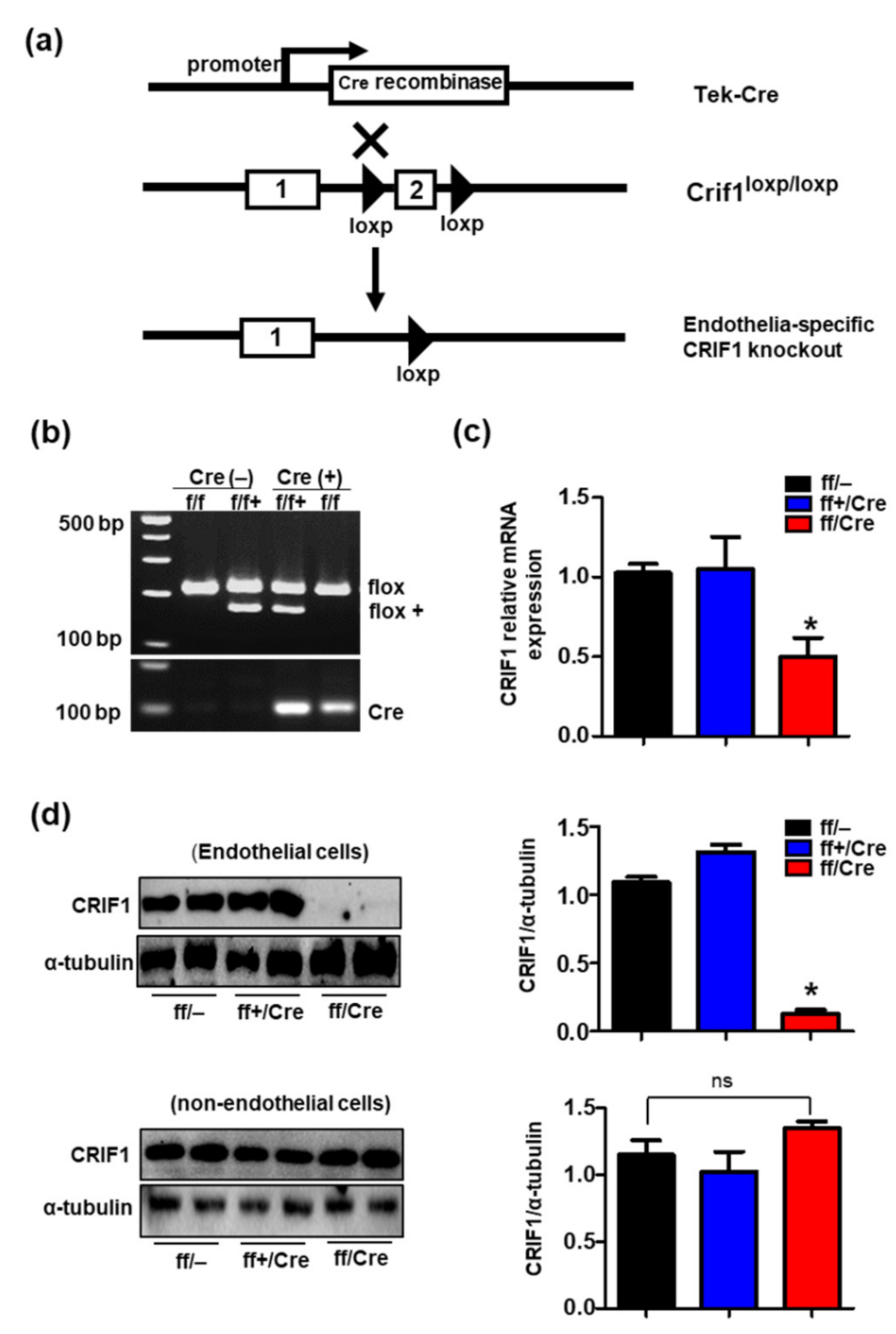
2.1. Mouse Studies
2.2. Western Blot
2.3. Quantitative RT-PCR
2.4. Histological Analysis
2.5. Echocardiography
2.6. SRT1720 Treatment
2.7. ATP Measurement
2.8. Measurement of Mitochondrial Membrane Potential and Mitochondrial ROS
2.9. SIRT1 Activity
2.10. Isolation of Mouse Coronary Endothelial Cells
2.11. Nitrite and Nitrate Measurements
2.12. ROS Measurement
2.13. Statistical Analysis
3. Results
3.1. Generation of Endothelial Cell-Specific Deletion of CRIF1 in Mice
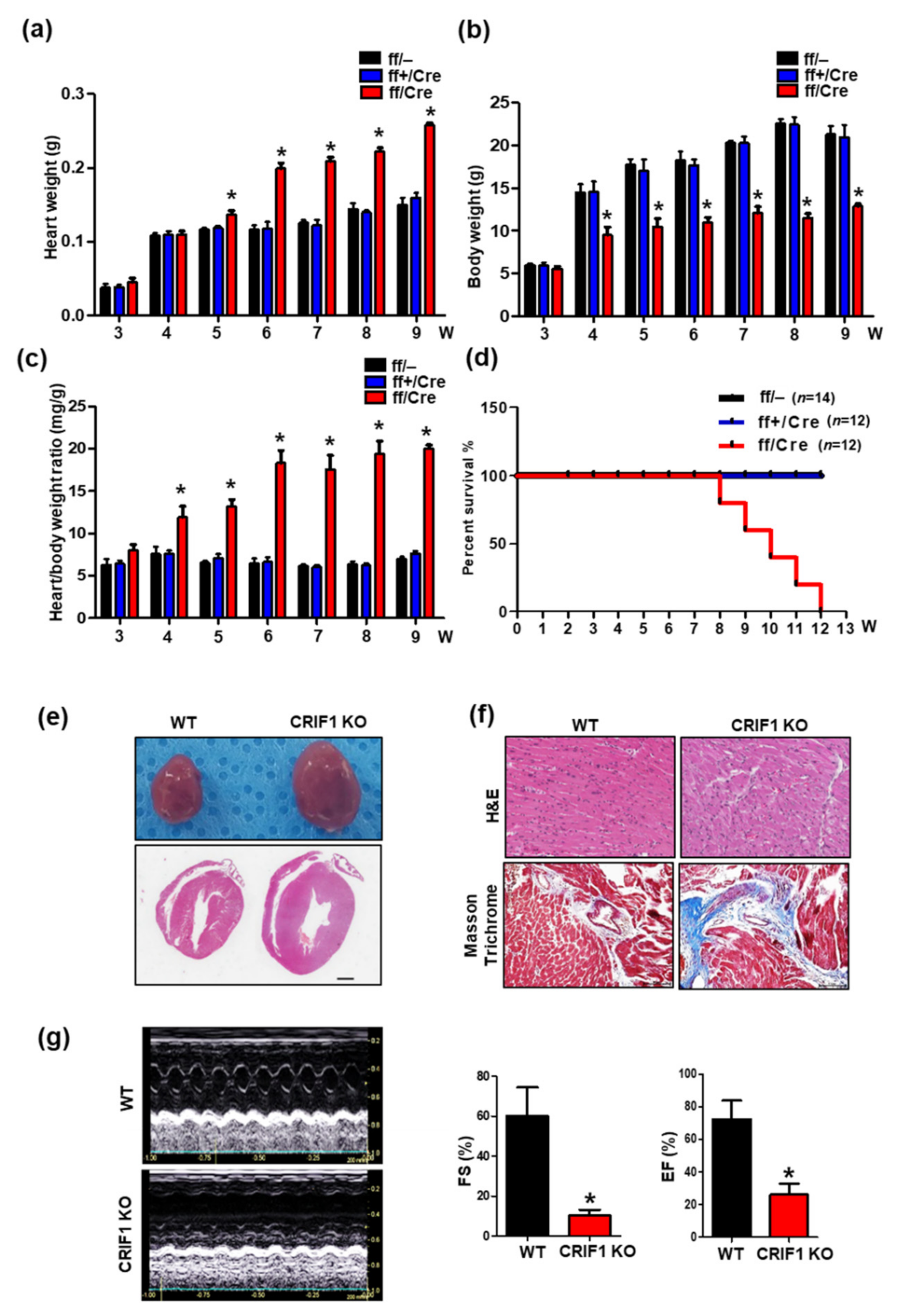
3.2. Endothelial Cell-Specific Deletion of CRIF1 Causes Cardiac Defects
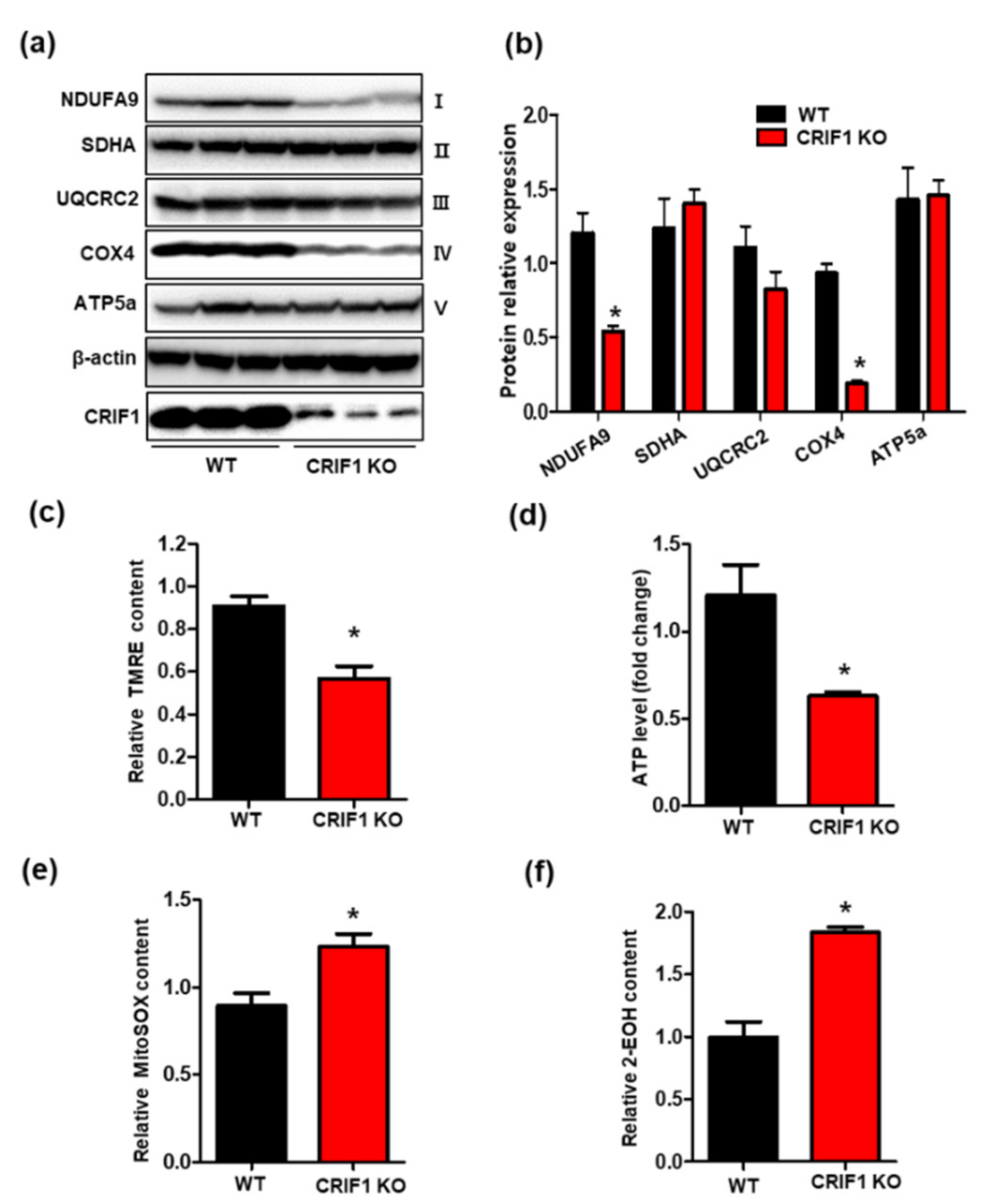
3.3. Endothelial Cell-Specific Deletion of CRIF1 Causes Mitochondrial Dysfunction in the Heart
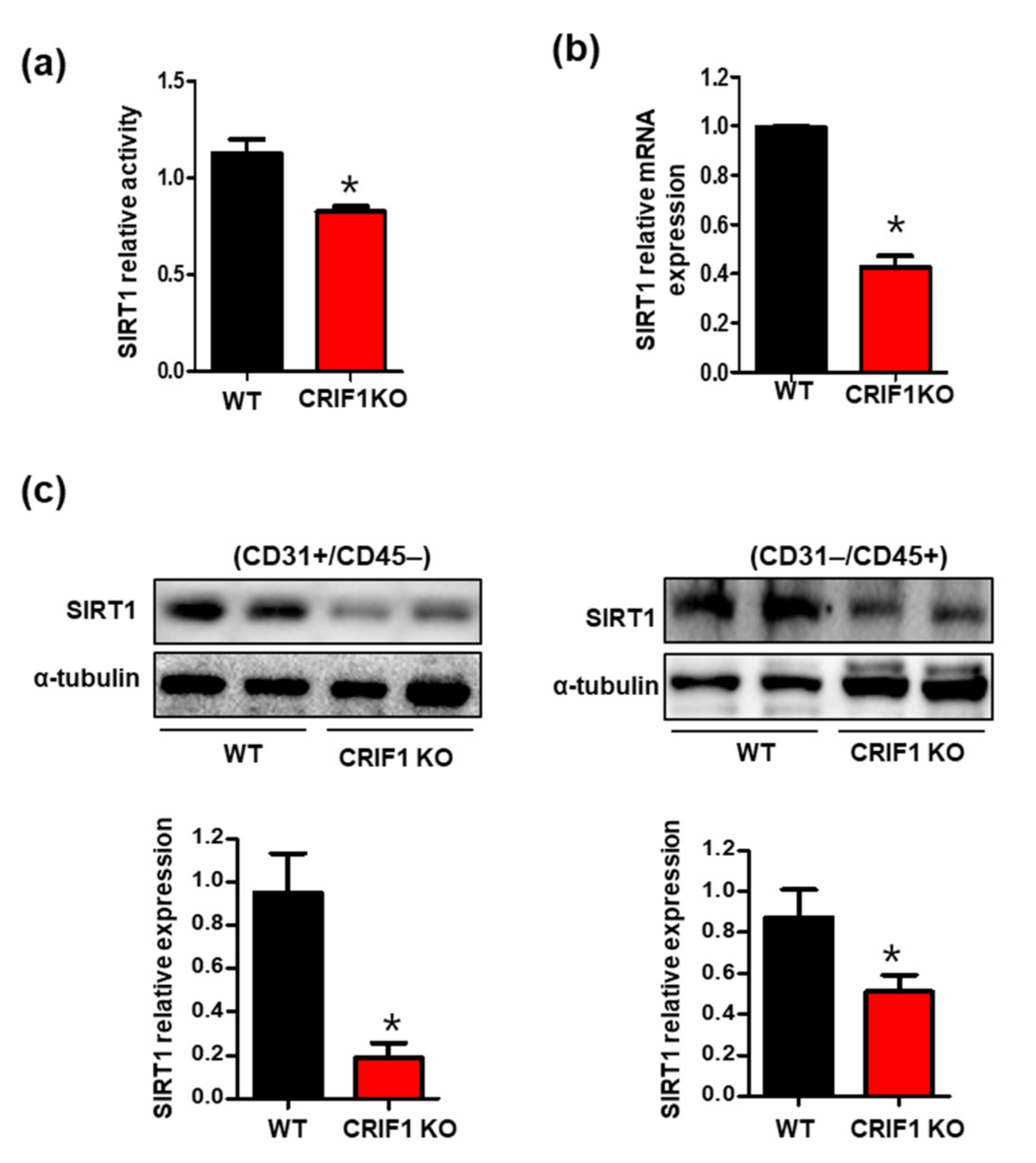
3.4. Increased SIRT1 Expression and Activation in CRIF1 EKO Mouse Hearts
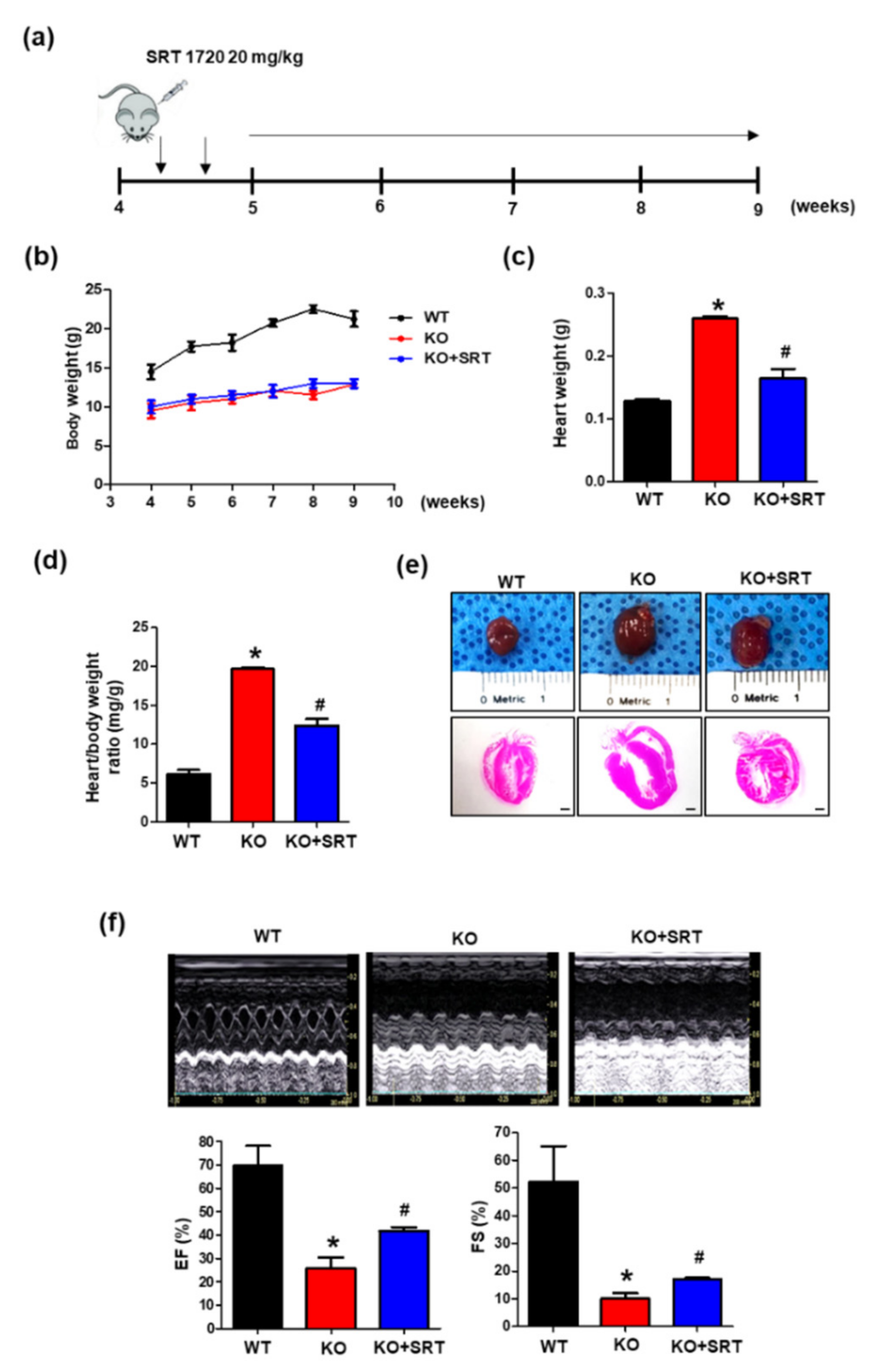
3.5. SIRT1 Activation Alleviates the Cardiac Dysfunction Induced by Endothelial Cell-Specific CRIF1 Deletion
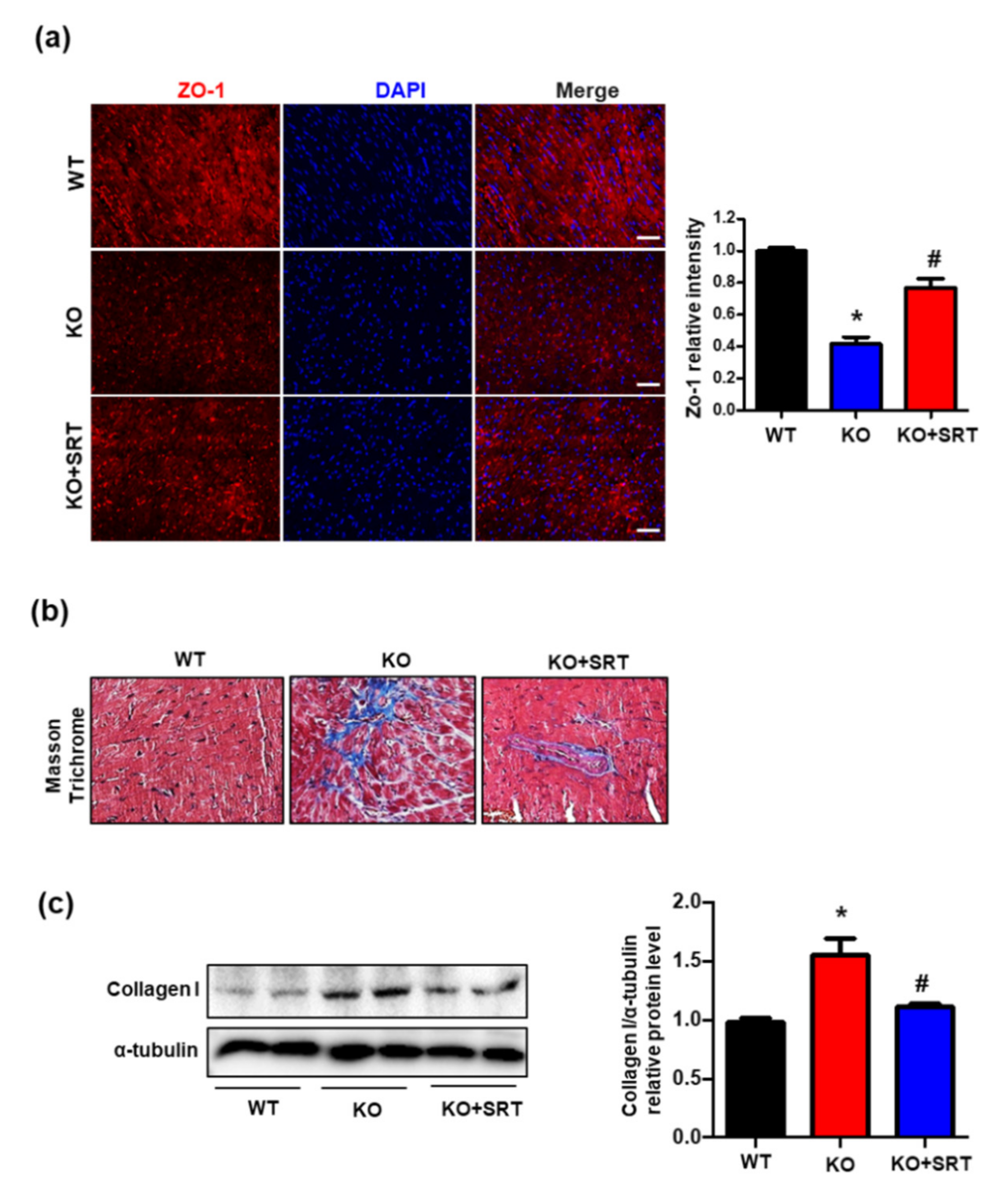
3.6. SIRT1 Activation Upregulates the Expression of Zonula Occludens-1 (ZO-1) and Collagen I
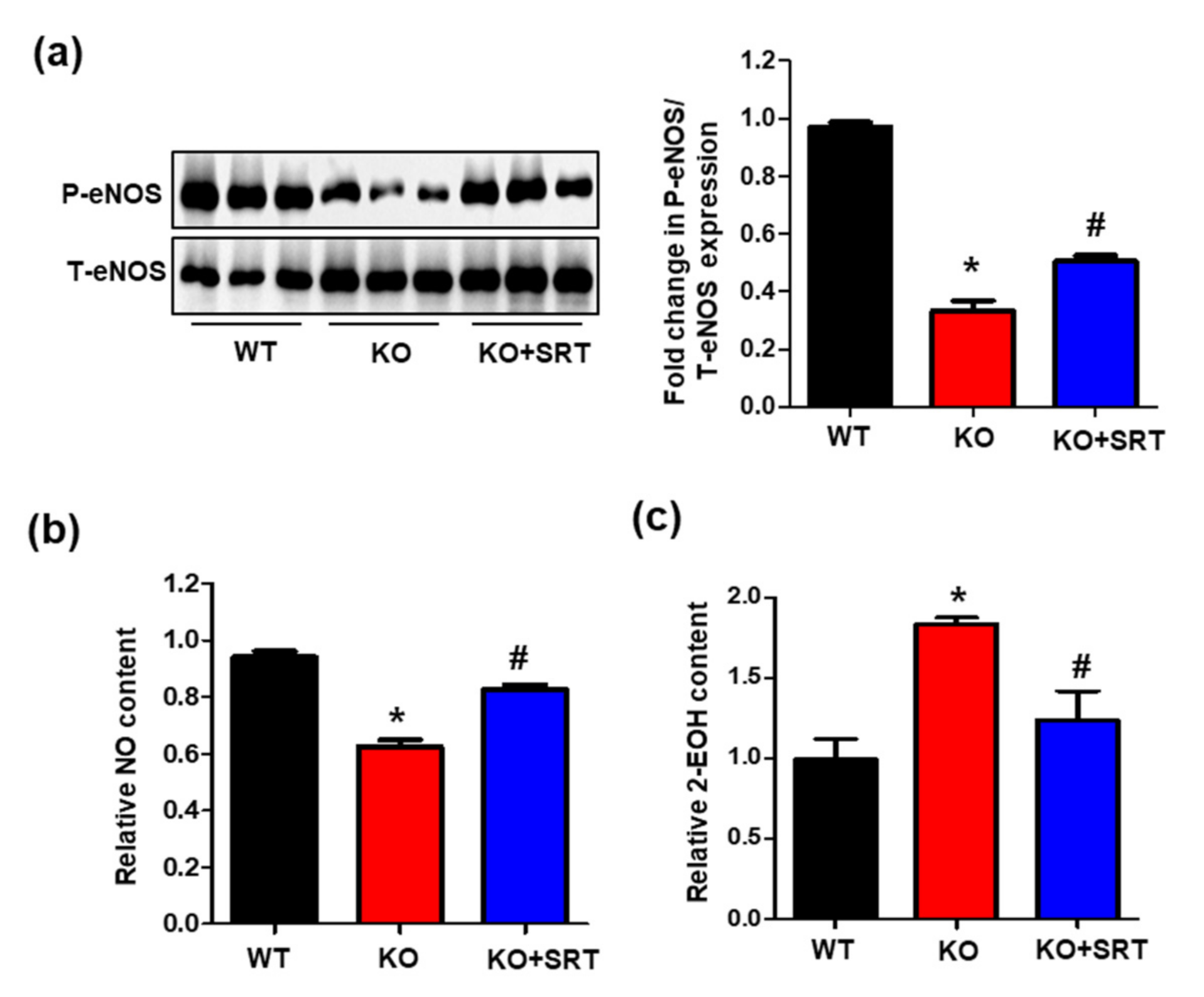
3.7. SIRT1 Activation Mediates eNOS/NO Activation and ROS Levels
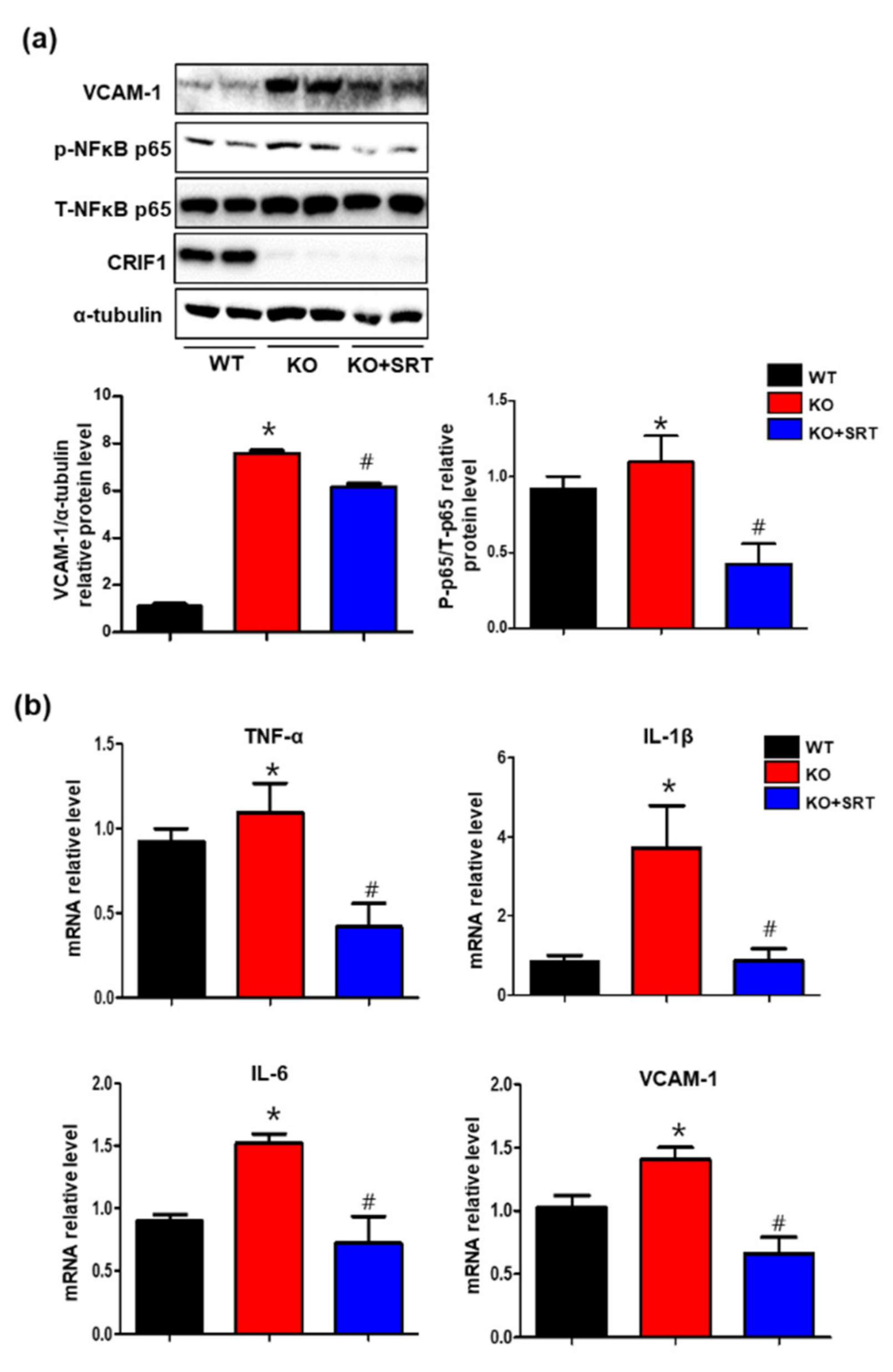
3.8. SIRT1 Activation Inhibits the Levels of Inflammatory Mediators in Heart Tissues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ziaeian, B.; Fonarow, G.C. Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 2016, 13, 368–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buja, L.M.; Ottaviani, G.; Mitchell, R.N. Pathobiology of cardiovascular diseases: An update. Cardiovasc. Pathol. 