Autophagy: Mechanisms and Therapeutic Potential of Flavonoids in Cancer


Abstract
:1. Introduction
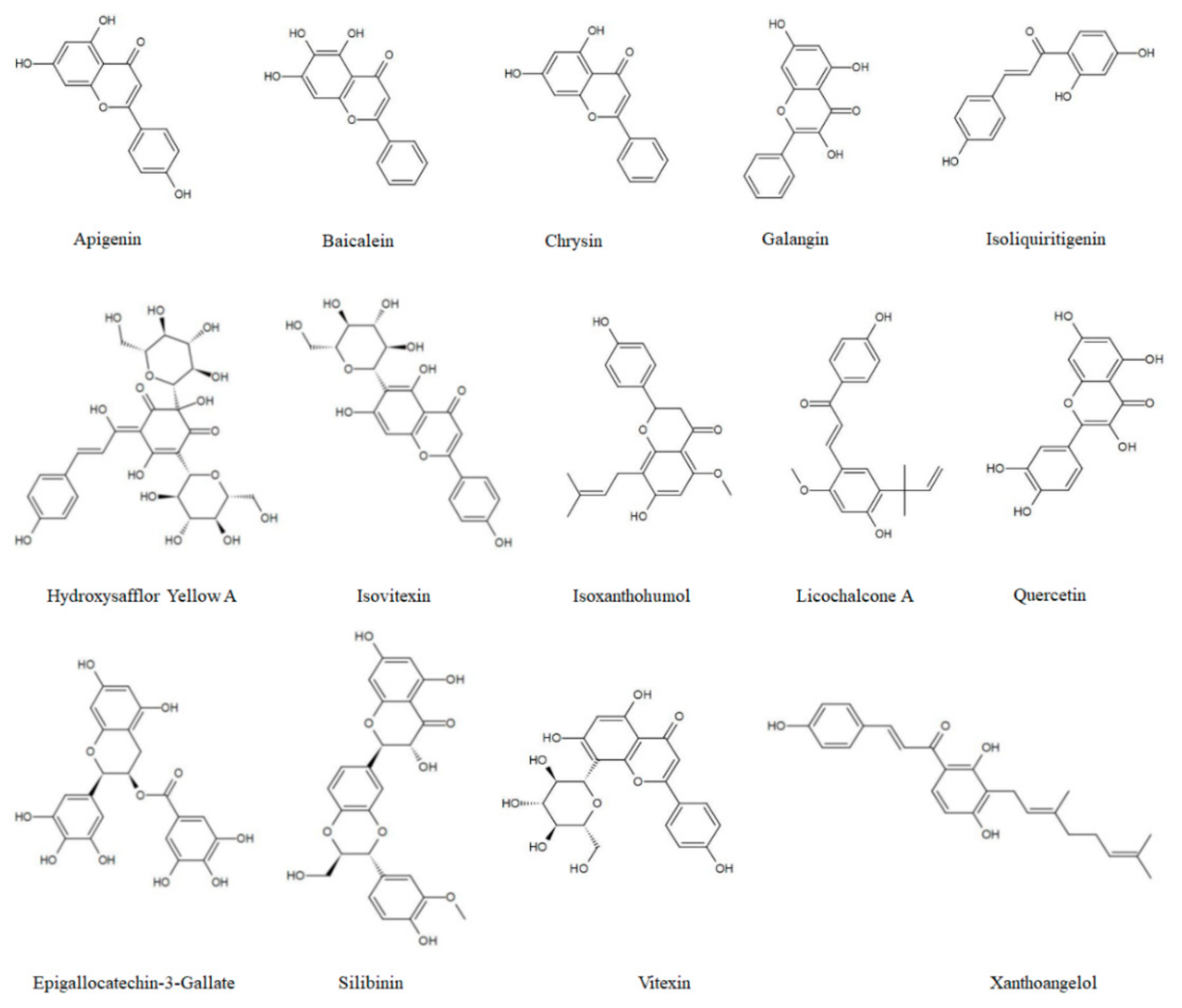
1.1. Flavonoids
1.2. Autophagy Mechanisms
1.3. Role of Autophagy in Cancer
2. Autophagic Cell Death
3. Interaction between Autophagy and Tumor Cell Migration, Invasion, and Angiogenesis
4. A Complex Landscape in Autophagy, the Cell Cycle, and Senescence
5. Crosstalk between Autophagy and Apoptosis
5.1. Synergism between Autophagy and Apoptosis
5.2. Antagonism Amid Autophagy and Apoptosis
5.3. Mutual Transformation in Autophagy and Apoptosis
5.4. Others
6. Targeting Autophagy Regulates MDR
7. Clinical Trials
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-MA | 3-methyladenine |
| 8-PN | 8-prenylnaringenin |
| ABC | ATP-binding cassette |
| ACC | Adenoid cystic carcinoma |
| ACD | Autophagic cell death |
| AKT | Protein kinase B |
| AML | Acute myeloid leukemia |
| AMPK | AMP-activated protein kinase |
| ATGs | Autophagy-related genes |
| BL | Burkitt’s lymphoma |
| BNIP3 | Bcl-2 adenovirus E1B 19-kDa-interacting protein 3 |
| CC | Cervical cancer |
| Cdc25C | Cell division cycle 25c |
| CDK | Cyclin-dependent kinase |
| CHOP | C/EBP homologous protein |
| CKI | Cyclin-dependent kinase inhibitor |
| CQ | chloroquine |
| CRC | Colorectal cancer |
| DAPK | Death-associated protein kinase |
| DDIT3 | DNA damage inducible transcript 3 |
| DRAM | Damage-regulated autophagy modulator |
| DRP-1 | DAPK-related protein kinase |
| DRR | DNA damage response |
| ECM | Extracellular matrix |
| EGCG | Epigallocatechin-3-Gallate |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial-mesenchymal transformation |
| ER | Endoplasmic reticulum |
| FAK | Focal adhesion kinase |
| FIP200 | FAK interacting protein 200kD |
| FLIP | Fas-associated death domain-like IL-1β-converting enzyme inhibitory protein |
| FoxO | Forkhead box class O |
| GC | Gastric cancer |
| GLUT1 | Glucose transporter1 |
| HCC | Hepatocellular carcinoma |
| HIF-1α | Hypoxia-induced factor 1α |
| HMGB1 | High-mobility group box 1 protein |
| HO-1 | Palatino Linotype |
| HSPs | Heat shock protein |
| HSYA | Hydroxysafflor yellow A |
| IGF-1 | Insulin-like growth factor-1 |
| ILK | Integrin-linked kinase |
| ISL | Isoliquiritigenin |
| IXN | Isoxanthohumol |
| Keap-1 | Kelch-like ECH-associated protein 1 |
| LA | Licochalcone A |
| LDHA | Lactate dehydrogenase A |
| LMP: | Lysosomal membrane permeabilization |
| lncRNA | Long non-coding RNA |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MDR | Multidrug resistance |
| MGMT | O6-methylguanine-DNA-methyltransferase |
| miRNAs | MicroRNAs |
| MMPs | Matrix metalloproteinases |
| mTOR | Mammalian target of rapamycin |
| NAC | N-acetylcysteine |
| NB | Neuroblastoma |
| NF-κB | Nuclear factor kappa B |
| Nrf2 | Nuclear factor erythroid 2 |
| NSLC | Non-small cell lung cancer |
| p-AKT | Phosphorylated Akt |
| PCGEM1 | Prostate cancer gene expression marker 1 |
| PE | Phosphatidylethanolamine |
| PEL | Primary effusion lymphoma |
| PI3K | Phosphoinositide 3-kinase |
| p-IκB | Phosphorylation of inhibitor of kappa B |
| PKM2 | Glycolysis-related proteins Pyruvate kinase M2 |
| PLKs | Polo-like kinases |
| p-mTOR | Phosphorylated mTOR |
| PXN | Paxillin |
| RAGE | Receptor for advanced glycation end products |
| RCC | Renal cell carcinoma |
| RCE | Rhus coriaria ethanolic extract |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| RPS6KB1 | Ribosomal protein S6 kinase B1 |
| SASP | Senescence-associated secretory phenotype |
| sEHi | Soluble epoxide hydrolase inhibitor |
| SIL | Silibinin |
| siRNA | Small interfering RNA |
| SIRT1 | Silent information regulator 2 homolog 1 |
| t-AUCB | trans-4[-4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid |
| TCA | Tricarboxylic acid |
| TGF | Transforming growth factor |
| TICs | Tumor-initiating stem cell-like cells |
| TMZ | Temozolomide |
| TNF | Tumor necrosis factor |
| TRAIL | TNF-related apoptosis-inducing ligand |
| TRIB3 | Tribbles pseudokinase 3 |
| UBL | Ubiquitin-like protein |
| ULK1/2 | Unc-51-like kinase |
| UVRAG | Ultra-violet radiation resistance-associated gene protein |
| XAG | Xanthoangelol |
| XBP-1 | X-box binding protein 1 |
| XN | Xanthohumol |
| zVAD-fmk | Benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone |
References
- Chahar, M.; Sharma, N.; Dobhal, M.; Joshi, Y.J.P.R. Flavonoids: A versatile source of anticancer drugs. Pharmacogn. Rev. 2011, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wu, M.; Cai, C.; Li, M.; Lu, J.J.J.O.E. Autophagy modulators from traditional Chinese medicine: Mechanisms and therapeutic potentials for cancer and neurodegenerative diseases. J. Ethnopharmacol. 2016, 194, 861–876. [Google Scholar] [CrossRef]
- Fang, D.; Xiong, Z.; Xu, J.; Yin, J.; Luo, R. Chemopreventive mechanisms of galangin against hepatocellular carcinoma: A review. Biomed. Pharmacother 2019, 109, 2054–2061. [Google Scholar] [CrossRef]
- Liao, C.; Lee, C.; Tsai, C.; Hsueh, C.; Wang, C.; Chen, I.; Tsai, M.; Liu, M.; Hsieh, A.; Su, K.; et al. Novel Investigations of Flavonoids as Chemopreventive Agents for Hepatocellular Carcinoma. BioMed Res. Int. 2015, 2015, 840542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
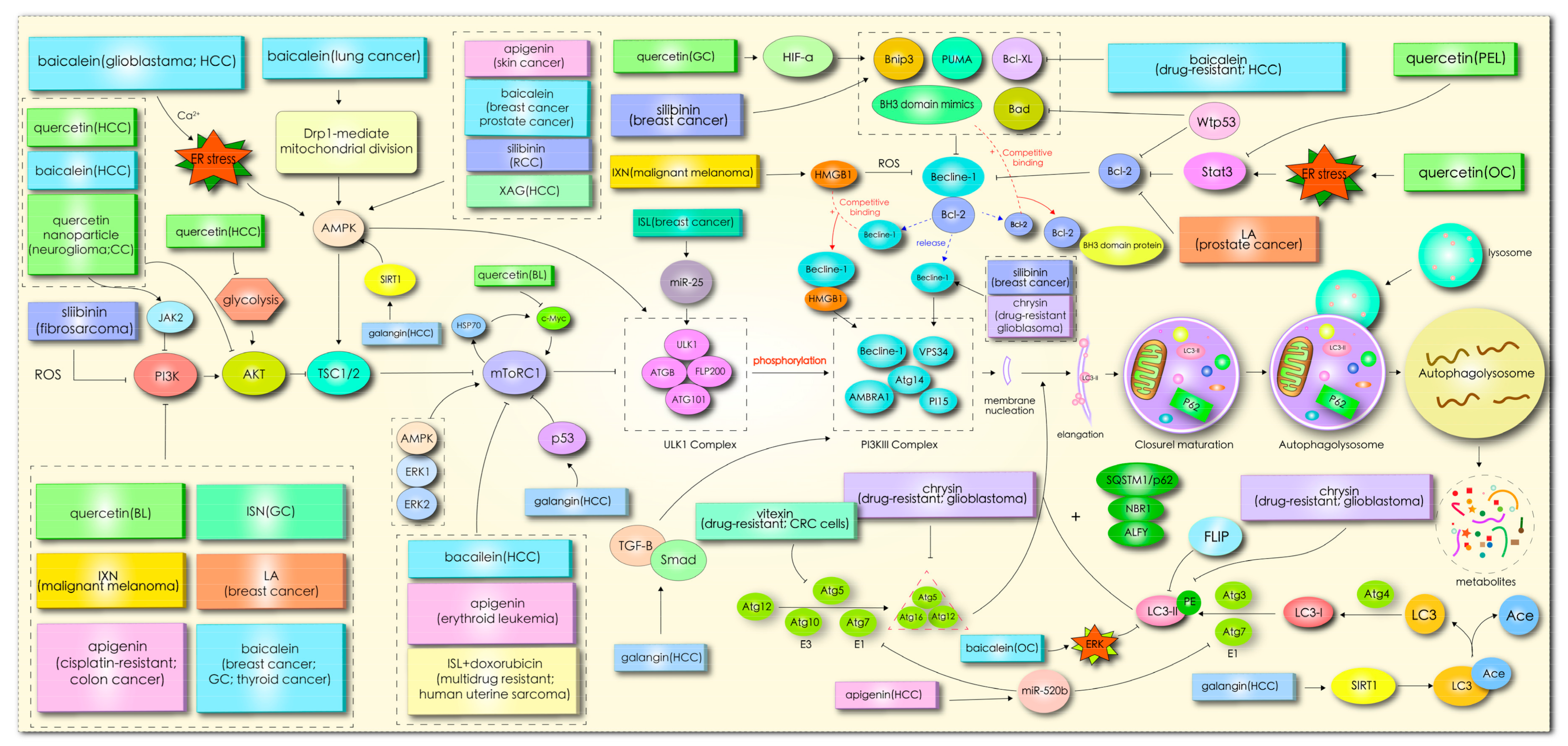
- Tong, X.; Bridgeman, B.B.; Smith, K.A.; Avram, M.J.; Pelling, J.C. AMPK-mTOR axis as key target for chemoprevention of UV-induced skin cancer by the bioflavonoid apigenin. Cancer Res. 2012, 72. [Google Scholar] [CrossRef]
- Bridgeman, B.B.; Wang, P.; Ye, B.; Pelling, J.C.; Volpert, O.V.; Tong, X. Inhibition of mTOR by apigenin in UVB-irradiated keratinocytes: A new implication of skin cancer prevention. Cell Signal. 2016, 28, 460–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiruthiga, C.; Devi, K.; Nabavi, S.; Bishayee, A.J.C. Autophagy: A Potential Therapeutic Target of Polyphenols in Hepatocellular Carcinoma. Cancers 2020, 12, 562. [Google Scholar] [CrossRef] [Green Version]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.P.; Salehi, B.; Sharifi-Rad, M.; Pezzani, R.; Kobarfard, F.; Sharifi-Rad, J.; Nigam, M. Programmed Cell Death, from a Cancer Perspective: An Overview. Mol. Diagn. Ther. 2018, 22, 281–295. [Google Scholar] [CrossRef]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Noda, T.; Ohsumi, Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 1999, 18, 3888–3896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumanomidou, T.; Mizushima, T.; Komatsu, M.; Suzuki, A.; Tanida, I.; Sou, Y.-S.; Ueno, T.; Kominami, E.; Tanaka, K.; Yamane, T. The crystal structure of human Atg4b, a processing and de-conjugating enzyme for autophagosome-forming modifiers. J. Mol. Biol. 2006, 355, 612–618. [Google Scholar] [CrossRef]
