
Deciphering the Role of Autophagy in Treatment of Resistance Mechanisms in Glioblastoma

 ,
,  , , and
, , and 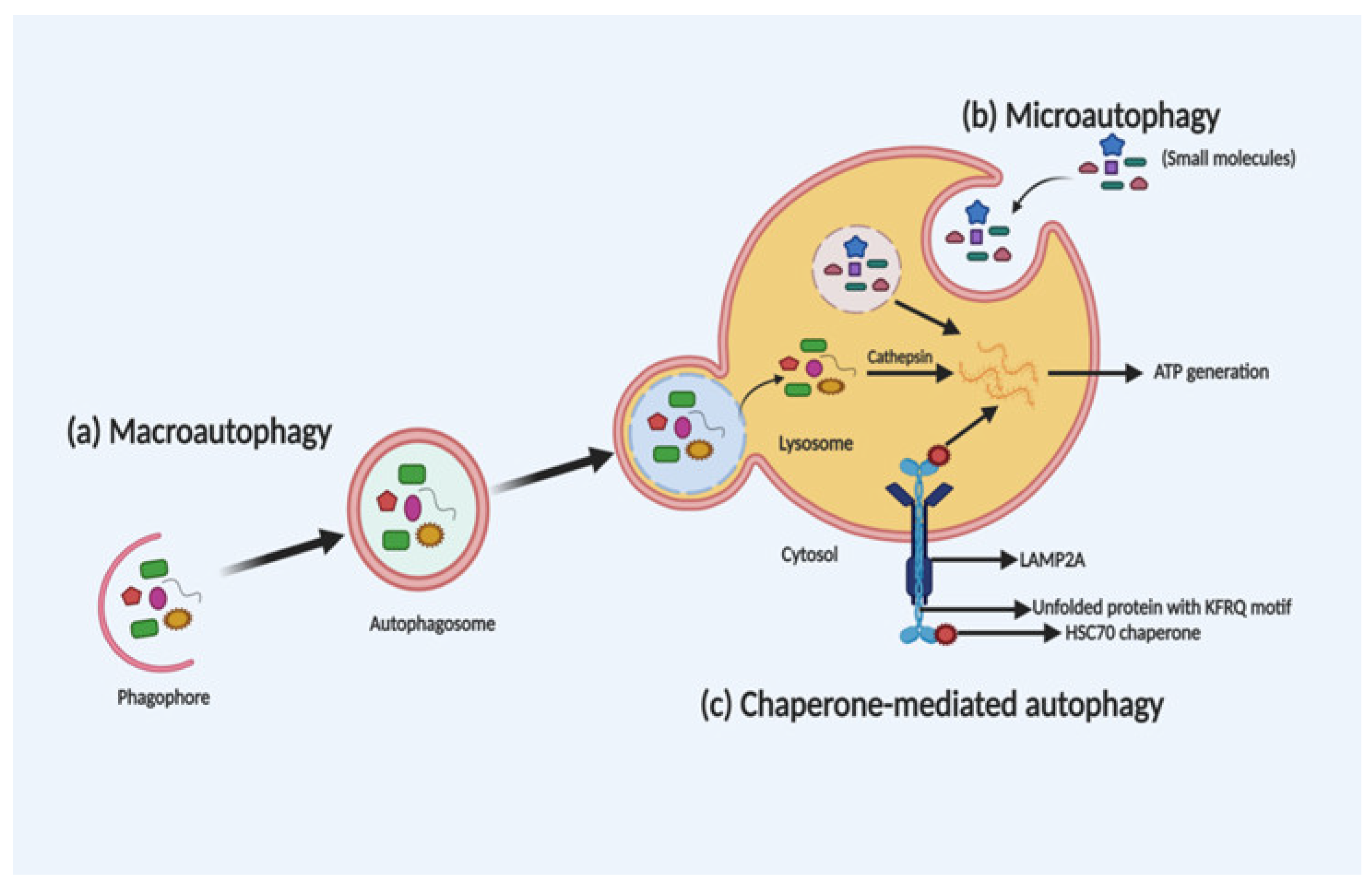{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Autophagy: The Process of Cellular Recycling
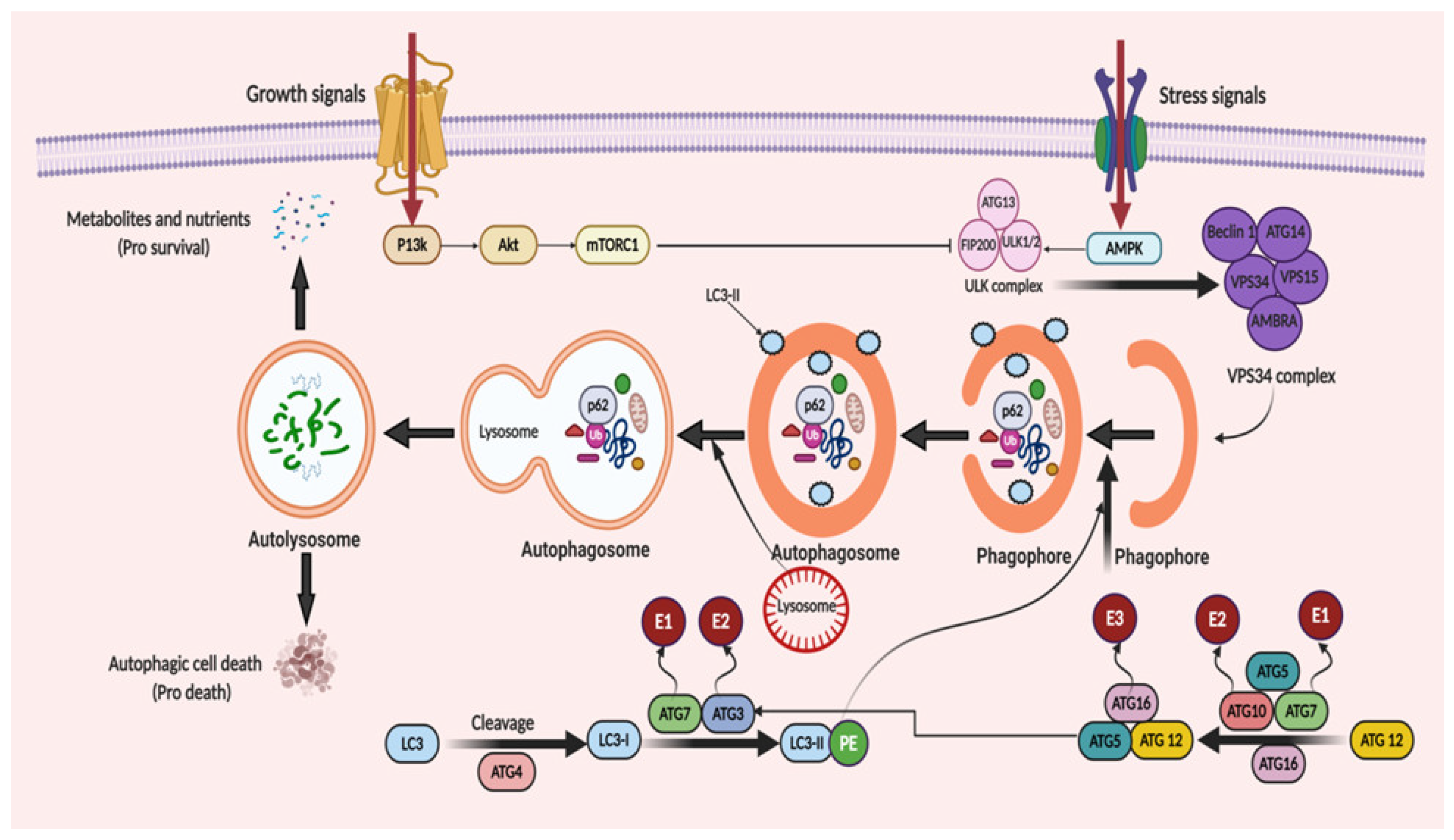
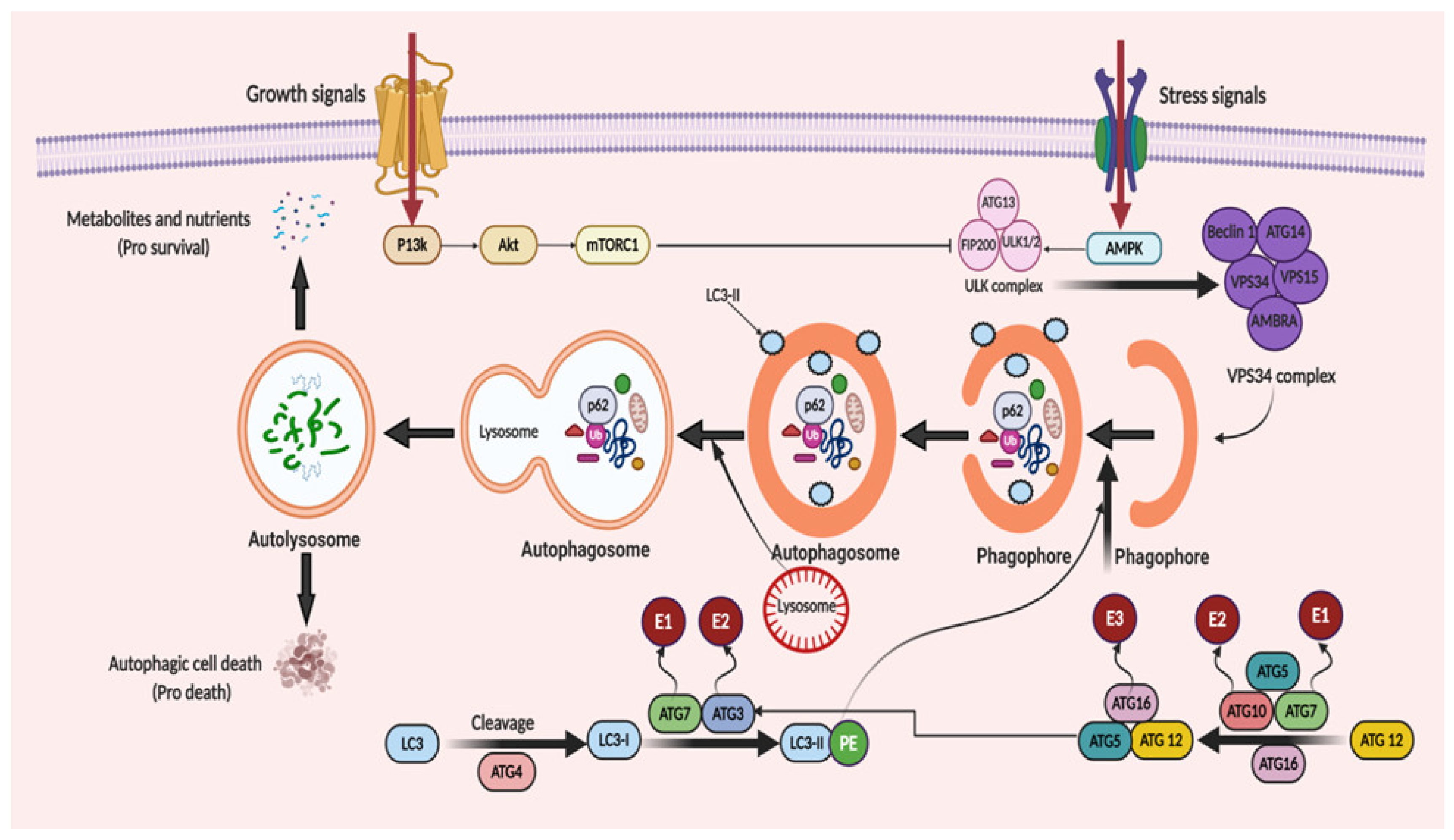
3. Molecular Mechanism of Autophagy: Sliding on the Edges of the Sword
3.1. Autophagy Promotes Tumor Progression in Gliomas
3.2. Autophagy Suppresses Tumor Progression in Gliomas
3.3. The Bipolar Role of Autophagy
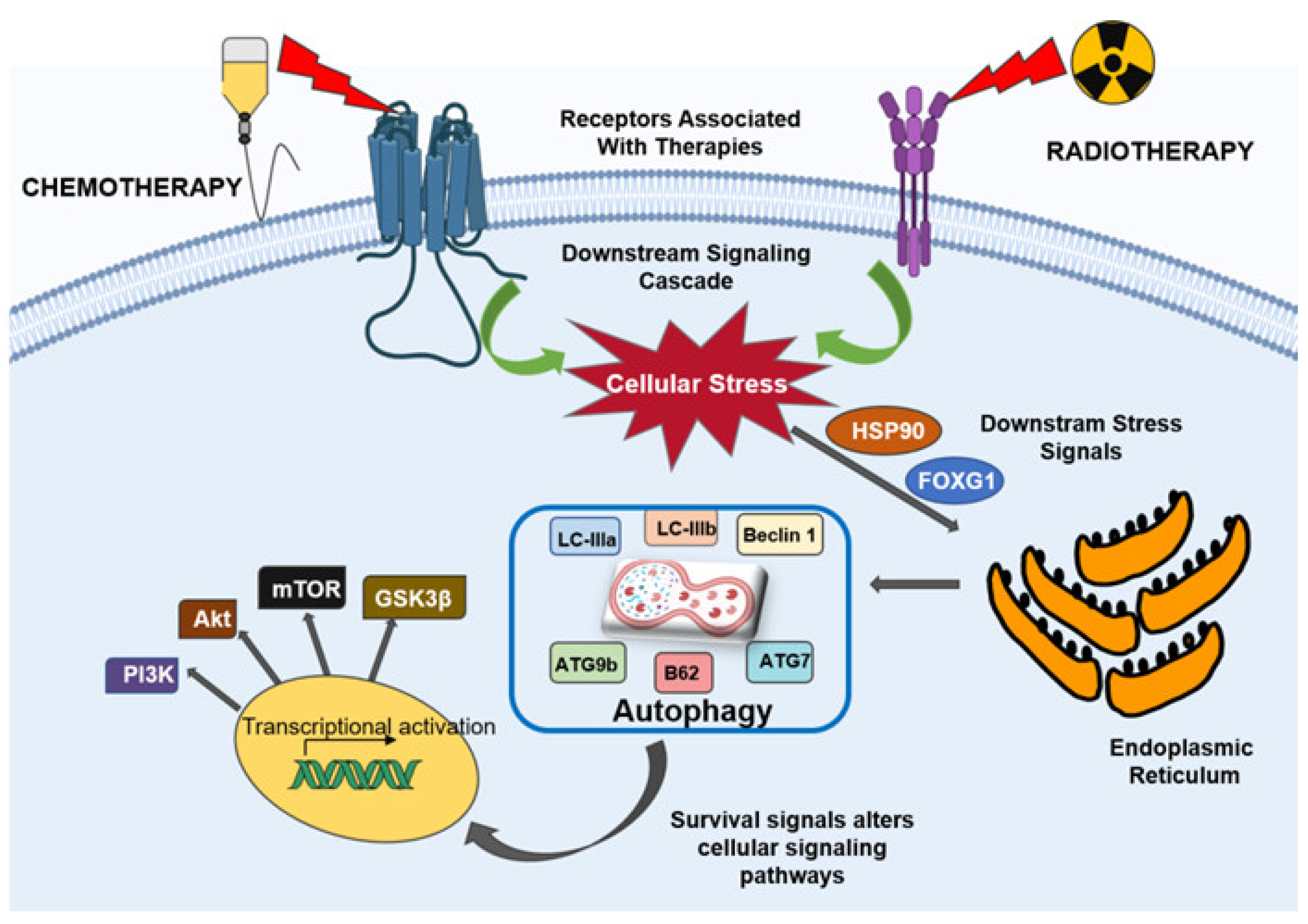
4. Autophagy Mediates Therapeutic Resistance in Glioblastoma
4.1. Autophagy in Chemoresistance
4.1.1. Cellular Pathways in Chemoresistance
4.1.2. Targeting Autophagy to Overcome Chemoresistance in Glioblastoma Cells
4.2. Autophagy in Radioresistance
Targeting Autophagy to Overcome Radioresistance in Glioblastoma Cells
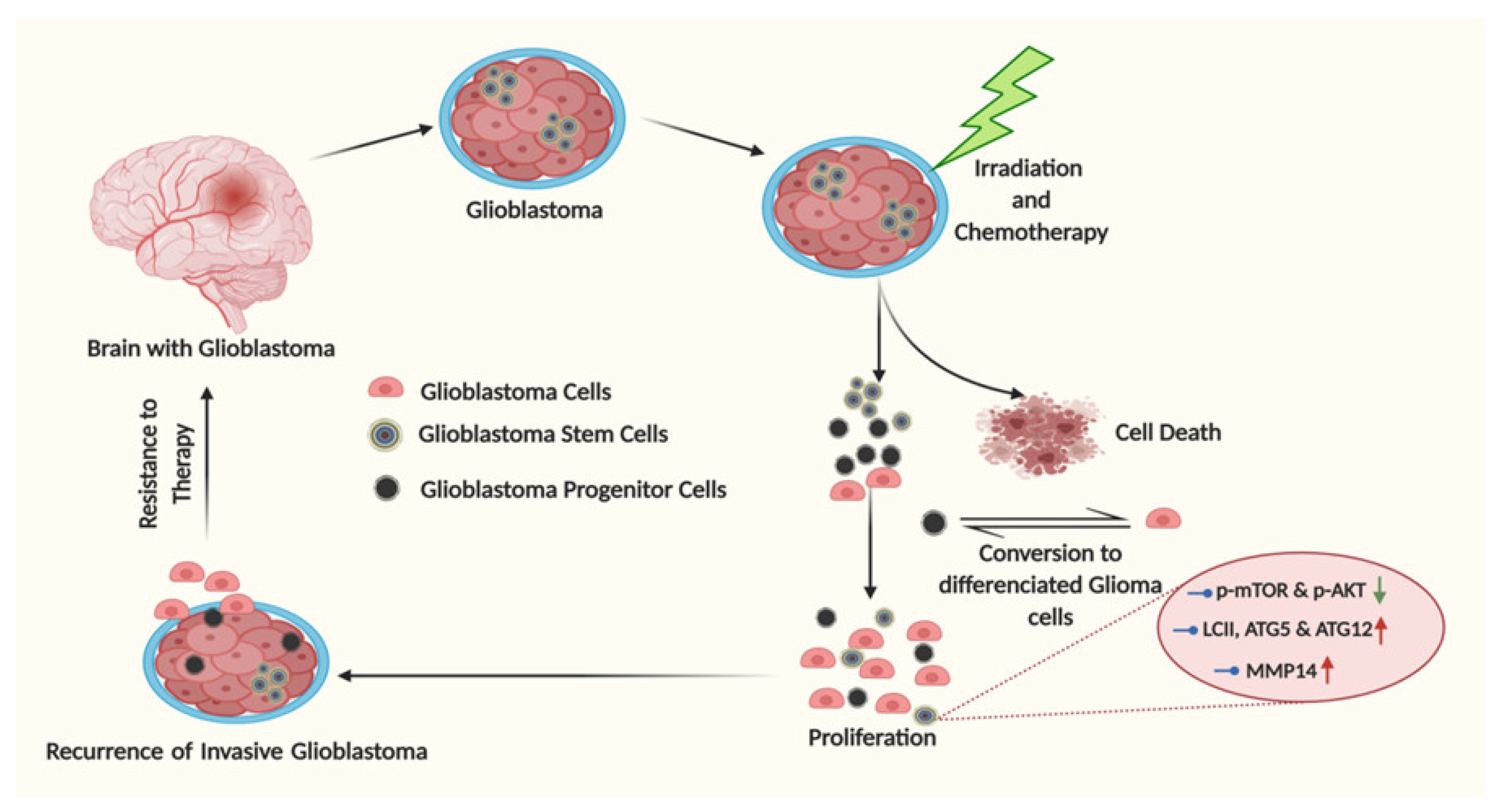
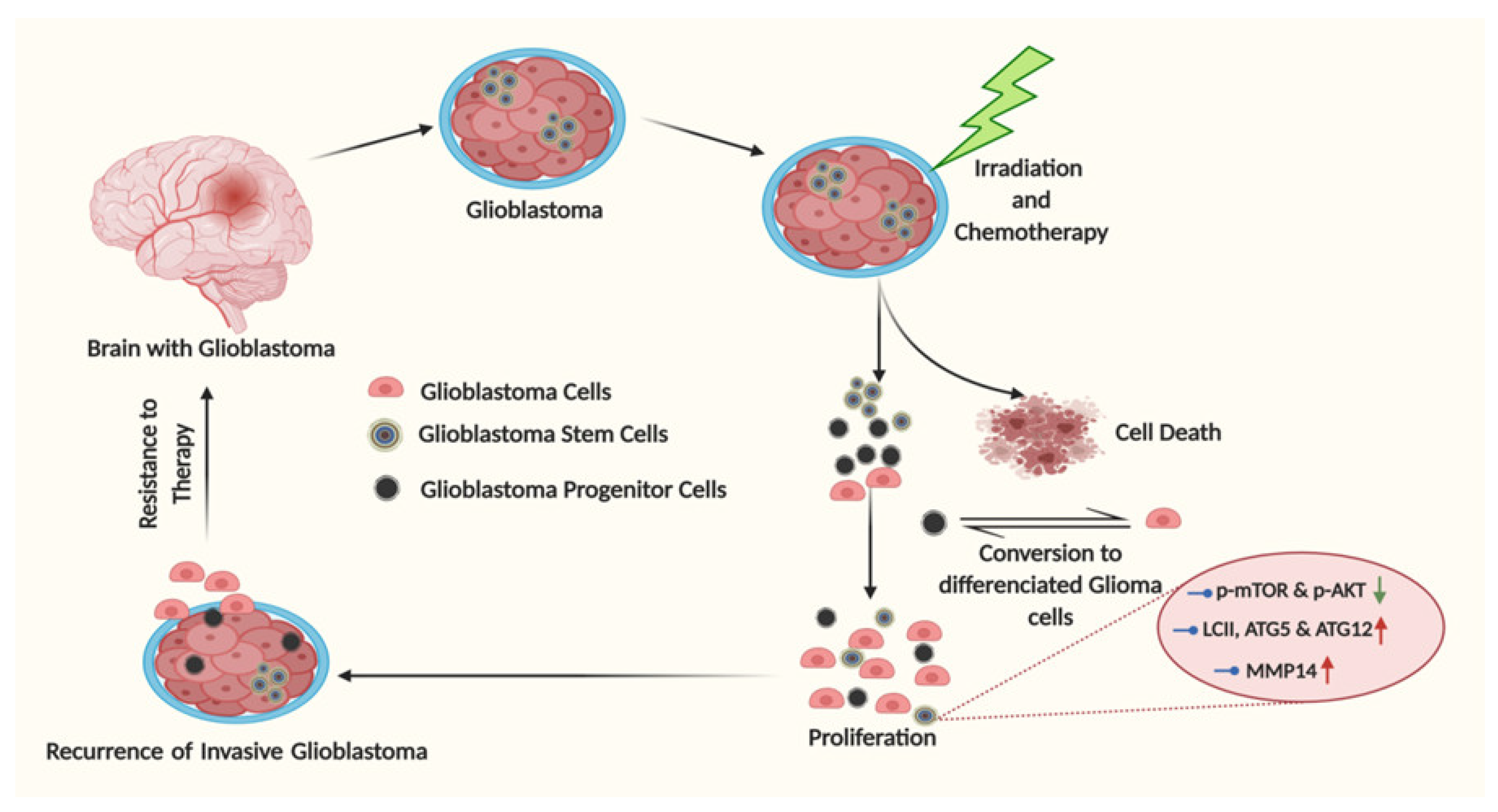
5. Glioma Stem Cells: The Undercover Resistance Mediators
Artificial Autophagy in Glioblastoma: A Tale of Intrigue
6. A Tail of Artificial Autophagy Signaling: Neural Stem Cells (NSCs), Astrocytes or Glioblastoma Mutations?
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hottinger, A.F.; Stupp, R.; Homicsko, K. Standards of care and novel approaches in the management of glioblastoma multiforme. Chin. J. Cancer 2014, 33, 32. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.E. Glioblastoma: Overview of disease and treatment. Clin. J. Oncol. Nurs. 2016, 20, S2. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhou, Y.; Li, Y.; Yang, L.; Ma, Y.; Peng, X.; Yang, S.; Liu, J.; Li, H. Autophagy: A novel mechanism of chemoresistance in cancers. Biomed. Pharmacother. 2019, 119, 109415. [Google Scholar] [CrossRef]
- Kaza, N.; Kohli, L.; Roth, K.A. Autophagy in brain tumors: A new target for therapeutic intervention. Brain Pathol. 2012, 22, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.G.; Macleod, K.F. Autophagy, cancer stem cells and drug resistance. J. Pathol. 2019, 247, 708–718. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W.; Cloonan, S.M.; Choi, A.M. Autophagy: A critical regulator of cellular metabolism and homeostasis. Mol. Cells 2013, 36, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.L.; Baehrecke, E.H. Autophagy functions in programmed cell death. Autophagy 2008, 4, 359–360. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.Y.; Ryan, K.M. Autophagy and cancer–Issues we need to digest. J. Cell Sci. 2012, 125, 2349–2358. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Zinn, P.O.; Singh, S.K.; Kotrotsou, A.; Abrol, S.; Thomas, G.; Mosley, J.; Elakkad, A.; Hassan, I.; Kumar, A.; Colen, R.R. Distinct radiomic phenotypes define glioblastoma TP53-PTEN-EGFR mutational landscape. Neurosurgery 2017, 64 (Suppl. 1), 203–210. [Google Scholar] [CrossRef]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal growth factor receptor in glioma: Signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Talukdar, S.; Pradhan, A.K.; Bhoopathi, P.; Shen, X.-N.; August, L.A.; Windle, J.J.; Sarkar, D.; Furnari, F.B.; Cavenee, W.K.; Das, S.K. MDA-9/Syntenin regulates protective autophagy in anoikis-resistant glioma stem cells. Proc. Natl. Acad. Sci. USA 2018, 115, 5768–5773. [Google Scholar] [CrossRef] [Green Version]