2019, 42, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting cardiac cellular composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, J.; Kong, Q.; Cheng, H.; Tu, F.; Yu, P.; Liu, Y.; Zhang, X.; Li, C.; Li, Y.; et al. Cardiomyocyte-specific deficiency of HSPB1 worsens cardiac dysfunction by activating NFkappaB-mediated leucocyte recruitment after myocardial infarction. Cardiovasc. Res. 2019, 115, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Lu, W.; Hashimoto, M.; Ono, N.; Muratani, M.; Nishikata, K.; Kim, J.D.; Ebihara, S.; Ishida, J.; Fukamizu, A. PRMT1 deficiency in mouse juvenile heart induces dilated cardiomyopathy and reveals cryptic alternative splicing products. iScience 2018, 8, 200–213. [Google Scholar] [CrossRef] [Green Version]
- Talman, V.; Kivela, R. Cardiomyocyte-endothelial cell interactions in cardiac remodeling and regeneration. Front. Cardiovasc. Med. 2018, 5, 101. [Google Scholar] [CrossRef] [Green Version]
- Matsuzawa, Y.; Lerman, A. Endothelial dysfunction and coronary artery disease: Assessment, prognosis, and treatment. Coron. Artery Dis. 2014, 25, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Alem, M.M. Endothelial dysfunction in chronic heart failure: Assessment, findings, significance, and potential therapeutic targets. Int. J. Mol. Sci. 2019, 20, 3198. [Google Scholar] [CrossRef] [Green Version]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.G.; Yi, H.S.; Choi, M.J.; Ryu, M.J.; Jung, S.; Chung, H.K.; Chang, J.Y.; Kim, Y.K.; Lee, S.E.; Kim, H.W.; et al. ANGPTL6 expression is coupled with mitochondrial OXPHOS function to regulate adipose FGF21. J. Endocrinol. 