Role of Bladder Cancer Metabolic Reprogramming in the Effectiveness of Immunotherapy

Abstract
:Simple Summary
Abstract
1. Introduction
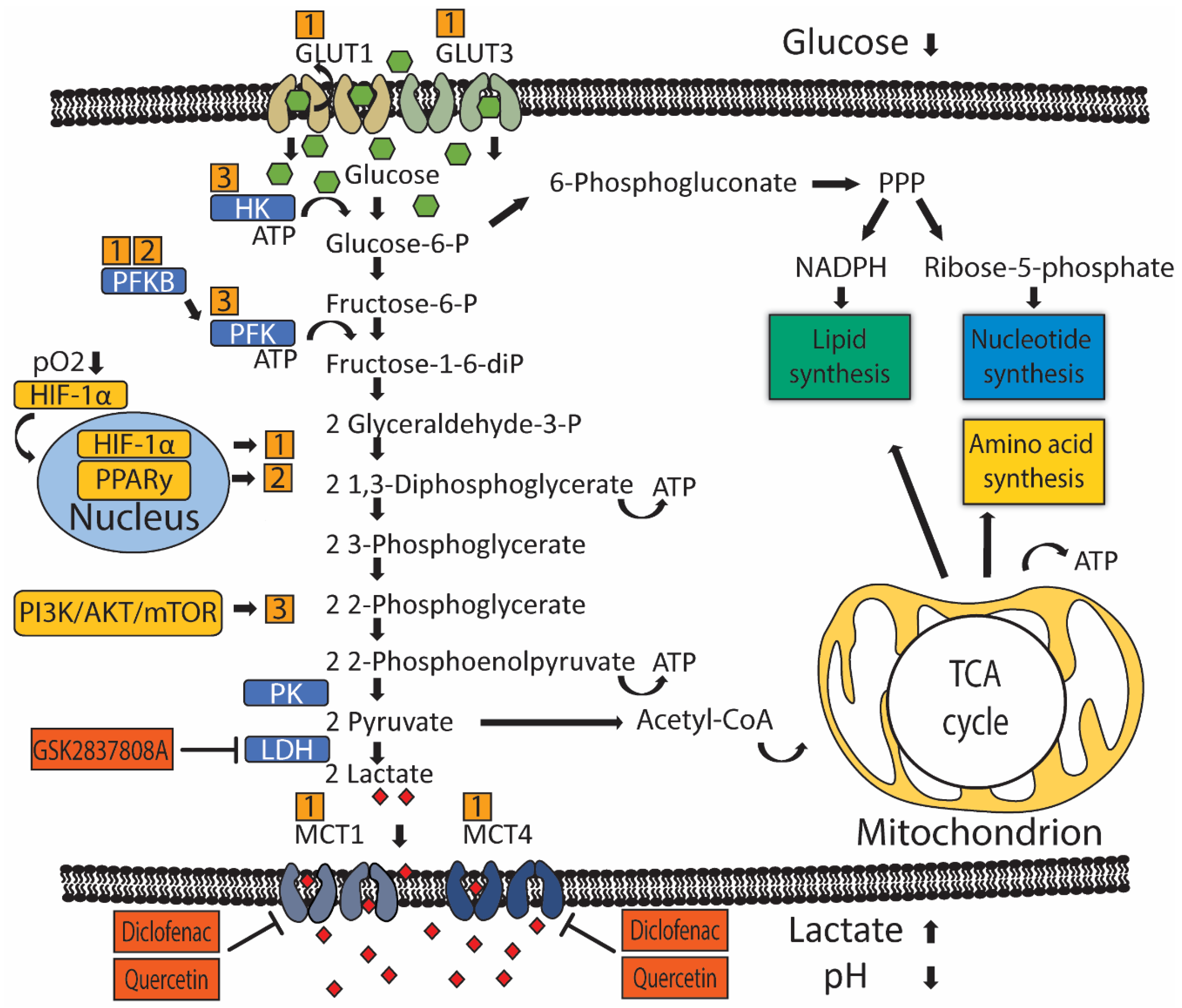
2. Glucose Metabolism in Urothelial Carcinoma
3. Regulation of Glucose Transport and Metabolism in UC
4. Impact of UC Metabolism on Cells of the Tumor Microenvironment and Immunotherapy
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babjuk, M.; Burger, M.; Compérat, E.M.; Gontero, P.; Mostafid, A.H.; Palou, J.; van Rhijn, B.W.G.; Rouprêt, M.; Shariat, S.F.; Sylvester, R.; et al. European Association of Urology Guidelines on Non-muscle-invasive Bladder Cancer (TaT1 and Carcinoma In Situ)—2019 Update. Eur. Urol. 2019, 76, 639–657. [Google Scholar] [CrossRef] [PubMed]
- Witjes, J.A.; Bruins, H.M.; Cathomas, R.; Comperat, E.M.; Cowan, N.C.; Gakis, G.; Hernandez, V.; Linares Espinos, E.; Lorch, A.; Neuzillet, Y.; et al. European Association of Urology Guidelines on Muscle-invasive and Metastatic Bladder Cancer: Summary of the 2020 Guidelines. Eur. Urol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Von der Maase, H.; Hansen, S.W.; Roberts, J.T.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Bodrogi, I.; Albers, P.; Knuth, A.; Lippert, C.M.; et al. Gemcitabine and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in advanced or metastatic bladder cancer: Results of a large, randomized, multinational, multicenter, phase III study. J. Clin. Oncol. 2000, 18, 3068–3077. [Google Scholar] [CrossRef] [PubMed]
- Dash, A.; Galsky, M.D.; Vickers, A.J.; Serio, A.M.; Koppie, T.M.; Dalbagni, G.; Bochner, B.H. Impact of renal impairment on eligibility for adjuvant cisplatin-based chemotherapy in patients with urothelial carcinoma of the bladder. Cancer 2006, 107, 506–513. [Google Scholar] [CrossRef]
- Galsky, M.D.; Hahn, N.M.; Rosenberg, J.; Sonpavde, G.; Hutson, T.; Oh, W.K.; Dreicer, R.; Vogelzang, N.; Sternberg, C.N.; Bajorin, D.F.; et al. Treatment of Patients With Metastatic Urothelial Cancer “Unfit” for Cisplatin-Based Chemotherapy. J. Clin. Oncol. 2011, 29, 2432–2438. [Google Scholar] [CrossRef] [Green Version]
- Galsky, M.D.; Chen, G.J.; Oh, W.K.; Bellmunt, J.; Roth, B.J.; Petrioli, R.; Dogliotti, L.; Dreicer, R.; Sonpavde, G. Comparative effectiveness of cisplatin-based and carboplatin-based chemotherapy for treatment of advanced urothelial carcinoma. Ann. Oncol. 2012, 23, 406–410. [Google Scholar] [CrossRef]
- Von der Maase, H.; Sengelov, L.; Roberts, J.T.; Ricci, S.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Zimmermann, A.; Arning, M. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J. Clin. Oncol. 2005, 23, 4602–4608. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Nair, V.S.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powles, T.; Duran, I.; van der Heijden, M.S.; Loriot, Y.; Vogelzang, N.J.; De Giorgi, U.; Oudard, S.; Retz, M.M.; Castellano, D.; Bamias, A.; et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2018, 391, 748–757. [Google Scholar] [CrossRef]