- Singh, S.S.; Vats, S.; Chia, A.Y.-Q.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- Yang, X.; Yu, D.-D.; Yan, F.; Jing, Y.-Y.; Han, Z.-P.; Sun, K.; Liang, L.; Hou, J.; Wei, L.-X. The role of autophagy induced by tumor microenvironment in different cells and stages of cancer. Cell Biosci. 2015, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Hippert, M.M.; O’Toole, P.S.; Thorburn, A. Autophagy in cancer: Good, bad, or both? Cancer Res. 2006, 66, 9349–9351. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; White, E. Role of tumor and host autophagy in cancer metabolism. Genes Dev. 2019, 33, 610–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Outschoorn, U.; Balliet, R.; Rivadeneira, D.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.; Lin, Z.; Witkiewicz, A.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010, 9, 3256–3276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.-W.; Feng, J.-N.; Cao, Y.; Meng, L.-P.; Wang, S.-L. Autophagy prevents autophagic cell death in Tetrahymena in response to oxidative stress. Zool. Res. 2015, 36, 167–173. [Google Scholar]
- Shimizu, S.; Yoshida, T.; Tsujioka, M.; Arakawa, S. Autophagic cell death and cancer. Int. J. Mol. Sci. 2014, 15, 3145–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, S.; Konishi, A.; Nishida, Y.; Mizuta, T.; Nishina, H.; Yamamoto, A.; Tsujimoto, Y. Involvement of JNK in the regulation of autophagic cell death. Oncogene 2010, 29, 2070–2082. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Li, X.; Guo, L.; Wu, X.; He, C.; Zhang, S.; Xiao, Y.; Yang, Y.; Hao, D. Combining radiation with autophagy inhibition enhances suppression of tumor growth and angiogenesis in esophageal cancer. Mol. Med. Rep. 2015, 12, 1645–1652. [Google Scholar] [CrossRef] [Green Version]
- Linder, B.; Kögel, D. Autophagy in Cancer Cell Death. Biology 2019, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Xiang, H.; Zhang, J.; Lin, C.; Zhang, L.; Liu, B.; Ouyang, L. Targeting autophagy-related protein kinases for potential therapeutic purpose. Acta Pharm. Sin. B 2020, 10, 569–581. [Google Scholar] [CrossRef]
- Dunlop, E.A.; Tee, A.R. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin. Cell Dev. Biol. 2014, 36, 121–129. [Google Scholar] [CrossRef]
- Sengupta, A.; Molkentin, J.D.; Yutzey, K.E. FoxO transcription factors promote autophagy in cardiomyocytes. J. Biol. Chem. 2009, 284, 28319–28331. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Lee, J.-s.; Inn, K.-s.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandesiri, M.; Chakilam, S.; Ivanovska, J.; Benderska, N.; Ocker, M.; Di Fazio, P.; Feoktistova, M.; Gali-Muhtasib, H.; Rave-Fränk, M.; Prante, O.; et al. Erratum to: DAPK plays an important role in panobinostat-induced autophagy and commits cells to apoptosis under autophagy deficient conditions. Apoptosis 2016, 21, 671–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, A.-M.; Zhang, X.-Y.; Hu, J.-N.; Ke, Z.-P. Apigenin sensitizes hepatocellular carcinoma cells to doxorubic through regulating miR-520b/ATG7 axis. Chem. Biol. Interact. 2018, 280, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cheng, X.; Gao, Y.; Zheng, J.; Xu, Q.; Sun, Y.; Guan, H.; Yu, H.; Sun, Z. Apigenin induces autophagic cell death in human papillary thyroid carcinoma BCPAP cells. Food Funct. 2015, 6, 3464–3472. [Google Scholar] [CrossRef]
- Cao, X.; Liu, B.; Cao, W.; Zhang, W.; Zhang, F.; Zhao, H.; Meng, R.; Zhang, L.; Niu, R.; Hao, X.; et al. Autophagy inhibition enhances apigenin-induced apoptosis in human breast cancer cells. Chin. J. Cancer Res. 2013, 25, 212–222. [Google Scholar] [CrossRef]
- Lee, Y.; Sung, B.; Kang, Y.J.; Kim, D.H.; Jang, J.-Y.; Hwang, S.Y.; Kim, M.; Lim, H.S.; Yoon, J.-H.; Chung, H.Y.; et al. Apigenin-induced apoptosis is enhanced by inhibition of autophagy formation in HCT116 human colon cancer cells. Int. J. Oncol. 2014, 44, 1599–1606. [Google Scholar] [CrossRef] [Green Version]
- Mohan, N.; Banik, N.L.; Ray, S.K. Combination of N-(4-hydroxyphenyl) retinamide and apigenin suppressed starvation-induced autophagy and promoted apoptosis in malignant neuroblastoma cells. Neurosci. Lett. 2011, 502, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Pi, C.; Wang, G. Inhibition of PI3K/Akt/mTOR pathway by apigenin induces apoptosis and autophagy in hepatocellular carcinoma cells. Biomed. Pharmacother. 2018, 103, 699–707. [Google Scholar] [CrossRef]
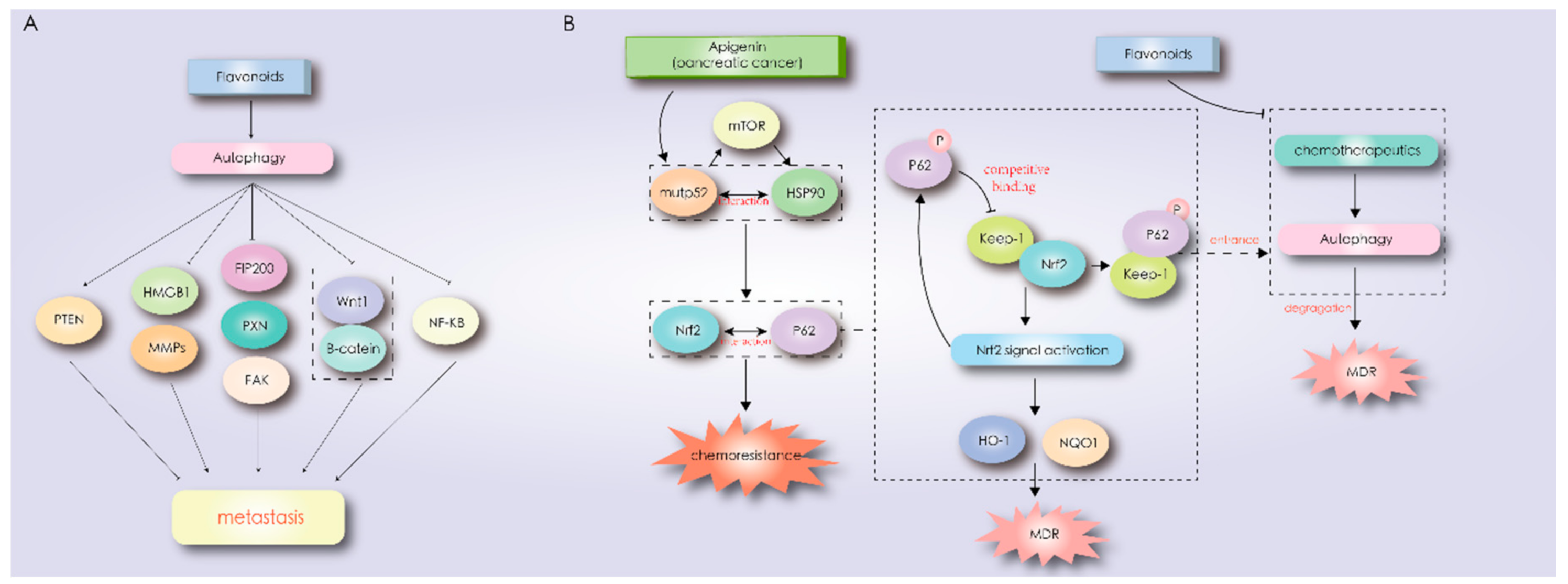
- Gilardini Montani, M.S.; Cecere, N.; Granato, M.; Romeo, M.A.; Falcinelli, L.; Ciciarelli, U.; D’Orazi, G.; Faggioni, A.; Cirone, M. Mutant p53, Stabilized by Its Interplay with HSP90, Activates a Positive Feed-Back Loop between NRF2 and p62 that Induces Chemo-Resistance to Apigenin in Pancreatic Cancer Cells. Cancers 2019, 11, 703. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Xu, H.; Yu, X.; Wang, X.; Zhu, X.; Xu, X. Apigenin inhibits in vitro and in vivo tumorigenesis in cisplatin-resistant colon cancer cells by inducing autophagy, programmed cell death and targeting m-TOR/PI3K/Akt signalling pathway. J. BUON 2019, 24, 488–493. [Google Scholar]
- Chen, Z.; Tian, D.; Liao, X.; Zhang, Y.; Xiao, J.; Chen, W.; Liu, Q.; Chen, Y.; Li, D.; Zhu, L.; et al. Apigenin Combined With Gefitinib Blocks Autophagy Flux and Induces Apoptotic Cell Death Through Inhibition of HIF-1α, c-Myc, p-EGFR, and Glucose Metabolism in EGFR L858R+T790M-Mutated H1975 Cells. Front. Pharmacol. 2019, 10, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhardwaj, M.; Cho, H.J.; Paul, S.; Jakhar, R.; Khan, I.; Lee, S.-J.; Kim, B.-Y.; Krishnan, M.; Khaket, T.P.; Lee, H.G.; et al. Vitexin induces apoptosis by suppressing autophagy in multi-drug resistant colorectal cancer cells. Oncotarget 2018, 9, 3278–3291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, S.-X.; Qiao, X. Isovitexin (IV) induces apoptosis and autophagy in liver cancer cells through endoplasmic reticulum stress. Biochem. Biophys. Res. Commun. 2018, 496, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Liu, G.; Wu, Y.; Liang, Q.; Li, L. Baicalein suppresses the growth of the human thyroid cancer cells by inducing mitotic catastrophe, apoptosis and autophagy via NF-kB signalling pathway. J. BUON 2020, 25, 389–394. [Google Scholar]