2017, 233, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.K.; Joung, K.H.; Ryu, M.J.; Kim, S.J.; Kim, H.; Chung, H.K.; Lee, M.H.; Lee, S.E.; Choi, M.J.; Chang, J.Y.; et al. Disruption of CR6-interacting factor-1 (CRIF1) in mouse islet beta cells leads to mitochondrial diabetes with progressive beta cell failure. Diabetologia 2015, 58, 771–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, M.J.; Kim, S.J.; Kim, Y.K.; Choi, M.J.; Tadi, S.; Lee, M.H.; Lee, S.E.; Chung, H.K.; Jung, S.B.; Kim, H.J.; et al. Crif1 deficiency reduces adipose OXPHOS capacity and triggers inflammation and insulin resistance in mice. PLoS Genet 2013, 9, e1003356. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.B.; Choi, M.J.; Ryu, D.; Yi, H.S.; Lee, S.E.; Chang, J.Y.; Chung, H.K.; Kim, Y.K.; Kang, S.G.; Lee, J.H.; et al. Reduced oxidative capacity in macrophages results in systemic insulin resistance. Nat. Commun. 2018, 9, 1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Kwon, M.C.; Ryu, M.J.; Chung, H.K.; Tadi, S.; Kim, Y.K.; Kim, J.M.; Lee, S.H.; Park, J.H.; Kweon, G.R.; et al. CRIF1 is essential for the synthesis and insertion of oxidative phosphorylation polypeptides in the mammalian mitochondrial membrane. Cell Metab. 2012, 16, 274–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.; Lee, S.H.; Kwon, M.C.; Yang, D.K.; Seo, H.R.; Kim, J.; Kim, Y.Y.; Im, S.K.; Abel, E.D.; Kim, K.T.; et al. Cardiomyocyte specific deletion of Crif1 causes mitochondrial cardiomyopathy in mice. PLoS ONE 2013, 8, e53577. [Google Scholar] [CrossRef] [PubMed]
- Piao, S.; Lee, J.W.; Nagar, H.; Jung, S.B.; Choi, S.; Kim, S.; Lee, I.; Kim, S.M.; Shin, N.; Lee, Y.R.; et al. CR6 interacting factor 1 deficiency promotes endothelial inflammation by SIRT1 downregulation. PLoS ONE 2018, 13, e0192693. [Google Scholar] [CrossRef] [Green Version]
- Nagar, H.; Jung, S.B.; Ryu, M.J.; Choi, S.J.; Piao, S.; Song, H.J.; Kang, S.K.; Shin, N.; Kim, D.W.; Jin, S.A.; et al. CR6-interacting factor 1 deficiency impairs vascular function by inhibiting the sirt1-endothelial nitric oxide synthase pathway. Antioxid. Redox Signal. 2017, 27, 234–249. [Google Scholar] [CrossRef]