- Sharma, P.; Retz, M.; Siefker-Radtke, A.; Baron, A.; Necchi, A.; Bedke, J.; Plimack, E.R.; Vaena, D.; Grimm, M.O.; Bracarda, S.; et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017, 18, 312–322. [Google Scholar] [CrossRef]
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Vuky, J.; Powles, T.; Plimack, E.R.; Hahn, N.M.; de Wit, R.; Pang, L.; et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): A multicentre, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 1483–1492. [Google Scholar] [CrossRef]
- Balar, A.V.; Galsky, M.D.; Rosenberg, J.E.; Powles, T.; Petrylak, D.P.; Bellmunt, J.; Loriot, Y.; Necchi, A.; Hoffman-Censits, J.; Perez-Gracia, J.L.; et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: A single-arm, multicentre, phase 2 trial. Lancet 2017, 389, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Pitt, M.J.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef] [Green Version]
- Afonso, J.; Santos, L.L.; Longatto-Filho, A.; Baltazar, F. Competitive glucose metabolism as a target to boost bladder cancer immunotherapy. Nat. Rev. Urol. 2020, 17, 77–106. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029. [Google Scholar] [CrossRef] [Green Version]
- Whyard, T.; Waltzer, W.C.; Waltzer, D.; Romanov, V. Metabolic alterations in bladder cancer: Applications for cancer imaging. Exp. Cell Res. 2016, 341, 77–83. [Google Scholar] [CrossRef]
- Sahu, D.; Lotan, Y.; Wittmann, B.; Neri, B.; Hansel, D.E. Metabolomics analysis reveals distinct profiles of nonmuscle-invasive and muscle-invasive bladder cancer. Cancer Med. 2017, 6, 2106–2120. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, M.M.; Balar, A.V. PD-1/PD-L1 Combinations in Advanced Urothelial Cancer: Rationale and Current Clinical Trials. Clin. Genitourin Cancer 2019, 17, e618–e626. [Google Scholar] [CrossRef] [PubMed]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Berra, E.; Benizri, E.; Ginouvès, A.; Volmat, V.; Roux, D.; Pouysségur, J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003, 22, 4082–4090. [Google Scholar] [CrossRef] [Green Version]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Chang, C.; Cui, Y.; Zhao, X.; Yang, J.; Shen, L.; Zhou, J.; Hou, Z.; Zhang, Z.; Ye, C.; et al. Steroid receptor coactivator-3 regulates glucose metabolism in bladder cancer cells through coactivation of hypoxia inducible factor 1α. J. Biol. Chem. 2014, 289, 11219–11229. [Google Scholar] [CrossRef] [Green Version]