- Caldera, V.; Mellai, M.; Annovazzi, L.; Valente, G.; Tessitore, L.; Schiffer, D. Stat3 expression and its correlation with proliferation and apoptosis/autophagy in gliomas. J. Oncol. 2008, 2008. [Google Scholar] [CrossRef] [Green Version]
- Tini, P.; Belmonte, G.; Toscano, M.; Miracco, C.; Palumbo, S.; Pastina, P.; Battaglia, G.; Nardone, V.; Butorano, M.A.G.M.; Masucci, A. Combined epidermal growth factor receptor and beclin1 autophagic protein expression analysis identifies different clinical presentations, responses to chemo-and radiotherapy, and prognosis in glioblastoma. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef]
- Müller-Greven, G.; Carlin, C.R.; Burgett, M.E.; Ahluwalia, M.S.; Lauko, A.; Nowacki, A.S.; Herting, C.J.; Qadan, M.A.; Bredel, M.; Toms, S.A. Macropinocytosis of bevacizumab by glioblastoma cells in the perivascular niche affects their survival. Clin. Cancer Res. 2017, 23, 7059–7071. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, H.; Kanzawa, T.; Kondo, Y.; Kondo, S. Inhibition of platelet-derived growth factor signalling induces autophagy in malignant glioma cells. Br. J. Cancer 2004, 90, 1069–1075. [Google Scholar] [CrossRef]
- Vehlow, A.; Cordes, N. DDR1 (discoidin domain receptor tyrosine kinase 1) drives glioblastoma therapy resistance by modulating autophagy. Autophagy 2019, 15, 1487–1488. [Google Scholar] [CrossRef]
- Shingu, T.; Holmes, L.; Henry, V.; Wang, Q.; Latha, K.; Gururaj, A.E.; Gibson, L.A.; Doucette, T.; Lang, F.F.; Rao, G. Suppression of RAF/MEK or PI3K synergizes cytotoxicity of receptor tyrosine kinase inhibitors in glioma tumor-initiating cells. J. Transl. Med. 2016, 14, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Q.-W.; Weiss, W.A. Inhibition of PI3K-Akt-mTOR signaling in glioblastoma by mTORC1/2 inhibitors. Methods Mol. Biol. 2012, 821, 349–359. [Google Scholar]
- Hu, Y.-L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azad, M.B.; Gibson, S.B. Role of BNIP3 in proliferation and hypoxia-induced autophagy: Implications for personalized cancer therapies. Ann. N. Y. Acad. Sci. 2010, 1210, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Womeldorff, M.; Gillespie, D.; Jensen, R.L. Hypoxia-inducible factor–1 and associated upstream and downstream proteins in the pathophysiology and management of glioblastoma. Neurosurg. Focus 2014, 37, E8. [Google Scholar] [CrossRef]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)—Chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017, 11, 781. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Xu, Z.; Dai, S.; Qian, L.; Sun, L.; Gong, Z. Targeting autophagy to sensitive glioma to temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 23. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.-Y.; Tsai, S.-Y.; Wu, C.-M.; Yen, C.-J.; Chuang, B.-F.; Chang, J.-Y. Novel phosphoinositide 3-kinase/mTOR dual inhibitor, NVP-BGT226, displays potent growth-inhibitory activity against human head and neck cancer cells in vitro and in vivo. Clin. Cancer Res. 2011, 17, 7116–7126. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Graham, P.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.; Kearsley, J.; Li, Y. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014, 5, e1437. [Google Scholar] [CrossRef]