- Aryal, P.; Kim, K.; Park, P.-H.; Ham, S.; Cho, J.; Song, K. Baicalein induces autophagic cell death through AMPK/ULK1 activation and downregulation of mTORC1 complex components in human cancer cells. FEBS J. 2014, 281, 4644–4658. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Duan, B.; Guo, Y.; Zhou, R.; Sun, J.; Bie, B.; Yang, S.; Huang, C.; Yang, J.; Li, Z. Baicalein sensitizes hepatocellular carcinoma cells to 5-FU and Epirubicin by activating apoptosis and ameliorating P-glycoprotein activity. Biomed. Pharmacother. 2018, 98, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Qiu, S.; Qin, J. Baicalein induced apoptosis and autophagy of undifferentiated thyroid cancer cells by the ERK/PI3K/Akt pathway. Am. J. Transl. Res. 2019, 11, 3341–3352. [Google Scholar]
- Yan, W.; Ma, X.; Zhao, X.; Zhang, S. Baicalein induces apoptosis and autophagy of breast cancer cells via inhibiting PI3K/AKT pathway in vivo and vitro. Drug Des. Dev. Ther. 2018, 12, 3961–3972. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Liu, J.; Liu, L.; Sun, X.; Huang, J.; Dong, J. Drp1-mediated mitochondrial fission contributes to baicalein-induced apoptosis and autophagy in lung cancer via activation of AMPK signaling pathway. Int. J. Biol. Sci. 2020, 16, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; He, J.; Zou, M.; Chen, W.; Lv, Y.; Li, Y. Small Interfering RNA Target for Long Non-coding RNA PCGEM1 Increases Sensitivity of LNCaP Cells to Baicalein. Anat. Rec. 2020. [Google Scholar] [CrossRef]
- Wu, R.; Murali, R.; Kabe, Y.; French, S.W.; Chiang, Y.-M.; Liu, S.; Sher, L.; Wang, C.C.; Louie, S.; Tsukamoto, H. Baicalein Targets GTPase-Mediated Autophagy to Eliminate Liver Tumor-Initiating Stem Cell-Like Cells Resistant to mTORC1 Inhibition. Hepatology 2018, 68, 1726–1740. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Fang, J.; Wang, H.; Fei, M.; Tang, T.; Liu, K.; Niu, W.; Zhou, Y. Baicalin suppresses proliferation, migration, and invasion in human glioblastoma cells via Ca-dependent pathway. Drug Des. Dev. Ther. 2018, 12, 3247–3261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Lu, M.; Jiang, X.-X.; Pan, M.-X.; Mao, J.-W.; Chen, M. Inhibiting reactive oxygen species-dependent autophagy enhanced baicalein-induced apoptosis in oral squamous cell carcinoma. J. Nat. Med. 2017, 71, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-F.; Li, T.; Tang, Z.-H.; Chang, L.-L.; Zhu, H.; Chen, X.-P.; Wang, Y.-T.; Lu, J.-J. Baicalein Triggers Autophagy and Inhibits the Protein Kinase B/Mammalian Target of Rapamycin Pathway in Hepatocellular Carcinoma HepG2 Cells. Phytother. Res. 2015, 29, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-F.; Xu, Y.-L.; Tang, Z.-H.; Li, T.; Zhang, L.-L.; Chen, X.; Lu, J.-H.; Leung, C.-H.; Ma, D.-L.; Qiang, W.-A.; et al. Baicalein Induces Beclin 1- and Extracellular Signal-Regulated Kinase-Dependent Autophagy in Ovarian Cancer Cells. Am. J. Chin. Med. 2017, 45, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.X.; Jiang, C.P.; Chen, W.B.; Zhang, G.; Luo, D.J.; Cao, Y.; Wu, J.H.; Ding, Y.T.; Liu, B.R. Baicalein Induces Apoptosis and Autophagy via Endoplasmic Reticulum Stress in Hepatocellular Carcinoma Cells. Biomed. Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, C.-L.; Chen, C.-M.; Chang, Y.-Z.; Liu, G.-Y.; Hung, H.-C.; Hsieh, T.-Y.; Lin, C.-L. Pine (Pinus morrisonicola Hayata) Needle Extracts Sensitize GBM8901 Human Glioblastoma Cells to Temozolomide by Downregulating Autophagy and O-6-Methylguanine-DNA Methyltransferase Expression. J. Agric. Food Chem. 2014, 62, 10458–10467. [Google Scholar] [CrossRef]
- Sameiyan, E.; Hayes, A.W.; Karimi, G. The effect of medicinal plants on multiple drug resistance through autophagy: A review of in vitro studies. Eur. J. Pharmacol. 2019, 852, 244–253. [Google Scholar] [CrossRef]
- Jia, L.; Huang, S.; Yin, X.; Zan, Y.; Guo, Y.; Han, L. Quercetin suppresses the mobility of breast cancer by suppressing glycolysis through Akt-mTOR pathway mediated autophagy induction. Life Sci. 2018, 208, 123–130. [Google Scholar] [CrossRef]
- Ji, Y.; Li, L.; Ma, Y.-X.; Li, W.-T.; Li, L.; Zhu, H.-Z.; Wu, M.-H.; Zhou, J.-R. Quercetin inhibits growth of hepatocellular carcinoma by apoptosis induction in part via autophagy stimulation in mice. J. Nutr. Biochem. 2019, 69, 108–119. [Google Scholar] [CrossRef]
- Tomas-Hernández, S.; Blanco, J.; Rojas, C.; Roca-Martínez, J.; Ojeda-Montes, M.J.; Beltrán-Debón, R.; Garcia-Vallvé, S.; Pujadas, G.; Arola, L.; Mulero, M. Resveratrol Potently Counteracts Quercetin Starvation-Induced Autophagy and Sensitizes HepG2 Cancer Cells to Apoptosis. Mol. Nutr. Food Res. 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Jakubowicz-Gil, J.; Langner, E.; Bądziul, D.; Wertel, I.; Rzeski, W. Silencing of Hsp27 and Hsp72 in glioma cells as a tool for programmed cell death induction upon temozolomide and quercetin treatment. Toxicol. Appl. Pharmacol. 2013, 273, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Jakubowicz-Gil, J.; Langner, E.; Wertel, I.; Piersiak, T.; Rzeski, W. Temozolomide, quercetin and cell death in the MOGGCCM astrocytoma cell line. Chem. Biol. Interact. 2010, 188, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tang, C.; Li, L.; Li, R.; Fan, Y. Quercetin blocks t-AUCB-induced autophagy by Hsp27 and Atg7 inhibition in glioblastoma cells in vitro. J. Neurooncol. 2016, 129, 39–45. [Google Scholar] [CrossRef]
- Jakubowicz-Gil, J.; Langner, E.; Bądziul, D.; Wertel, I.; Rzeski, W. Quercetin and sorafenib as a novel and effective couple in programmed cell death induction in human gliomas. Neurotox. Res. 2014, 26, 64–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Li, J.; Liu, T.; Li, S.; Feng, J.; Yu, Q.; Zhang, J.; Chen, J.; Zhou, Y.; Ji, J.; et al. Quercetin shows anti-tumor effect in hepatocellular carcinoma LM3 cells by abrogating JAK2/STAT3 signaling pathway. Cancer Med. 2019, 8, 4806–4820. [Google Scholar] [CrossRef]
- Liu, Y.; Gong, W.; Yang, Z.Y.; Zhou, X.S.; Gong, C.; Zhang, T.R.; Wei, X.; Ma, D.; Ye, F.; Gao, Q.L. Quercetin induces protective autophagy and apoptosis through ER stress via the p-STAT3/Bcl-2 axis in ovarian cancer. Apoptosis 2017, 22, 544–557. [Google Scholar] [CrossRef]
- Granato, M.; Rizzello, C.; Gilardini Montani, M.S.; Cuomo, L.; Vitillo, M.; Santarelli, R.; Gonnella, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Quercetin induces apoptosis and autophagy in primary effusion lymphoma cells by inhibiting PI3K/AKT/mTOR and STAT3 signaling pathways. J. Nutr. Biochem. 2017, 41, 124–136. [Google Scholar] [CrossRef]
- Granato, M.; Rizzello, C.; Romeo, M.A.; Yadav, S.; Santarelli, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Concomitant reduction of c-Myc expression and PI3K/AKT/mTOR signaling by quercetin induces a strong cytotoxic effect against Burkitt’s lymphoma. Int. J. Biochem. Cell Biol. 2016, 79, 393–400. [Google Scholar] [CrossRef]