- Wan, W.; Peng, K.; Li, M.; Qin, L.; Tong, Z.; Yan, J.; Shen, B.; Yu, C. Histone demethylase JMJD1A promotes urinary bladder cancer progression by enhancing glycolysis through coactivation of hypoxia inducible factor 1α. Oncogene 2017, 36, 3868–3877. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cheng, Y.; Li, P.; Tao, J.; Deng, X.; Zhang, X.; Gu, M.; Lu, Q.; Yin, C. A lentiviral sponge for miRNA-21 diminishes aerobic glycolysis in bladder cancer T24 cells via the PTEN/PI3K/AKT/mTOR axis. Tumour. Biol. 2015, 36, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, X.; Wu, S.; Xue, M.; Chen, W. Long non-coding RNA UCA1 promotes glycolysis by upregulating hexokinase 2 through the mTOR-STAT3/microRNA143 pathway. Cancer Sci. 2014, 105, 951–955. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, J.N.; Akbani, R.; Broom, B.M.; Wang, W.; Verhaak, R.G.W.; McConkey, D.; Lerner, S.; Morgan, M.; Creighton, C.J.; Smith, C.; et al. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Lu, Q.; Wu, D.; Li, P.; Xu, B.; Qing, W.; Wang, M.; Zhang, Z.; Zhang, W. microRNA-21 modulates cell proliferation and sensitivity to doxorubicin in bladder cancer cells. Oncol. Rep. 2011, 25, 1721–1729. [Google Scholar] [CrossRef]
- Shu, Y.; Lu, Y.; Pang, X.; Zheng, W.; Huang, Y.; Li, J.; Ji, J.; Zhang, C.; Shen, P. Phosphorylation of PPARγ at Ser84 promotes glycolysis and cell proliferation in hepatocellular carcinoma by targeting PFKFB4. Oncotarget 2016, 7, 76984–76994. [Google Scholar] [CrossRef] [Green Version]
- Shashni, B.; Sakharkar, K.R.; Nagasaki, Y.; Sakharkar, M.K. Glycolytic enzymes PGK1 and PKM2 as novel transcriptional targets of PPARγ in breast cancer pathophysiology. J. Drug Target 2013, 21, 161–174. [Google Scholar] [CrossRef]
- Hurst, C.D.; Alder, O.; Platt, F.M.; Droop, A.; Stead, L.F.; Burns, J.E.; Burghel, G.J.; Jain, S.; Klimczak, L.J.; Lindsay, H.; et al. Genomic Subtypes of Non-invasive Bladder Cancer with Distinct Metabolic Profile and Female Gender Bias in KDM6A Mutation Frequency. Cancer Cell 2017, 32, 701–715.e707. [Google Scholar] [CrossRef]
- Rochel, N.; Krucker, C.; Coutos-Thévenot, L.; Osz, J.; Zhang, R.; Guyon, E.; Zita, W.; Vanthong, S.; Hernandez, O.A.; Bourguet, M.; et al. Recurrent activating mutations of PPARγ associated with luminal bladder tumors. Nat. Commun. 2019, 10, 253. [Google Scholar] [CrossRef]
- Kamoun, A.; de Reyniès, A.; Allory, Y.; Sjödahl, G.; Robertson, A.G.; Seiler, R.; Hoadley, K.A.; Groeneveld, C.S.; Al-Ahmadie, H.; Choi, W.; et al. A Consensus Molecular Classification of Muscle-invasive Bladder Cancer. Eur. Urol. 2020, 77, 420–433. [Google Scholar] [CrossRef]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, R.A.; Owen, G.I. Glucose transporters: Expression, regulation and cancer. Biol. Res. 2002, 35, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Yang, X.; Cheng, Y.; Zhang, X.; Yang, C.; Deng, X.; Li, P.; Tao, J.; Yang, H.; Wei, J.; et al. MicroRNA-218 Increases the Sensitivity of Bladder Cancer to Cisplatin by Targeting Glut1. Cell Physiol. Biochem. 2017, 41, 921–932. [Google Scholar] [CrossRef]