- Golden, E.B.; Cho, H.-Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg. Focus 2014, 37, E12. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.-U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Jacobi, A.; Vater, C.; Zou, L.; Zou, X.; Stiehler, M. Icariin promotes angiogenic differentiation and prevents oxidative stress-induced autophagy in endothelial progenitor cells. Stem Cells 2015, 33, 1863–1877. [Google Scholar] [CrossRef] [PubMed]
- Gatica, D.; Chiong, M.; Lavandero, S.; Klionsky, D.J. Molecular mechanisms of autophagy in the cardiovascular system. Circ. Res. 2015, 116, 456–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Z.; Wang, T.; Zhu, H.; Zhang, P.; Han, R.; Liu, Y.; Ni, P.; Shen, H.; Xu, W.; Xu, H. HMGB1 modulates Lewis cell autophagy and promotes cell survival via RAGE-HMGB1-Erk1/2 positive feedback during nutrient depletion. Immunobiology 2015, 220, 539–544. [Google Scholar] [CrossRef]
- Pan, H.; Wang, Z.; Jiang, L.; Sui, X.; You, L.; Shou, J.; Jing, Z.; Xie, J.; Ge, W.; Cai, X. Autophagy inhibition sensitizes hepatocellular carcinoma to the multikinase inhibitor linifanib. Sci. Rep. 2014, 4, 6683. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Masui, K.; Gini, B.; Wykosky, J.; Zanca, C.; Mischel, P.S.; Furnari, F.B.; Cavenee, W.K. A tale of two approaches: Complementary mechanisms of cytotoxic and targeted therapy resistance may inform next-generation cancer treatments. Carcinogenesis 2013, 34, 725–738. [Google Scholar] [CrossRef]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef]
- Kimmelman, A.C. The dynamic nature of autophagy in cancer. Genes Dev. 2011, 25, 1999–2010. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. Electron tomography reveals the endoplasmic reticulum as a membrane source for autophagosome formation. Autophagy 2010, 6, 301–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burman, C.; Ktistakis, N.T. Autophagosome formation in mammalian cells. Semin. Immunopathol. 2010, 32, 397–413. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J. Regulated self-cannibalism. Nature 2004, 431, 31–32. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.Y.; Kir, S.; Tooze, S.A. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 2007, 282, 25464–25474. [Google Scholar] [CrossRef] [Green Version]
- Young, A.R.; Chan, E.Y.; Hu, X.W.; Köchl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888–3900. [Google Scholar] [CrossRef] [Green Version]
- O’ Farrell, F.; Rusten, T.E.; Stenmark, H. Phosphoinositide 3-kinases as accelerators and brakes of autophagy. FEBS J. 2013, 280, 6322–6337. [Google Scholar] [CrossRef] [PubMed]
- Proikas-Cezanne, T.; Robenek, H. Freeze-fracture replica immunolabelling reveals human WIPI-1 and WIPI-2 as membrane proteins of autophagosomes. J. Cell. Mol. Med. 2011, 15, 2007–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Schulman, B.A. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat. Struct. Mol. Biol. 2014, 21, 336. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.D.; Nakatogawa, H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. 2013, 55, 39–50. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef]