- Jung, S.B.; Kim, C.S.; Naqvi, A.; Yamamori, T.; Mattagajasingh, I.; Hoffman, T.A.; Cole, M.P.; Kumar, A.; Dericco, J.S.; Jeon, B.H.; et al. Histone deacetylase 3 antagonizes aspirin-stimulated endothelial nitric oxide production by reversing aspirin-induced lysine acetylation of endothelial nitric oxide synthase. Circ. Res. 2010, 107, 877–887. [Google Scholar] [CrossRef]
- Fink, B.; Laude, K.; McCann, L.; Doughan, A.; Harrison, D.G.; Dikalov, S. Detection of intracellular superoxide formation in endothelial cells and intact tissues using dihydroethidium and an HPLC-based assay. Am. J. Physiol.-Cell Physiol. 2004, 287, C895–C902. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.M.; Huang, A.; Kaley, G.; Sun, D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am. J. Physiol.-Heart Circ. Physiol. 2009, 297, H1829–H1836. [Google Scholar] [CrossRef] [Green Version]
- Zielonka, J.; Hardy, M.; Kalyanaraman, B. HPLC study of oxidation products of hydroethidine in chemical and biological systems: Ramifications in superoxide measurements. Free Radic. Biol. Med. 2009, 46, 329–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planavila, A.; Dominguez, E.; Navarro, M.; Vinciguerra, M.; Iglesias, R.; Giralt, M.; Lope-Piedrafita, S.; Ruberte, J.; Villarroya, F. Dilated cardiomyopathy and mitochondrial dysfunction in Sirt1-deficient mice: A role for Sirt1-Mef2 in adult heart. J. Mol. Cell. Cardiol. 2012, 53, 521–531. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Wang, S.; Shang, Y.C.; Maiese, K. Targeting cardiovascular disease with novel SIRT1 pathways. Future Cardiol. 