- Fei, X.; Qi, M.; Wu, B.; Song, Y.; Wang, Y.; Li, T. MicroRNA-195-5p suppresses glucose uptake and proliferation of human bladder cancer T24 cells by regulating GLUT3 expression. FEBS Lett. 2012, 586, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Yongzhi, H.; Chen, S.; Luo, X.; Lin, Y.; Zhou, Y.; Jin, H.; Hou, B.; Deng, Y.; Tu, L.; et al. The prognostic value of GLUT1 in cancers: A systematic review and meta-analysis. Oncotarget 2017, 8, 43356–43367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, H.; Tschirdewahn, S.; Szarvas, T.; Rübben, H.; Schmid, K.W.; Grabellus, F. Expression of GLUT1 is associated with increasing grade of malignancy in non-invasive and invasive urothelial carcinomas of the bladder. Oncol. Lett. 2011, 2, 1149–1153. [Google Scholar] [CrossRef] [Green Version]
- Hoskin, P.J.; Sibtain, A.; Daley, F.M.; Wilson, G.D. GLUT1 and CAIX as intrinsic markers of hypoxia in bladder cancer: Relationship with vascularity and proliferation as predictors of outcome of ARCON. Br. J. Cancer 2003, 89, 1290–1297. [Google Scholar] [CrossRef] [Green Version]
- Boström, P.J.; Thoms, J.; Sykes, J.; Ahmed, O.; Evans, A.; van Rhijn, B.W.; Mirtti, T.; Stakhovskyi, O.; Laato, M.; Margel, D.; et al. Hypoxia Marker GLUT-1 (Glucose Transporter 1) is an Independent Prognostic Factor for Survival in Bladder Cancer Patients Treated with Radical Cystectomy. Bladder Cancer 2016, 2, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef]
- Sun, C.M.; Xiong, D.B.; Yan, Y.; Geng, J.; Liu, M.; Yao, X.D. Genetic alteration in phosphofructokinase family promotes growth of muscle-invasive bladder cancer. Int. J. Biol. Markers 2016, 31, e286–e293. [Google Scholar] [CrossRef]
- Conde, V.R.; Oliveira, P.F.; Nunes, A.R.; Rocha, C.S.; Ramalhosa, E.; Pereira, J.A.; Alves, M.G.; Silva, B.M. The progression from a lower to a higher invasive stage of bladder cancer is associated with severe alterations in glucose and pyruvate metabolism. Exp. Cell Res. 2015, 335, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Zeng, J.; Xie, R.; Schulz, M.J.; Tedesco, R.; Qu, J.; Erhard, K.F.; Mack, J.F.; Raha, K.; Rendina, A.R.; et al. Discovery of a Novel 2,6-Disubstituted Glucosamine Series of Potent and Selective Hexokinase 2 Inhibitors. ACS Med. Chem. Lett. 2016, 7, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, K.Y.; Wang de, G.; Liu, P.F.; Cao, Y.W.; Wang, Y.H.; Yang, X.C.; Hu, C.X.; Sun, L.J.; Niu, H.T. Targeting of MCT1 and PFKFB3 influences cell proliferation and apoptosis in bladder cancer by altering the tumor microenvironment. Oncol. Rep. 2016, 36, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lu, C.; Fang, M.; Yan, W.; Chen, M.; Ji, Y.; He, S.; Liu, T.; Chen, T.; Xiao, J. HIF-1α activates hypoxia-induced PFKFB4 expression in human bladder cancer cells. Biochem. Biophys. Res. Commun. 2016, 476, 146–152. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, F.; Wu, X.R. Inhibition of Pyruvate Kinase M2 Markedly Reduces Chemoresistance of Advanced Bladder Cancer to Cisplatin. Sci. Rep. 2017, 7, 45983. [Google Scholar] [CrossRef] [Green Version]