- Ao, X.; Zou, L.; Wu, Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014, 21, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Nara, A.; Mizushima, N.; Yamamoto, A.; Kabeya, Y.; Ohsumi, Y.; Yoshimori, T. SKD1 AAA ATPase-dependent endosomal transport is involved in autolysosome formation. Cell Struct. Funct. 2002, 27, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Gammoh, N.; Fraser, J.; Puente, C.; Syred, H.M.; Kang, H.; Ozawa, T.; Lam, D.; Acosta, J.C.; Finch, A.J.; Holland, E.; et al. Suppression of autophagy impedes glioblastoma development and induces senescence. Autophagy 2016, 12, 1431–1439. [Google Scholar] [CrossRef]
- Tamrakar, S.; Yashiro, M.; Kawashima, T.; Uda, T.; Terakawa, Y.; Kuwae, Y.; Ohsawa, M.; Ohata, K. Clinicopathological Significance of Autophagy-related Proteins and its Association with Genetic Alterations in Gliomas. Anticancer Res. 2019, 39, 1233–1242. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Sivridis, E.; Mitrakas, A.; Kalamida, D.; Zois, C.E.; Haider, S.; Piperidou, C.; Pappa, A.; Gatter, K.C.; Harris, A.L. Autophagy and lysosomal related protein expression patterns in human glioblastoma. Cancer Biol. Ther. 2014, 15, 1468–1478. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.-P.; Zeng, W.-J.; Chen, Y.-H.; Li, H.; Wang, J.-Y.; Cheng, Q.; Yu, J.; Zhou, H.-H.; Liu, Z.-Z.; Xiao, J.; et al. Knockdown ATG4C inhibits gliomas progression and promotes temozolomide chemosensitivity by suppressing autophagic flux. J. Exp. Clin. Cancer Res. 2019, 38, 298. [Google Scholar] [CrossRef] [PubMed]
- Jennewein, L.; Ronellenfitsch, M.W.; Antonietti, P.; Ilina, E.I.; Jung, J.; Stadel, D.; Flohr, L.-M.; Zinke, J.; von Renesse, J.; Drott, U.; et al. Diagnostic and clinical relevance of the autophago-lysosomal network in human gliomas. Oncotarget 2016, 7, 20016–20032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Luo, W.; Wang, J.; Peng, T.; Sun, G.; Shi, J.; Li, Z.; Zhang, B. Malat1 activates autophagy and promotes cell proliferation by sponging miR-101 and upregulating STMN1, RAB5A and ATG4D expression in glioma. Biochem. Biophys. Res. Commun. 2017, 492, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Mazure, N.M.; Pouysségur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Pellegrino, M.; Santolla, M.F.; Lappano, R.; Ricchio, E.; Abonante, S.; Maggiolini, M. GPER mediates activation of HIF1α/VEGF signaling by estrogens. Cancer Res. 2014, 74, 4053–4064. [Google Scholar] [CrossRef] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Jawhari, S.; Ratinaud, M.-H.; Verdier, M. Glioblastoma, hypoxia and autophagy: A survival-prone ‘menage-a-trois’. Cell Death Dis. 2016, 7, e2434. [Google Scholar] [CrossRef]
- Yang, L.; Lin, C.; Wang, L.; Guo, H.; Wang, X. Hypoxia and hypoxia-inducible factors in glioblastoma multiforme progression and therapeutic implications. Exp. Cell Res. 2012, 318, 2417–2426. [Google Scholar] [CrossRef]
- Wu, H.B.; Yang, S.; Weng, H.Y.; Chen, Q.; Zhao, X.L.; Fu, W.J.; Niu, Q.; Ping, Y.F.; Wang, J.M.; Zhang, X.; et al. Autophagy-induced KDR/VEGFR-2 activation promotes the formation of vasculogenic mimicry by glioma stem cells. Autophagy 2017, 13, 1528–1542. [Google Scholar] [CrossRef] [Green Version]
- Lamoral-Theys, D.; Le Mercier, M.; Le Calvé, B.; Rynkowski, M.A.; Bruyère, C.; Decaestecker, C.; Haibe-Kains, B.; Bontempi, G.; Dubois, J.; Lefranc, F.; et al. Long-term temozolomide treatment induces marked amino metabolism modifications and an increase in TMZ sensitivity in Hs683 oligodendroglioma cells. Neoplasia 2010, 12, 69–79. [Google Scholar] [CrossRef]