2012, 8, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tornavaca, O.; Chia, M.; Dufton, N.; Almagro, L.O.; Conway, D.E.; Randi, A.M.; Schwartz, M.A.; Matter, K.; Balda, M.S. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J. Cell Biol. 2015, 208, 821–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segers, V.F.M.; Brutsaert, D.L.; De Keulenaer, G.W. Cardiac remodeling: Endothelial cells have more to say than just no. Front. Physiol. 2018, 9, 382. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The extracellular matrix in ischemic and nonischemic heart failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef]
- Godecke, A.; Heinicke, T.; Kamkin, A.; Kiseleva, I.; Strasser, R.H.; Decking, U.K.; Stumpe, T.; Isenberg, G.; Schrader, J. Inotropic response to beta-adrenergic receptor stimulation and anti-adrenergic effect of ACh in endothelial NO synthase-deficient mouse hearts. J. Physiol. 2001, 532, 195–204. [Google Scholar] [CrossRef]
- Jones, S.P.; Greer, J.J.; van Haperen, R.; Duncker, D.J.; de Crom, R.; Lefer, D.J. Endothelial nitric oxide synthase overexpression attenuates congestive heart failure in mice. Proc. Natl. Acad. Sci. USA 2003, 100, 4891–4896. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.P.; Greer, J.J.; Kakkar, A.K.; Ware, P.D.; Turnage, R.H.; Hicks, M.; van Haperen, R.; de Crom, R.; Kawashima, S.; Yokoyama, M.; et al. Endothelial nitric oxide synthase overexpression attenuates myocardial reperfusion injury. Am. J. Physiol.-Heart Circ. Physiol. 2004, 286, H276–H282. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.P.; Bolli, R. The ubiquitous role of nitric oxide in cardioprotection. J. Mol. Cell. Cardiol. 2006, 40, 16–23. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Q.; Zeng, Z.; Wu, J.; Zhang, Y.; Chen, Z. Sirt1 inhibits oxidative stress in vascular endothelial cells. Oxid. Med. Cell. Longev. 