- Wang, K.; Liu, R.; Li, J.; Mao, J.; Lei, Y.; Wu, J.; Zeng, J.; Zhang, T.; Wu, H.; Chen, L.; et al. Quercetin induces protective autophagy in gastric cancer cells: Involvement of Akt-mTOR- and hypoxia-induced factor 1α-mediated signaling. Autophagy 2011, 7, 966–978. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Moon, J.Y.; Ahn, K.S.; Cho, S.K. Quercetin induces mitochondrial mediated apoptosis and protective autophagy in human glioblastoma U373MG cells. Oxid. Med. Cell Longev. 2013, 2013, 596496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, C.-Y.; Chen, S.-Y.; Kuo, C.-W.; Lu, C.-C.; Yen, G.-C. Quercetin facilitates cell death and chemosensitivity through RAGE/PI3K/AKT/mTOR axis in human pancreatic cancer cells. J. Food Drug Anal. 2019, 27, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Ha, E.J.; Kim, K.Y.; Kim, C.E.; Jun, D.Y.; Kim, Y.H. Enhancement of Quercetin-Induced Apoptosis by Cotreatment with Autophagy Inhibitor Is Associated with Augmentation of BAK-Dependent Mitochondrial Pathway in Jurkat T Cells. Oxid. Med. Cell Longev. 2019, 2019, 7989276. [Google Scholar] [CrossRef] [Green Version]
- Wen, M.; Wu, J.; Luo, H.; Zhang, H.J.P. Galangin induces autophagy through upregulation of p53 in HepG2 cells. Pharmacology 2012, 89, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tang, C.J.O.R. Galangin suppresses human laryngeal carcinoma via modulation of caspase-3 and AKT signaling pathways. Oncol. Rep. 2017, 38, 703–714. [Google Scholar] [CrossRef]
- Enkhbat, T.; Nishi, M.; Yoshikawa, K.; Jun, H.; Tokunaga, T.; Takasu, C.; Kashihara, H.; Ishikawa, D.; Tominaga, M.; Shimada, M. Epigallocatechin-3-gallate Enhances Radiation Sensitivity in Colorectal Cancer Cells Through Nrf2 Activation and Autophagy. Anticancer Res. 2018, 38, 6247–6252. [Google Scholar] [CrossRef]
- Tsai, C.-Y.; Chen, C.-Y.; Chiou, Y.-H.; Shyu, H.-W.; Lin, K.-H.; Chou, M.-C.; Huang, M.-H.; Wang, Y.-F. Epigallocatechin-3-Gallate Suppresses Human Herpesvirus 8 Replication and Induces ROS Leading to Apoptosis and Autophagy in Primary Effusion Lymphoma Cells. Int. J. Mol. Sci. 2017, 19, 16. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Chang, C.; Chen, Y.; Bi, F.; Ji, C.; Liu, W. EGCG overcomes gefitinib resistance by inhibiting autophagy and augmenting cell death through targeting ERK phosphorylation in NSCLC. Onco Targets Ther. 2019, 12, 6033–6043. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Xie, J.; Liu, X.; Li, Z.; Fang, K.; Zhang, L.; Han, M.; Zhang, Z.; Gong, Z.; Lin, X.; et al. Autophagy induction by xanthoangelol exhibits anti-metastatic activities in hepatocellular carcinoma. Cell Biochem. Funct. 2019, 37, 128–138. [Google Scholar] [CrossRef]
- Delmulle, L.; Vanden Berghe, T.; Keukeleire, D.D.; Vandenabeele, P. Treatment of PC-3 and DU145 prostate cancer cells by prenylflavonoids from hop (Humulus lupulus L.) induces a caspase-independent form of cell death. Phytother. Res. 2008, 22, 197–203. [Google Scholar] [CrossRef]
- Krajnović, T.; Drača, D.; Kaluđerović, G.N.; Dunđerović, D.; Mirkov, I.; Wessjohann, L.A.; Maksimović-Ivanić, D.; Mijatović, S. The hop-derived prenylflavonoid isoxanthohumol inhibits the formation of lung metastasis in B16-F10 murine melanoma model. Food Chem. Toxicol. 2019, 129, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Krajnović, T.; Kaluđerović, G.N.; Wessjohann, L.A.; Mijatović, S.; Maksimović-Ivanić, D. Versatile antitumor potential of isoxanthohumol: Enhancement of paclitaxel activity in vivo. Pharmacol. Res. 2016, 105, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qi, Q.; Zhou, W.; Feng, Z.; Huang, B.; Chen, A.; Zhang, D.; Li, W.; Zhang, Q.; Jiang, Z.; et al. Inhibition of glioma growth by flavokawain B is mediated through endoplasmic reticulum stress induced autophagy. Autophagy 2018, 14, 2007–2022. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.; Zhang, W.-J.; Fan, Q.-X.; Wang, L.-X. Licochalcone A inhibits PI3K/Akt/mTOR signaling pathway activation and promotes autophagy in breast cancer cells. Oncol. Lett. 2018, 15, 1869–1873. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-C.; Wu, C.-H.; Shieh, T.-M.; Chen, H.-Y.; Huang, T.-C.; Hsia, S.-M. The licorice dietary component isoliquiritigenin chemosensitizes human uterine sarcoma cells to doxorubicin and inhibits cell growth by inducing apoptosis and autophagy via inhibition of m-TOR signaling. J. Funct. Foods 2017, 33, 332–344. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, N.; Liu, P.; Chen, Q.; Situ, H.; Xie, T.; Zhang, J.; Peng, C.; Lin, Y.; Chen, J. MicroRNA-25 regulates chemoresistance-associated autophagy in breast cancer cells, a process modulated by the natural autophagy inducer isoliquiritigenin. Oncotarget 2014, 5, 7013–7026. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-R.; Wang, S.-Y.; Sun, W.; Wei, C. Isoliquiritigenin inhibits proliferation and metastasis of MKN28 gastric cancer cells by suppressing the PI3K/AKT/mTOR signaling pathway. Mol. Med. Rep. 2018, 18, 3429–3436. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Liu, L.; Liu, Y.; Wang, S.; Zhang, S.; Dong, R.; Xu, M.; Ma, Y.; Wang, J.; Zhang, Q.; et al. Hydroxysafflor yellow A induces autophagy in human liver cancer cells by regulating Beclin 1 and ERK expression. Exp. Ther. Med. 2020, 19, 2989–2996. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Jin, X.; Li, Q.; Tashiro, S.-I.; Onodera, S.; Ikejima, T. Silibinin induced autophagic and apoptotic cell death in HT1080 cells through a reactive oxygen species pathway. J. Pharmacol. Sci. 2010, 113, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.-J.; Li, Q.-S.; Xia, M.-Y.; Tashiro, S.-I.; Onodera, S.; Ikejima, T. Silibinin activated p53 and induced autophagic death in human fibrosarcoma HT1080 cells via reactive oxygen species-p38 and c-Jun N-terminal kinase pathways. Biol. Pharm. Bull. 2011, 34, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Jiang, K.; Wang, W.; Jin, X.; Wang, Z.; Ji, Z.; Meng, G. Silibinin, a natural flavonoid, induces autophagy via ROS-dependent mitochondrial dysfunction and loss of ATP involving BNIP3 in human MCF7 breast cancer cells. Oncol. Rep. 2015, 33, 2711–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, S.; Li, L.; Chen, S.; Yu, Y.; Qi, M.; Tashiro, S.-I.; Onodera, S.; Ikejima, T. Silibinin induced-autophagic and apoptotic death is associated with an increase in reactive oxygen and nitrogen species in HeLa cells. Free Radic. Res. 2011, 45, 1307–1324. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ma, Z.; Guan, Z.; Chen, Y.; Wu, K.; Guo, P.; Wang, X.; He, D.; Zeng, J. Autophagy induction by silibinin positively contributes to its anti-metastatic capacity via AMPK/mTOR pathway in renal cell carcinoma. Int. J. Mol. Sci. 2015, 16, 8415–8429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prajapati, V.; Kale, R.K.; Singh, R.P. Silibinin combination with arsenic strongly inhibits survival and invasiveness of human prostate carcinoma cells. Nutr. Cancer 2015, 67, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Jin, S.; Jiang, Z.; Wang, J. Inhibitory effects of silibinin on proliferation and lung metastasis of human high metastasis cell line of salivary gland adenoid cystic carcinoma via autophagy induction. Onco Targets Ther. 2016, 9, 6609–6618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, B.; Chung, H.Y.; Kim, N.D. Role of Apigenin in Cancer Prevention via the Induction of Apoptosis and Autophagy. J. Cancer Prev. 2016, 21, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Psahoulia, F.H.; Moumtzi, S.; Roberts, M.L.; Sasazuki, T.; Shirasawa, S.; Pintzas, A. Quercetin mediates preferential degradation of oncogenic Ras and causes autophagy in Ha-RAS-transformed human colon cells. Carcinogenesis 2007, 28, 1021–1031. [Google Scholar] [CrossRef]
- Chi, S.; Kitanaka, C.; Noguchi, K.; Mochizuki, T.; Nagashima, Y.; Shirouzu, M.; Fujita, H.; Yoshida, M.; Chen, W.; Asai, A.; et al. Oncogenic Ras triggers cell suicide through the activation of a caspase-independent cell death program in human cancer cells. Oncogene 1999, 18, 2281–2290. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Li, L.; Zeng, J.; Gao, Y.; Chen, Y.-L.; Wang, Z.-Q.; Wang, X.-Y.; Chang, L.S.; He, D. Inhibitory effect of silibinin on EGFR signal-induced renal cell carcinoma progression via suppression of the EGFR/MMP-9 signaling pathway. Oncol. Rep. 2012, 28. [Google Scholar] [CrossRef] [Green Version]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J. 2018, 285, 1751–1766. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.; Li, C.; Zheng, W.; Guo, Q.; Zhang, Y.; Kang, M.; Zhang, B.; Yang, B.; Li, B.; Yang, H.; et al. Inhibition of autophagy promotes metastasis and glycolysis by inducing ROS in gastric cancer cells. Oncotarget 2015, 6, 39839–39854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.-T.; Liu, H.; Mao, M.-J.; Tan, Y.; Mo, X.-Q.; Meng, X.-J.; Cao, M.-T.; Zhong, C.-Y.; Liu, Y.; Shan, H.; et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol. Cancer 2019, 18, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gugnoni, M.; Sancisi, V.; Manzotti, G.; Gandolfi, G.; Ciarrocchi, A. Autophagy and epithelial-mesenchymal transition: An intricate interplay in cancer. Cell Death Dis. 2016, 7, e2520. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef] [Green Version]
- Sachdev, U.; Cui, X.; Hong, G.; Namkoong, S.; Karlsson, J.M.; Baty, C.J.; Tzeng, E. High mobility group box 1 promotes endothelial cell angiogenic behavior in vitro and improves muscle perfusion in vivo in response to ischemic injury. J. Vasc. Surg. 2012, 55, 180–191. [Google Scholar] [CrossRef] [Green Version]
- De, A.; De, A.; Papasian, C.; Hentges, S.; Banerjee, S.; Haque, I.; Banerjee, S.K. Emblica officinalis extract induces autophagy and inhibits human ovarian cancer cell proliferation, angiogenesis, growth of mouse xenograft tumors. PLoS ONE 2013, 8, e72748. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Hou, T.; Dan, W.; Liu, T.; Luan, J.; Liu, B.; Li, L.; Zeng, J. Silibinin inhibits epithelial-mesenchymal transition of renal cell carcinoma through autophagy-dependent Wnt/β-catenin signaling. Int. J. Mol. Med. 2020, 45, 1341–1350. [Google Scholar] [CrossRef] [Green Version]
- Coca-Pelaz, A.; Rodrigo, J.P.; Bradley, P.J.; Vander Poorten, V.; Triantafyllou, A.; Hunt, J.L.; Strojan, P.; Rinaldo, A.; Haigentz, M.; Takes, R.P.; et al. Adenoid cystic carcinoma of the head and neck—An update. Oral Oncol. 2015, 51, 652–661. [Google Scholar] [CrossRef]
- Mathiassen, S.G.; De Zio, D.; Cecconi, F. Autophagy and the Cell Cycle: A Complex Landscape. Front. Oncol. 2017, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.; He, Z.; Kitazato, K.; Wang, Y. Selective Autophagy Regulates Cell Cycle in Cancer Therapy. Theranostics 2019, 9, 104–125. [Google Scholar] [CrossRef]
- Anand, S.K.; Sharma, A.; Singh, N.; Kakkar, P. Entrenching role of cell cycle checkpoints and autophagy for maintenance of genomic integrity. DNA Repair 2020, 86, 102748. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Alzahrani, A.M.; Hanieh, H.N.; Kumar, S.A.; Ben Ammar, R.; Rengarajan, T.; Alhoot, M.A. Autophagy and senescence: A new insight in selected human diseases. J. Cell Physiol. 2019, 234, 21485–21492. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.J.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.J.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gewirtz, D.A. The four faces of autophagy: Implications for cancer therapy. Cancer Res. 2014, 74, 647–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, Y.; Wei, J.; Zhao, C.; Li, G. Natural Polyphenols Targeting Senescence: A Novel Prevention and Therapy Strategy for Cancer. Int. J. Mol. Sci. 2020, 21, 684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruela-de-Sousa, R.R.; Fuhler, G.M.; Blom, N.; Ferreira, C.V.; Aoyama, H.; Peppelenbosch, M.P. Cytotoxicity of apigenin on leukemia cell lines: Implications for prevention and therapy. Cell Death Dis. 2010, 1, e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Hasasna, H.; Athamneh, K.; Al Samri, H.; Karuvantevida, N.; Al Dhaheri, Y.; Hisaindee, S.; Ramadan, G.; Al Tamimi, N.; AbuQamar, S.; Eid, A.; et al. Rhus coriaria induces senescence and autophagic cell death in breast cancer cells through a mechanism involving p38 and ERK1/2 activation. Sci. Rep. 2015, 5, 13013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewinska, A.; Adamczyk-Grochala, J.; Kwasniewicz, E.; Deregowska, A.; Wnuk, M. Diosmin-induced senescence, apoptosis and autophagy in breast cancer cells of different p53 status and ERK activity. Toxicol. Lett. 2017, 265, 117–130. [Google Scholar] [CrossRef]
- Corcelle, E.; Djerbi, N.; Mari, M.; Nebout, M.; Fiorini, C.; Fénichel, P.; Hofman, P.; Poujeol, P.; Mograbi, B. Control of the autophagy maturation step by the MAPK ERK and p38: Lessons from environmental carcinogens. Autophagy 2007, 3, 57–59. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Pei, F.; Yang, F.; Li, L.; Amin, A.D.; Liu, S.; Buchan, J.R.; Cho, W.C. Role of Autophagy and Apoptosis in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2017, 18, 367. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: Where do they meet? Apoptosis 2014, 19, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Gao, P.; Zhang, J. Crosstalk between Autophagy and Apoptosis: Potential and Emerging Therapeutic Targets for Cardiac Diseases. Int. J. Mol. Sci. 2016, 17, 332. [Google Scholar] [CrossRef] [PubMed]