- Tiram, G.; Ferber, S.; Ofek, P.; Eldar-Boock, A.; Ben-Shushan, D.; Yeini, E.; Krivitsky, A.; Blatt, R.; Almog, N.; Henkin, J.; et al. Reverting the molecular fingerprint of tumor dormancy as a therapeutic strategy for glioblastoma. FASEB J. 2018, 32, 5835–5850. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Patric, I.R.P.; Patil, V.; Shwetha, S.D.; Hegde, A.S.; Chandramouli, B.A.; Arivazhagan, A.; Santosh, V.; Somasundaram, K. Methylation silencing of ULK2, an autophagy gene, is essential for astrocyte transformation and tumor growth. J. Biol. Chem. 2014, 289, 22306–22318. [Google Scholar] [CrossRef] [Green Version]
- Miracco, C.; Cosci, E.; Oliveri, G.; Luzi, P.; Pacenti, L.; Monciatti, I.; Mannucci, S.; De Nisi, M.C.; Toscano, M.; Malagnino, V. Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours. Int. J. Oncol. 2007, 30, 429–436. [Google Scholar] [PubMed]
- Aoki, H.; Kondo, Y.; Aldape, K.; Yamamoto, A.; Iwado, E.; Yokoyama, T.; Hollingsworth, E.F.; Kobayashi, R.; Hess, K.; Shinojima, N. Monitoring autophagy in glioblastoma with antibody against isoform B of human microtubule-associated protein 1 light chain 3. Autophagy 2008, 4, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013, 32, 699–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in glioblastoma: Rationale and preclinical/clinical evidence. Dis. Markers 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-Y.; Zhang, L.-Q.; Zhang, X.-G.; Li, X.; Ren, Y.-B.; Ma, X.-Y.; Li, X.-G.; Wang, L.-X. Association between AKT/mTOR signalling pathway and malignancy grade of human gliomas. J. Neuro-Oncol. 2011, 103, 453–458. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Jeevan, D.; Neil, J.; Shannon, C.; Albert, L.; Murali, R. Deconstructing mTOR complexes in regulation of Glioblastoma Multiforme and its stem cells. Adv. Biol. Regul. 2013, 53, 202–210. [Google Scholar] [CrossRef]
- Guo, X.; Xue, H.; Guo, X.; Gao, X.; Xu, S.; Yan, S.; Han, X.; Li, T.; Shen, J.; Li, G. MiR224-3p inhibits hypoxia-induced autophagy by targeting autophagy-related genes in human glioblastoma cells. Oncotarget 2015, 6, 41620–41637. [Google Scholar] [CrossRef] [Green Version]
- Azad, M.B.; Chen, Y.; Henson, E.S.; Cizeau, J.; McMillan-Ward, E.; Israels, S.J.; Gibson, S.B. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy 2008, 4, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Jiao, Y.; Wang, Z.; Li, T.; Liu, Y.; Li, Y.; Zhao, X.; Wang, D. Sinomenine Hydrochloride Inhibits Human Glioblastoma Cell Growth through Reactive Oxygen Species Generation and Autophagy-Lysosome Pathway Activation: An In Vitro and In Vivo Study. Int. J. Mol. Sci. 2017, 18, 1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Xue, X.; Guo, R.B.; Sun, X.L.; Hu, G. Resveratrol enhances the antitumor effects of temozolomide in glioblastoma via ROS-dependent AMPK-TSC-mTOR signaling pathway. CNS Neurosci. Ther. 2012, 18, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qi, Q.; Zhou, W.; Feng, Z.; Huang, B.; Chen, A.; Zhang, D.; Li, W.; Zhang, Q.; Jiang, Z.; et al. Inhibition of glioma growth by flavokawain B is mediated through endoplasmic reticulum stress induced autophagy. Autophagy 2018, 14, 2007–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lépine, S.; Allegood, J.C.; Edmonds, Y.; Milstien, S.; Spiegel, S. Autophagy induced by deficiency of sphingosine-1-phosphate phosphohydrolase 1 is switched to apoptosis by calpain-mediated autophagy-related gene 5 (Atg5) cleavage. J. Biol. Chem. 2011, 286, 44380–44390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Zhao, L.; Liu, L.; Gao, P.; Tian, W.; Wang, X.; Jin, H.; Xu, H.; Chen, Q. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell 2010, 1, 468–477. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Qi, Q.; Hua, X.; Li, X.; Zhang, W.; Sun, H.; Li, S.; Wang, X.; Li, B. Beclin 1, an autophagy-related gene, augments apoptosis in U87 glioblastoma cells. Oncol. Rep. 2014, 31, 1761–1767. [Google Scholar] [CrossRef] [Green Version]
- Feng, F.; Zhang, M.; Yang, C.; Heng, X.; Wu, X. The dual roles of autophagy in gliomagenesis and clinical therapy strategies based on autophagic regulation mechanisms. Biomed. Pharmacother. 2019, 120, 109441. [Google Scholar] [CrossRef]
- Su, M.; Mei, Y.; Sinha, S. Role of the crosstalk between autophagy and apoptosis in cancer. J. Oncol. 2013, 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against Fatal Sindbis Virus Encephalitis by Beclin, a Novel Bcl-2-Interacting Protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohn, T.T.; Wirawan, E.; Brown, R.J.; Harris, J.R.; Masliah, E.; Vandenabeele, P. Depletion of Beclin-1 due to proteolytic cleavage by caspases in the Alzheimer’s disease brain. Neurobiol. Dis. 2011, 43, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Cao, L.; Peng, Y.; Tan, Y.; Xie, M.; Kang, R.; Livesey, K.M.; Tang, D. A critical role for UVRAG in apoptosis. Autophagy 2011, 7, 1242–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radoshevich, L.; Murrow, L.; Chen, N.; Fernandez, E.; Roy, S.; Fung, C.; Debnath, J. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 2010, 142, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinstein, A.D.; Eisenstein, M.; Ber, Y.; Bialik, S.; Kimchi, A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol. Cell 2011, 44, 698–709. [Google Scholar] [CrossRef] [Green Version]
- Pyo, J.O.; Jang, M.H.; Kwon, Y.K.; Lee, H.J.; Jun, J.I.; Woo, H.N.; Cho, D.H.; Choi, B.; Lee, H.; Kim, J.H.; et al. Essential roles of Atg5 and FADD in autophagic cell death: Dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 2005, 280, 20722–20729. [Google Scholar] [CrossRef] [Green Version]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Zhang, Y.; Li, Y.; Liu, Z.; Zuo, J.; Fang, F. Two domains are critical for the nuclear localization of soluble adenylyl cyclase. Biochimie 2006, 88, 319–328. [Google Scholar] [CrossRef]
- Gramatzki, D.; Dehler, S.; Rushing, E.J.; Zaugg, K.; Hofer, S.; Yonekawa, Y.; Bertalanffy, H.; Valavanis, A.; Korol, D.; Rohrmann, S. Glioblastoma in the Canton of Zurich, Switzerland revisited: 2005 to 2009. Cancer 2016, 122, 2206–2215. [Google Scholar] [CrossRef]
- Voldborg, B.R.; Damstrup, L.; Spang-Thomsen, M.; Poulsen, H.S. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 1997, 8, 1197–1206. [Google Scholar] [CrossRef]
- Katz, A.M.; Amankulor, N.M.; Pitter, K.; Helmy, K.; Squatrito, M.; Holland, E.C. Astrocyte-specific expression patterns associated with the PDGF-induced glioma microenvironment. PLoS ONE 2012, 7, e32453. [Google Scholar] [CrossRef] [Green Version]