2017, 2017, 7543973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arunachalam, G.; Yao, H.; Sundar, I.K.; Caito, S.; Rahman, I. SIRT1 regulates oxidant- and cigarette smoke-induced eNOS acetylation in endothelial cells: Role of resveratrol. Biochem. Biophys. Res. Commun. 2010, 393, 66–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, E.; Tonnessen, T.; Lunde, P.K.; Semb, S.O.; Wasserstrom, J.A.; Sejersted, O.M.; Christensen, G. Mechanisms of cardiomyocyte dysfunction in heart failure following myocardial infarction in rats. J. Mol. Cell. Cardiol. 1998, 30, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Fortuno, M.A.; Querejeta, R.; Ravassa, S.; Lopez, B.; Lopez, N.; Diez, J. Cardiomyocyte apoptosis in hypertensive cardiomyopathy. Cardiovasc. Res. 2003, 59, 549–562. [Google Scholar] [CrossRef]
- Premer, C.; Kanelidis, A.J.; Hare, J.M.; Schulman, I.H. Rethinking endothelial dysfunction as a crucial target in fighting heart failure. Mayo Clin. Proc. Innov. Qual. Outcomes 2019, 3, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Leucker, T.M.; Jones, S.P. Endothelial dysfunction as a nexus for endothelial cell-cardiomyocyte miscommunication. Front. Physiol. 2014, 5, 328. [Google Scholar] [CrossRef] [Green Version]
- Luu, A.Z.; Chowdhury, B.; Al-Omran, M.; Teoh, H.; Hess, D.A.; Verma, S. Role of endothelium in doxorubicin-induced cardiomyopathy. JACC Basic Transl. Sci. 2018, 3, 861–870. [Google Scholar] [CrossRef]
- Hofmann, J.J.; Briot, A.; Enciso, J.; Zovein, A.C.; Ren, S.; Zhang, Z.W.; Radtke, F.; Simons, M.; Wang, Y.; Iruela-Arispe, M.L. Endothelial deletion of murine Jag1 leads to valve calcification and congenital heart defects associated with Alagille syndrome. Development 2012, 139, 4449–4460. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.; Chung, J.I.; King, D.A.; D’Amato, G.; Paik, D.T.; Duan, A.; Chang, A.; Nagelberg, D.; Sharma, B.; Jeong, Y.; et al. Endothelial deletion of Ino80 disrupts coronary angiogenesis and causes congenital heart disease. Nat. Commun. 2018, 9, 368. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wu, J.; Zhu, C.; Liu, J.; Chen, X.; Zhuang, T.; Kuang, Y.; Wang, Y.; Hu, H.; Yu, P.; et al. Endothelial S1pr1 regulates pressure overload-induced cardiac remodelling through AKT-eNOS pathway. J. Cell. Mol. Med. 2020, 24, 2013–2026. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.L. Sirt1 and the Mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, M.N.; Grimbert, L.; Moulin, M.; Gressette, M.; Rucker-Martin, C.; Lemaire, C.; Mericskay, M.; Veksler, V.; Ventura-Clapier, R.; Garnier, A.; et al. Inducible cardiac-specific deletion of Sirt1 in male mice reveals progressive cardiac dysfunction and sensitization of the heart to pressure overload. Int. J. Mol. Sci. 2019, 20, 5005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Feng, J.; Zhang, R.; Chen, J.; Han, D.; Li, X.; Yang, B.; Li, X.; Fan, M.; Li, C.; et al. SIRT1 Activation by resveratrol alleviates cardiac dysfunction via mitochondrial regulation in diabetic cardiomyopathy mice. Oxid. Med. Cell. Longev. 2017, 2017, 4602715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gano, L.B.; Donato, A.J.; Pasha, H.M.; Hearon, C.M., Jr.; Sindler, A.L.; Seals, D.R. The SIRT1 activator SRT1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1754–H1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, K.; Matsushita, T.; Takayama, K.; Tanaka, T.; Miyaji, N.; Ibaraki, K.; Araki, D.; Kanzaki, N.; Matsumoto, T.; Kuroda, R. Intraperitoneal injection of the SIRT1 activator SRT1720 attenuates the progression of experimental osteoarthritis in mice. Bone Joint Res. 2018, 7, 252–262. [Google Scholar] [CrossRef] [Green Version]