- Oral, O.; Oz-Arslan, D.; Itah, Z.; Naghavi, A.; Deveci, R.; Karacali, S.; Gozuacik, D. Cleavage of Atg3 protein by caspase-8 regulates autophagy during receptor-activated cell death. Apoptosis 2012, 17, 810–820. [Google Scholar] [CrossRef]
- White, E. Autophagy and p53. Cold Spring Harb. Perspect Med. 2016, 6, a026120. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- D’Orazi, G.; Cirone, M. Mutant p53 and Cellular Stress Pathways: A Criminal Alliance That Promotes Cancer Progression. Cancers 2019, 11, 614. [Google Scholar] [CrossRef] [Green Version]
- Sisinni, L.; Pietrafesa, M.; Lepore, S.; Maddalena, F.; Condelli, V.; Esposito, F.; Landriscina, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Breast Cancer: The Balance between Apoptosis and Autophagy and Its Role in Drug Resistance. Int. J. Mol. Sci. 2019, 20, 857. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.-H.; Eo, S.K.; Lee, J.H.; Park, S.-Y. Quercetin-induced autophagy flux enhances TRAIL-mediated tumor cell death. Oncol. Rep. 2015, 34, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Kwak, M.; Yang, K.; Park, J.; Lee, S.; Park, Y.; Hong, E.; Sun, E.; An, H.; Park, S.; Pang, K.; et al. Galangin enhances TGF-β1-mediated growth inhibition by suppressing phosphorylation of threonine 179 residue in Smad3 linker region. Biochem. Biophys. Res. Commun. 2017, 494, 706–713. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, J.; Lin, B.; Li, X.; Zhang, H.; Ding, H.; Chen, X.; Lan, L.; Luo, H.J.T. Galangin suppresses HepG2 cell proliferation by activating the TGF-β receptor/Smad pathway. Toxicology 2014, 326, 9–17. [Google Scholar] [CrossRef]
- Zhang, H.; Li, N.; Wu, J.; Su, L.; Chen, X.; Lin, B.; Luo, H.J.E.J.O.P. Galangin inhibits proliferation of HepG2 cells by activating AMPK via increasing the AMP/TAN ratio in a LKB1-independent manner. Eur. J. Pharmacol. 2013, 718, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.; Xiong, Y.; Wu, J.; Ding, H.; Chen, X.; Lan, L.; Zhang, H.J.S.R. Galangin Induces Autophagy via Deacetylation of LC3 by SIRT1 in HepG2 Cells. Sci. Rep. 2016, 6, 30496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.-L.; Chow, J.-M.; Chang, J.-H.; Wen, Y.-C.; Lin, Y.-W.; Yang, S.-F.; Lee, W.-J.; Chien, M.-H. Quercetin simultaneously induces G /G -phase arrest and caspase-mediated crosstalk between apoptosis and autophagy in human leukemia HL-60 cells. Environ. Toxicol. 2017, 32, 1857–1868. [Google Scholar] [CrossRef] [PubMed]
- Calgarotto, A.K.; Maso, V.; Junior, G.C.F.; Nowill, A.E.; Filho, P.L.; Vassallo, J.; Saad, S.T.O. Antitumor activities of Quercetin and Green Tea in xenografts of human leukemia HL60 cells. Sci. Rep. 2018, 8, 3459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holczer, M.; Besze, B.; Zámbó, V.; Csala, M.; Bánhegyi, G.; Kapuy, O. Epigallocatechin-3-Gallate (EGCG) Promotes Autophagy-Dependent Survival via Influencing the Balance of mTOR-AMPK Pathways upon Endoplasmic Reticulum Stress. Oxid. Med. Cell Longev. 2018, 2018, 6721530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Zhou, Y.; Fan, J.; Cao, S.; Cao, T.; Huang, F.; Zhuang, S.; Wang, Y.; Yu, X.; Mao, H. Heat shock protein 72 enhances autophagy as a protective mechanism in lipopolysaccharide-induced peritonitis in rats. Am. J. Pathol. 2011, 179, 2822–2834. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Fröhlich, L.J.B. p53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, J.C.; Lu, Q.-Y.; Li, G.; Moro, A.; Takahashi, H.; Chen, M.; Go, V.L.W.; Reber, H.A.; Eibl, G.; Hines, O.J. Evidence for activation of mutated p53 by apigenin in human pancreatic cancer. Biochim. Biophys. Acta 2012, 1823, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.A.; Khathayer, F.; Ray, S.K. Quercetin and Sodium Butyrate Synergistically Increase Apoptosis in Rat C6 and Human T98G Glioblastoma Cells Through Inhibition of Autophagy. Neurochem. Res. 2019, 44, 1715–1725. [Google Scholar] [CrossRef]
- Chakrabarti, M.; Ray, S.K. Anti-tumor activities of luteolin and silibinin in glioblastoma cells: Overexpression of miR-7-1-3p augmented luteolin and silibinin to inhibit autophagy and induce apoptosis in glioblastoma in vivo. Apoptosis 2016, 21, 312–328. [Google Scholar] [CrossRef]
- Iwamoto, F.; Kreisl, T.; Kim, L.; Duic, J.; Butman, J.; Albert, P.; Fine, H.J.C. Phase 2 trial of talampanel, a glutamate receptor inhibitor, for adults with recurrent malignant gliomas. Cancer Int. Int. J. Am. Cancer Soc. 2010, 116, 1776–1782. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Shen, C.; Li, C.; Liu, Y.; Gao, D.; Shi, C.; Peng, F.; Liu, Z.; Zhao, B.; Zheng, Z.; et al. Inhibition of autophagy induced by quercetin at a late stage enhances cytotoxic effects on glioma cells. Tumour. Biol. 2016, 37, 3549–3560. [Google Scholar] [CrossRef] [PubMed]
- Qiao, D.; Li, Y.; Xing, J.; Sun, P.; Wang, Y.; Zhang, Y.; Chen, L.; Ren, X.; Lin, Z.; Jin, J.; et al. Baicalein inhibits PI3K/AKT signaling pathway and induces autophagy of MGC-803 cells. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2019, 35, 613–618. [Google Scholar] [PubMed]
- Liu, B.Y.; Ding, L.L.; Zhang, L.; Wang, S.; Wang, Y.; Wang, B.; Li, L. Baicalein Induces Autophagy and Apoptosis through AMPK Pathway in Human Glioma Cells. Am. J. Chin. Med. 2019, 47, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Yo, Y.T.; Shieh, G.S.; Hsu, K.F.; Wu, C.L.; Shiau, A.L. Licorice and licochalcone-A induce autophagy in LNCaP prostate cancer cells by suppression of Bcl-2 expression and the mTOR pathway. J. Agric. Food Chem. 2009, 57, 8266–8273. [Google Scholar] [CrossRef]
- Lou, M.; Zhang, L.N.; Ji, P.G.; Feng, F.Q.; Liu, J.H.; Yang, C.; Li, B.F.; Wang, L. Quercetin nanoparticles induced autophagy and apoptosis through AKT/ERK/Caspase-3 signaling pathway in human neuroglioma cells: In vitro and in vivo. Biomed. Pharmacother. 2016, 84, 1–9. [Google Scholar] [CrossRef]
- Luo, C.L.; Liu, Y.Q.; Wang, P.; Song, C.H.; Wang, K.J.; Dai, L.P.; Zhang, J.Y.; Ye, H. The effect of quercetin nanoparticle on cervical cancer progression by inducing apoptosis, autophagy and anti-proliferation via JAK2 suppression. Biomed. Pharmacother. 2016, 82, 595–605. [Google Scholar] [CrossRef]
- Li, Y.-J.; Lei, Y.-H.; Yao, N.; Wang, C.-R.; Hu, N.; Ye, W.-C.; Zhang, D.-M.; Chen, Z.-S. Autophagy and multidrug resistance in cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, Z.; Nie, Y.; Shi, Y.; Fan, D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett. 2014, 347, 159–166. [Google Scholar] [CrossRef]