- Sjöström, S.; Wibom, C.; Andersson, U.; Brännström, T.; Broholm, H.; Johansen, C.; Collatz-Laier, H.; Liu, Y.; Bondy, M.; Henriksson, R. Genetic variations in VEGF and VEGFR2 and glioblastoma outcome. J. Neuro-Oncol. 2011, 104, 523–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sathornsumetee, S.; Cao, Y.; Marcello, J.E.; Herndon, J.E. Tumor angiogenic and hypoxic profiles predict radiographic response and survival in malignant astrocytoma patients treated with bevacizumab and irinotecan. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 271. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A.R. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061. [Google Scholar]
- Soomro, S.H.; Ting, L.R.; Qing, Y.Y.; Ren, M. Molecular biology of glioblastoma: Classification and mutational locations. J. Pak. Med. Assoc. 2017, 67, 1410–1414. [Google Scholar] [PubMed]
- Trejo-Solís, C.; Jimenez-Farfan, D.; Rodriguez-Enriquez, S.; Fernandez-Valverde, F.; Cruz-Salgado, A.; Ruiz-Azuara, L.; Sotelo, J. Copper compound induces autophagy and apoptosis of glioma cells by reactive oxygen species and JNK activation. BMC Cancer 2012, 12, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, M.; Bajpai, V.K.; Sahasrabuddhe, A.A.; Kumar, A.; Sinha, R.A.; Behari, S.; Godbole, M.M. Inhibition of N-(4-hydroxyphenyl) retinamide-induced autophagy at a lower dose enhances cell death in malignant glioma cells. Carcinogenesis 2008, 29, 600–609. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vicencio, J.M.; Kepp, O.; Tasdemir, E.; Maiuri, M.C.; Kroemer, G. To die or not to die: That is the autophagic question. Curr. Mol. Med. 2008, 8, 78. [Google Scholar]
- Thayyullathil, F.; Rahman, A.; Pallichankandy, S.; Patel, M.; Galadari, S. ROS-dependent prostate apoptosis response-4 (Par-4) up-regulation and ceramide generation are the prime signaling events associated with curcumin-induced autophagic cell death in human malignant glioma. FEBS Open Bio 2014, 4, 763–776. [Google Scholar] [CrossRef] [Green Version]
- Keshmiri-Neghab, H.; Goliaei, B.; Nikoofar, A. Gossypol enhances radiation induced autophagy in glioblastoma multiforme. Gen. Physiol. Biophys. 2014, 33, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Butera, G.; Pacchiana, R.; Donadelli, M. Molecular interplay between mutant p53 proteins and autophagy in cancer cells. Biochim. Biophys. Acta (BBA) Rev. Cancer 2017, 1867, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Kakudo, Y.; Takahashi, S.; Sakamoto, Y.; Kato, S.; Ishioka, C. Overexpression of DRAM enhances p53-dependent apoptosis. Cancer Med. 2013, 2, 1–10. [Google Scholar] [CrossRef]
- Chollat-Namy, M.; Safta-Saadoun, T.B.; Haferssas, D.; Meurice, G.; Chouaib, S.; Thiery, J. The pharmalogical reactivation of p53 function improves breast tumor cell lysis by granzyme B and NK cells through induction of autophagy. Cell Death Dis. 2019, 10, 1–20. [Google Scholar] [CrossRef]
- White, E. Autophagy and p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026120. [Google Scholar] [CrossRef]
- Chen, J.-H.; Zhang, P.; Chen, W.-D.; Li, D.-D.; Wu, X.-Q.; Deng, R.; Jiao, L.; Li, X.; Ji, J.; Feng, G.-K. ATM-mediated PTEN phosphorylation promotes PTEN nuclear translocation and autophagy in response to DNA-damaging agents in cancer cells. Autophagy 2015, 11, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Webb, A.E.; Brunet, A. FOXO transcription factors: Key regulators of cellular quality control. Trends Biochem. Sci. 2014, 39, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z. The FoxO–Autophagy Axis in health and disease. Trends Endocrinol. Metab. 2019, 30, 658–671. [Google Scholar] [CrossRef]