- Massari, F.; Ciccarese, C.; Santoni, M.; Iacovelli, R.; Mazzucchelli, R.; Piva, F.; Scarpelli, M.; Berardi, R.; Tortora, G.; Lopez-Beltran, A.; et al. Metabolic phenotype of bladder cancer. Cancer Treat Rev. 2016, 45, 46–57. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Meredith, D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflug. Arch. 2004, 447, 619–628. [Google Scholar] [CrossRef]
- Parks, S.K.; Chiche, J.; Pouyssegur, J. pH control mechanisms of tumor survival and growth. J. Cell. Physiol. 2011, 226, 299–308. [Google Scholar] [CrossRef]
- Halestrap, A.P. The SLC16 gene family—Structure, role and regulation in health and disease. Mol. Asp. Med. 2013, 34, 337–349. [Google Scholar] [CrossRef]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.-W.; Kim, Y.; Lee, J.-H.; Kim, Y.-S. Prognostic Significance of Lactate/Proton Symporters MCT1, MCT4, and Their Chaperone CD147 Expressions in Urothelial Carcinoma of the Bladder. Urology 2014, 84, 245.e215–245.e249. [Google Scholar] [CrossRef] [PubMed]
- Todenhöfer, T.; Seiler, R.; Stewart, C.; Moskalev, I.; Gao, J.; Ladhar, S.; Kamjabi, A.; Al Nakouzi, N.; Hayashi, T.; Choi, S.; et al. Selective Inhibition of the Lactate Transporter MCT4 Reduces Growth of Invasive Bladder Cancer. Mol. Cancer Ther. 2018, 17, 2746–2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.W.; Kim, W.-J.; Yun, S.J. The role of the tumor microenvironment in bladder cancer development and progression. Transl. Cancer Res. 2017, 6, S744–S758. [Google Scholar] [CrossRef]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Feijoo-Carnero, C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, P.S.; Chamoto, K.; Kumar, A.; Honjo, T. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8+ T Cells and Facilitates Anti–PD-1 Therapy. Cancer Immunol. Res. 2018, 6, 1375. [Google Scholar] [CrossRef] [PubMed]
- Renner, K.; Bruss, C.; Schnell, A.; Koehl, G.; Becker, H.M.; Fante, M.; Menevse, A.-N.; Kauer, N.; Blazquez, R.; Hacker, L.; et al. Restricting Glycolysis Preserves T Cell Effector Functions and Augments Checkpoint Therapy. Cell Rep. 2019, 29, 135–150.e139. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Gnanaprakasam, J.N.R.; Chen, X.; Kang, S.; Xu, X.; Sun, H.; Liu, L.; Rodgers, H.; Miller, E.; Cassel, T.A.; et al. Inosine is an alternative carbon source for CD8(+)-T-cell function under glucose restriction. Nat. Metab. 2020, 2, 635–647. [Google Scholar] [CrossRef]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8(+) T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 2017, 32, 377–391.e379. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Chi, H. mTOR signaling in the differentiation and function of regulatory and effector T cells. Curr. Opin. Immunol. 2017, 46, 103–111. [Google Scholar] [CrossRef]