- Li, J.; Hou, N.; Faried, A.; Tsutsumi, S.; Kuwano, H. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Eur. J. Cancer 2010, 46, 1900–1909. [Google Scholar] [CrossRef]
- O’Donovan, T.R.; O’Sullivan, G.C.; McKenna, S.L. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011, 7, 509–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, L.S.; Cheng, J.C.; Beckham, T.H.; Keane, T.E.; Norris, J.S.; Liu, X. Autophagy is increased in prostate cancer cells overexpressing acid ceramidase and enhances resistance to C6 ceramide. Prostate Cancer Prostatic Dis. 2011, 14, 30–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Yang, M.; Kang, R.; Wang, Z.; Zhao, Y.; Yu, Y.; Xie, M.; Yin, X.; Livesey, K.M.; Lotze, M.T.; et al. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia 2011, 25, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yu, Y.; Kang, R.; Yang, M.; Xie, M.; Wang, Z.; Tang, D.; Zhao, M.; Liu, L.; Zhang, H.; et al. Up-regulated autophagy by endogenous high mobility group box-1 promotes chemoresistance in leukemia cells. Leuk. Lymphoma 2012, 53, 315–322. [Google Scholar] [CrossRef]
- Yu, Y.; Yang, L.; Zhao, M.; Zhu, S.; Kang, R.; Vernon, P.; Tang, D.; Cao, L. Targeting microRNA-30a-mediated autophagy enhances imatinib activity against human chronic myeloid leukemia cells. Leukemia 2012, 26, 1752–1760. [Google Scholar] [CrossRef]
- Zou, Z.; Wu, L.; Ding, H.; Wang, Y.; Zhang, Y.; Chen, X.; Chen, X.; Zhang, C.-Y.; Zhang, Q.; Zen, K. MicroRNA-30a sensitizes tumor cells to cis-platinum via suppressing beclin 1-mediated autophagy. J. Biol. Chem. 2012, 287, 4148–4156. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Tang, J.; Li, C.; Kong, J.; Wang, J.; Wu, Y.; Xu, E.; Lai, M. MiR-22 regulates 5-FU sensitivity by inhibiting autophagy and promoting apoptosis in colorectal cancer cells. Cancer Lett. 2015, 356, 781–790. [Google Scholar] [CrossRef]
- Pennati, M.; Lopergolo, A.; Profumo, V.; De Cesare, M.; Sbarra, S.; Valdagni, R.; Zaffaroni, N.; Gandellini, P.; Folini, M. miR-205 impairs the autophagic flux and enhances cisplatin cytotoxicity in castration-resistant prostate cancer cells. Biochem. Pharmacol. 2014, 87, 579–597. [Google Scholar] [CrossRef]
- Wei, Y.; Zou, Z.; Becker, N.; Anderson, M.; Sumpter, R.; Xiao, G.; Kinch, L.; Koduru, P.; Christudass, C.S.; Veltri, R.W.; et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013, 154, 1269–1284. [Google Scholar] [CrossRef] [Green Version]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, Y.; Waguri, S.; Sou, Y.-S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferry, D.R.; Smith, A.; Malkhandi, J.; Fyfe, D.W.; de Takats, P.G.; Anderson, D.; Baker, J.; Kerr, D.J. Phase I clinical trial of the flavonoid quercetin: Pharmacokinetics and evidence for in vivo tyrosine kinase inhibition. Clin. Cancer Res. 1996, 2, 659–668. [Google Scholar] [PubMed]
- Yan, X.; Qi, M.; Li, P.; Zhan, Y.; Shao, H. Apigenin in cancer therapy: Anti-cancer effects and mechanisms of action. Cell Biosci. 2017, 7, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talib, W.H.; Alsalahat, I.; Daoud, S.; Abutayeh, R.F.; Mahmod, A.I. Plant-Derived Natural Products in Cancer Research: Extraction, Mechanism of Action, and Drug Formulation. Molecules 2020, 25, 5319. [Google Scholar] [CrossRef]
- Storka, A.; Vcelar, B.; Klickovic, U.; Gouya, G.; Weisshaar, S.; Aschauer, S.; Bolger, G.; Helson, L.; Wolzt, M. Safety, tolerability and pharmacokinetics of liposomal curcumin in healthy humans. Int. J. Clin. Pharmacol. Ther. 2015, 53, 54–65. [Google Scholar] [CrossRef]
- Bayet-Robert, M.; Kwiatkowski, F.; Leheurteur, M.; Gachon, F.; Planchat, E.; Abrial, C.; Mouret-Reynier, M.-A.; Durando, X.; Barthomeuf, C.; Chollet, P. Phase I dose escalation trial of docetaxel plus curcumin in patients with advanced and metastatic breast cancer. Cancer Biol. Ther. 2010, 9. [Google Scholar] [CrossRef] [Green Version]
- Belcaro, G.; Hosoi, M.; Pellegrini, L.; Appendino, G.; Ippolito, E.; Ricci, A.; Ledda, A.; Dugall, M.; Cesarone, M.R.; Maione, C.; et al. A controlled study of a lecithinized delivery system of curcumin (Meriva®) to alleviate the adverse effects of cancer treatment. Phytother. Res. 2014, 28, 444–450. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, Y.; Zhang, Y.; Wan, X.; Li, J.; Liu, K.; Wang, F.; Liu, K.; Liu, Q.; Yang, C.; et al. Anti-cancer activities of tea epigallocatechin-3-gallate in breast cancer patients under radiotherapy. Curr. Mol. Med. 2012, 12, 163–176. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Flavonoids | Cancer Model Cell Line/Animal | Mechanisms | Pharmacological Effects | Ref. | |
|---|---|---|---|---|---|
| Flavones | Apigenin | Skin cancer SKH-1 mouse | Autophagy induced by AMPK-mTOR axis to prevent UV-mediated skin cancer | - | [6,7] |
| Hepatocellular carcinoma doxorubicin-resistant cell BEL-7402/ADM Nude mice | Sensitizes drug-resistant cells to doxorubic through suppressing miR-520b/ATG7 axis | Autophagy promotes the occurrence of MDR | [36] | ||
| Papillary thyroid carcinoma BCPAP cells |
| Induction of autophagic cell death | [37] | ||
| Breast cancer T47D, MDA-MB-231 Colorectal cancer HCT116 Hepatocellular carcinoma Hepg2 Male BALB/c nude mice Neuroblastoma SH-SY5Y, SK-N-BE2, IMR-32 (N-(4-hydroxyphenyl) retinamide + apigenin) |
| Induction of protective autophagy and inhibition of apoptosis | [38,39,40,41] | ||
| Pancreatic cancer Panc1, PaCa44 |
| - | [42] | ||
| Colorectal cancer cisplatin-resistant cell HT-29 BALB/c nude mice | Induces autophagic cell death and inhibits the growth of cells by targeting m-TOR/PI3K/AKT signaling pathway | Autophagy inhibits the occurrence of MDR | [43] | ||
| Non-small cell lung cancer EGFR-TKIs-resistant NCI-H1975 (Apigenin + Gefitinib) |
| Inhibition of protective autophagy and induction of apoptosis | [44] | ||
| Vitexin | Colorectal cancer Multidrug-resistant cell line HCT-116DR BALB/c pathogen-free athymic nude mice | Decreases the expression of ATG5 and BECN1 and the transformation of LC3-I to LC3-II to inhibit autophagy, enhance apoptosis, and overcome MDR | Autophagy inhibition promotes apoptosis and overcomes MDR | [45] | |
| Isovitexin | Hepatocellular carcinoma Hepg2, SK-Hep1 Hepg2 tumor-bearing mouse |
| Induction of autophagy promotes apoptosis | [46] | |
| Baicalein | Thyroid carcinoma MDA-T68 |
| Induction of autophagic cell death | [47] | |
| Prostate cancer PC-3, DU145 Breast cancer MDA-MB-231 |
| Induction of autophagic cell death | [48] | ||