- Davidson, S.M. Endothelial mitochondria and heart disease. Cardiovasc. Res. 2010, 88, 58–66. [Google Scholar] [CrossRef]
- Man, A.W.C.; Li, H.; Xia, N. The role of Sirtuin1 in regulating endothelial function, arterial remodeling and vascular aging. Front. Physiol. 2019, 10, 1173. [Google Scholar] [CrossRef] [Green Version]
- Kitada, M.; Ogura, Y.; Koya, D. The protective role of Sirt1 in vascular tissue: Its relationship to vascular aging and atherosclerosis. Aging (Albany NY) 2016, 8, 2290–2307. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piao, S.; Lee, I.; Jin, S.-A.; Kim, S.; Nagar, H.; Choi, S.-j.; Jeon, B.H.; Kim, C.-S. SIRT1 Activation Attenuates the Cardiac Dysfunction Induced by Endothelial Cell-Specific Deletion of CRIF1. Biomedicines 2021, 9, 52. https://doi.org/10.3390/biomedicines9010052
Piao S, Lee I, Jin S-A, Kim S, Nagar H, Choi S-j, Jeon BH, Kim C-S. SIRT1 Activation Attenuates the Cardiac Dysfunction Induced by Endothelial Cell-Specific Deletion of CRIF1. Biomedicines. 2021; 9(1):52. https://doi.org/10.3390/biomedicines9010052
Chicago/Turabian StylePiao, Shuyu, Ikjun Lee, Seon-Ah Jin, Seonhee Kim, Harsha Nagar, Su-jeong Choi, Byeong Hwa Jeon, and Cuk-Seong Kim. 2021. "SIRT1 Activation Attenuates the Cardiac Dysfunction Induced by Endothelial Cell-Specific Deletion of CRIF1" Biomedicines 9, no. 1: 52. https://doi.org/10.3390/biomedicines9010052
APA StylePiao, S., Lee, I., Jin, S.-A., Kim, S., Nagar, H., Choi, S.-j., Jeon, B. H., & Kim, C.-S. (2021). SIRT1 Activation Attenuates the Cardiac Dysfunction Induced by Endothelial Cell-Specific Deletion of CRIF1. Biomedicines, 9(1), 52. https://doi.org/10.3390/biomedicines9010052