- Linke, M.; Fritsch, S.D.; Sukhbaatar, N.; Hengstschläger, M.; Weichhart, T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. 2017, 591, 3089–3103. [Google Scholar] [CrossRef]
- Domblides, C.; Lartigue, L.; Faustin, B. Control of the Antitumor Immune Response by Cancer Metabolism. Cells 2019, 8, 104. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Tabernero, J.; Joshua, A.; Desai, J.; Varga, A.I.; Moreno, V.; Gomez-Roca, C.A.; Markman, B.; De Braud, F.G.; Patel, S.P.; et al. BMS-986205, an indoleamine 2, 3-dioxygenase 1 inhibitor (IDO1i), in combination with nivolumab (nivo): Updated safety across all tumor cohorts and efficacy in advanced bladder cancer (advBC). J. Clin. Oncol. 2019, 37, 358. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol. Res. 2017, 5, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Guo, P.; Zhang, Y.; Xiong, H.; Yu, X.; Xu, S.; Wang, X.; He, D.; Jin, X. The antidiabetic drug metformin inhibits the proliferation of bladder cancer cells in vitro and in vivo. Int. J. Mol. Sci. 2013, 14, 24603–24618. [Google Scholar] [CrossRef]
- Baltazar, F.; Afonso, J.; Costa, M.; Granja, S. Lactate beyond a Waste Metabolite: Metabolic Affairs and Signaling in Malignancy. Front. Oncol. 2020, 10, 231. [Google Scholar] [CrossRef] [Green Version]
- Walenta, S.; Mueller-Klieser, W.F. Lactate: Mirror and motor of tumor malignancy. Semin. Radiat. Oncol. 2004, 14, 267–274. [Google Scholar] [CrossRef]
- Phypers, B.; Pierce, J.M.T. Lactate physiology in health and disease. Contin. Educ. Anaesth. Crit. Care Pain 2006, 6, 128–132. [Google Scholar] [CrossRef]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 2011, 39, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef]
- Husain, Z.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate and myeloid-derived suppressor cells: Linking metabolism to cancer immunology. Oncoimmunology 2013, 2, e26383. [Google Scholar] [CrossRef] [Green Version]
- Baumann, T.; Dunkel, A.; Schmid, C.; Schmitt, S.; Hiltensperger, M.; Lohr, K.; Laketa, V.; Donakonda, S.; Ahting, U.; Lorenz-Depiereux, B.; et al. Regulatory myeloid cells paralyze T cells through cell–cell transfer of the metabolite methylglyoxal. Nat. Immunol. 2020, 21, 555–566. [Google Scholar] [CrossRef]
- Komohara, Y.; Fujiwara, Y.; Ohnishi, K.; Takeya, M. Tumor-associated macrophages: Potential therapeutic targets for anti-cancer therapy. Adv. Drug Deliv. Rev. 2016, 99, 180–185. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, D.; Xu, T.; Liu, P.; Cao, Y.; Wang, Y.; Yang, X.; Xu, X.; Wang, X.; Niu, H. Bladder cancer cells re-educate TAMs through lactate shuttling in the microfluidic cancer microenvironment. Oncotarget 2015, 6, 39196–39210. [Google Scholar] [CrossRef] [Green Version]