| Hepatocellular carcinoma 5-FU-resistant cell BEL-7402/5-FU |
| Induction of autophagy to overcome MDR | [49] | ||
| Breast cancer MCF-7, MDA-MB-231 Female BALB/c nude mice Thyroid cancer Fro |
| - | [50,51] | ||
| Non-small cell lung cancer A549, H1299 Male C57BL/6 mice |
| - | [52] | ||
| Prostate cancer LNCaP | siRNA targeted for PCGEM1 increases sensitivity of LNCaP cells to baicalein | - | [53] | ||
| Hepatocellular carcinoma TICDR, Huh7 Male NSGTM mice (NOD-SCID-Il2rg−/− mice) with HCC cells obtained from a patient |
| Inhibition of autophagy and overcoming MDR | [54] | ||
| Glioblastoma U87, U251 Male BALB/c athymic nude mice |
| Autophagy enhances the proportion of apoptotic cells | [55] | ||
| Oral squamous cell carcinoma Cal27 | Induction of protective autophagy by promoting ROS pathway and apoptosis | Autophagy reduces the proportion of apoptotic cells | [56] | ||
| Hepatocellular carcinoma Hepg2 |
| Induction of protective autophagy | [57] | ||
| Ovarian cancer HEY, A2780 | The activation of ERK might cause protective autophagy and apoptosis | Autophagy reduces the proportion of apoptotic cells | [58] | ||
| Hepatocellular carcinoma SMMC-7721, Bel-7402 |
| Autophagy induction reduces cell apoptosis | [59] | ||
| Dihydroflavone | Chrysin | Glioblastoma TMZ-resistant cell GBM8901 |
| Autophagy triggers the occurrence of MDR | [60,61] |
| Flavonols | Quercetin | Breast cancer MCF-7, MDA-MB-231 Female BALB/c nude mice |
| Induction of autophagy to inhibit cell migration and invasion | [62] |
| Hepatocellular carcinoma SMMC7721, HepG2 Male BALB/c nude mice |
| Induction of autophagy to stimulate apoptotic cell death | [63] | ||
| Hepatocellular carcinoma HepG2 (resveratrol + quercetin) |
| Induction of protective autophagy | [64] | ||
| Glioblastoma multiforme T98G (quercetin + temozolomide) Anaplastic astrocytoma MOGGCCM (quercetin + temozolomide) |
| High doses of drugs reduce autophagic cell death and increase apoptotic death | [65,66] | ||
| Glioblastoma U251, U87 Male Sprague Dawley rats |
| Inhibition of protective autophagy | [67] | ||
| Glioblastoma multiforme T98G (quercetin + sorafenib) Anaplastic astrocytoma MOGGCCM (quercetin + sorafenib) |
| - | [68] | ||
| Hepatocellular carcinoma LM3 Nude mice tumor model |
| - | [69] | ||
| Ovarian cancer CaOV3, P#1 Female nude athymic NOD/SCID mice |
| Induction of protective autophagy | [70] | ||
| Primary effusion lymphoma BC3, BCBL1, BC1 |
| Induction of pro-survival autophagy | [71] | ||
| Burkitt’s lymphoma Akata, 2A8, Ramos | Induces autophagy by inhibiting PI3K/AKT/mTOR pathway and partially degraded mutant c-Myc | Induction of autophagic cell death | [72] | ||
| Gastric cancer AGS, MKN28 Female BALB/c nude mice |
| Induction of protective autophagy | [73] | ||
| Glioblastoma U373MG |
| Induction of protective autophagy | [74] | ||
| Pancreatic cancer MIA PACA-2 GEM-resistant MIA PACA-2 GEMR |
| Induction of autophagy to overcome MDR | [75] | ||
| Human T cell acute lymphoblastic leukemia J/Neo, J/BCL-XL | Induces autophagy resulting from attenuating the AKT-mTOR pathway and BCL-XL-sensitive mitochondrial apoptosis | Induction of protective autophagy | [76] | ||
| Galangin | Hepatocellular carcinoma Hepg2 | Induces autophagy through the activation of p53 signal pathway | - | [77] | |
| Laryngeal carcinoma TU212, HEP-2 SPF male BALB/c nude mice |
| - | [78] | ||
| Flavanols | EGCG | Colorectal cancer HCT-116 | Enhances radiation sensitivity through Nrf2 activation and autophagy | - | [79] |
| Primary effusion lymphoma BCBL-1, BC-1 | Induces apoptosis and autophagy through ROS generation | - | [80] | ||
| Non-small cell lung cancer A549 (gefitinib-resistant cell)/ BALB/C male nude mice |
| Inhibition of autophagy to overcome MDR | [81] | ||
| Chalcone | Xanthoangelol | Hepatocellular carcinoma Hep3B, HuH7 Male athymic BALB/c nu/nu SPF mice |
| Suppression of HCC cell metastasis by inducing autophagic cell death | [82] |
| Isoxanthohumol | Prostate cancer PC3, DU145 | Induces autophagic cell death and cell death is not rescued by caspase inhibitor zVAD-fmk | Induction of autophagic cell death | [83] | |
| Malignant melanoma B16-F10 Lung metastatic model (female syngeneic C57BL/6 mice) |
| - | [84] | ||
| Malignant melanoma B16, A375 Syngeneic C57BL/6 mice |
| Induction of protective autophagy | [85] | ||
| Flavokawain B | Glioblastoma multiforme U251, U87, T98, P3 Male athymic mice |
| Induction of autophagy to promote senescence | [86] | |
| Licochalcone A | Breast cancer MCF-7 |
| - | [87] | |
| Isoliquiritigenin | Human uterine sarcoma Drug-resistant cell MES-SA/Dx5, MES-SA/Dx5-R (doxorubicin-resistant cell) |
| Inhibition of autophagy to overcome MDR | [88] | |
| Breast cancer Epirubicin-resistant cell MCF-7/ADR Female NOD/SCID mice |
| Induction of autophagic cell death to overcome MDR | [89] | ||
| Gastric cancer MKN28 |
| Suppression of cell metastasis by inducing autophagy | [90] | ||
| Hydroxysafflor yellow A | Hepatocellular carcinoma Hepg2 | Induces autophagy through upregulation of Beclin-1 expression, and inhibition of ERK phosphorylation | Induction of autophagy inhibits cell proliferation | [91] | |
| Flavonolignans | Silibinin | Fibrosarcoma HT1080 |
| Induction of autophagic cell death | [92,93] |
| Breast cancer MCF-7 |
| Induction of autophagic cell death | [94] | ||
| Cervix carcinoma HeLa |
| Induction of autophagic cell death | [95] | ||
| Renal cell carcinoma ACHN, 786-O |
| Autophagy induction inhibits cancer cell metastasis | [96] | ||
| Prostate carcinoma DU14 (silibinin + arsenic) |
| - | [97] | ||
| Salivary gland adenoid cystic carcinoma GDC066 Lung metastasis model (BALB/c nu/nu mice) |
| Autophagy induction inhibits cancer cell proliferation | [98] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, X.; Zhang, X.; Jiang, Y.; Su, Q.; Li, Q.; Li, Z. Autophagy: Mechanisms and Therapeutic Potential of Flavonoids in Cancer. Biomolecules 2021, 11, 135. https://doi.org/10.3390/biom11020135
Pang X, Zhang X, Jiang Y, Su Q, Li Q, Li Z. Autophagy: Mechanisms and Therapeutic Potential of Flavonoids in Cancer. Biomolecules. 2021; 11(2):135. https://doi.org/10.3390/biom11020135
Chicago/Turabian StylePang, Xuening, Xiaoyi Zhang, Yuhuan Jiang, Quanzhong Su, Qun Li, and Zichao Li. 2021. "Autophagy: Mechanisms and Therapeutic Potential of Flavonoids in Cancer" Biomolecules 11, no. 2: 135. https://doi.org/10.3390/biom11020135
APA StylePang, X., Zhang, X., Jiang, Y., Su, Q., Li, Q., & Li, Z. (2021). Autophagy: Mechanisms and Therapeutic Potential of Flavonoids in Cancer. Biomolecules, 11(2), 135. https://doi.org/10.3390/biom11020135