- Haas, R.; Smith, J.; Rocher-Ros, V.; Nadkarni, S.; Montero-Melendez, T.; D’Acquisto, F.; Bland, E.J.; Bombardieri, M.; Pitzalis, C.; Perretti, M.; et al. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol. 2015, 13, e1002202. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Pilon-Thomas, S.; Kodumudi, K.N.; El-Kenawi, A.E.; Russell, S.; Weber, A.M.; Luddy, K.; Damaghi, M.; Wojtkowiak, J.W.; Mulé, J.J.; Ibrahim-Hashim, A.; et al. Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy. Cancer Res. 2016, 76, 1381–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robey, I.F.; Roe, D.J. Safety and Tolerability of LongTerm Sodium Bicarbonate Consumption in Cancer Care. J. Integr Oncol. 2015, 4, 128. [Google Scholar]
- Daneshmandi, S.; Wegiel, B.; Seth, P. Blockade of Lactate Dehydrogenase-A (LDH-A) Improves Efficacy of Anti-Programmed Cell Death-1 (PD-1) Therapy in Melanoma. Cancers 2019, 11, 450. [Google Scholar] [CrossRef]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A.; et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018, 27, 977–987.e974. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Drug | Target | Combination Therapy | Setting | Identifier |
|---|---|---|---|---|
| INCB001158 | Arginase | Pembrolizumab | Solid tumors, including UC | NCT02903914 |
| L-NMMA | Nitric oxide synthase | Pembrolizumab | Solid tumors, including UC | NCT03236935 |
| BMS-986205 | IDO | Nivolumab | mUC | NCT02658890 |
| KHK2455 | IDO | Avelumab | Platinum-relapsed mUC | NCT03915405 |
| Epacadostat | IDO | Pembrolizumab | Platinum-relapsed mUC | NCT03374488, NCT03361865 |
| Epacadostat | IDO | Pembrolizumab | Neoadjuvant MIUC | NCT03832673, |
| Epacadostat | IDO | Retifanlimab | Neoadjuvant MIUC | NCT04586244 |
| Epacadostat, INCB001158 | Arginase, IDO | Pembrolizumab | Solid tumors, including UC | NCT03361228 |
| Metformin | Hypoxia, oxidative phosphorylation | Nivolumab | NSCLC | NCT03048500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scholtes, M.P.; de Jong, F.C.; Zuiverloon, T.C.M.; Theodorescu, D. Role of Bladder Cancer Metabolic Reprogramming in the Effectiveness of Immunotherapy. Cancers 2021, 13, 288. https://doi.org/10.3390/cancers13020288
Scholtes MP, de Jong FC, Zuiverloon TCM, Theodorescu D. Role of Bladder Cancer Metabolic Reprogramming in the Effectiveness of Immunotherapy. Cancers. 2021; 13(2):288. https://doi.org/10.3390/cancers13020288
Chicago/Turabian StyleScholtes, Mathijs P., Florus C. de Jong, Tahlita C. M. Zuiverloon, and Dan Theodorescu. 2021. "Role of Bladder Cancer Metabolic Reprogramming in the Effectiveness of Immunotherapy" Cancers 13, no. 2: 288. https://doi.org/10.3390/cancers13020288