- Hou, T.; Li, Z.; Zhao, Y.; Zhu, W.-G. Mechanisms controlling the anti-neoplastic functions of FoxO proteins. Semin. Cancer Biol. 2018, 50, 101–114. [Google Scholar] [CrossRef]
- Zhang, J.; Ng, S.; Wang, J.; Zhou, J.; Tan, S.-H.; Yang, N.; Lin, Q.; Xia, D.; Shen, H.-M. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy 2015, 11, 629–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C. Transcriptional regulation of autophagy: Mechanisms and diseases. Front. Cell Dev. Biol. 2019, 7, 114. [Google Scholar]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Nàger, M.; Sallán, M.C.; Visa, A.; Pushparaj, C.; Santacana, M.; Macià, A.; Yeramian, A.; Cantí, C.; Herreros, J. Inhibition of WNT-CTNNB1 signaling upregulates SQSTM1 and sensitizes glioblastoma cells to autophagy blockers. Autophagy 2018, 14, 619–636. [Google Scholar] [CrossRef]
- Yeldag, G.; Rice, A.; del Río Hernández, A. Chemoresistance and the self-maintaining tumor microenvironment. Cancers 2018, 10, 471. [Google Scholar] [CrossRef] [Green Version]
- Hombach-Klonisch, S.; Mehrpour, M.; Shojaei, S.; Harlos, C.; Pitz, M.; Hamai, A.; Siemianowicz, K.; Likus, W.; Wiechec, E.; Toyota, B.D. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol. Ther. 2018, 184, 13–41. [Google Scholar] [CrossRef]
- Wojton, J.; Meisen, W.H.; Kaur, B. How to train glioma cells to die: Molecular challenges in cell death. J. Neuro-Oncol. 2016, 126, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.A.; Das, B.C.; Ray, S.K. Targeting autophagy for combating chemoresistance and radioresistance in glioblastoma. Apoptosis 2018, 23, 563–575. [Google Scholar] [CrossRef]
- Li, Y.-J.; Lei, Y.-H.; Yao, N.; Wang, C.-R.; Hu, N.; Ye, W.-C.; Zhang, D.-M.; Chen, Z.-S. Autophagy and multidrug resistance in cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef]
- Liu, X.; Sun, K.; Wang, H.; Dai, Y. Knockdown of retinoblastoma protein may sensitize glioma cells to cisplatin through inhibition of autophagy. Neurosci. Lett. 2016, 620, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shang, Z.; Zhou, Y.; Hu, X.; Chen, Y.; Fan, Y.; Wei, X.; Wu, L.; Liang, Q.; Zhang, J. Autophagy mediates glucose starvation-induced glioblastoma cell quiescence and chemoresistance through coordinating cell metabolism, cell cycle, and survival. Cell Death Dis. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chédeville, A.L.; Lourdusamy, A.; Monteiro, A.R.; Hill, R.; Madureira, P.A. Investigating Glioblastoma Response to Hypoxia. Biomedicines 2020, 8, 310. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zheng, H.; Ruan, J.; Fang, W.; Li, A.; Tian, G.; Niu, X.; Luo, S.; Zhao, P. Autophagy inhibition induces enhanced proapoptotic effects of ZD6474 in glioblastoma. Br. J. Cancer 2013, 109, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.-W.; Cheng, C.; Hackett, C.; Feldman, M.; Houseman, B.T.; Nicolaides, T.; Haas-Kogan, D.; James, C.D.; Oakes, S.A.; Debnath, J.; et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci. Signal. 2010, 3, ra81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Li, J.; Chen, L.; Qi, S.; Yu, S.; Weng, Z.; Hu, Z.; Zhou, Q.; Xin, Z.; Shi, L. HERC3-mediated SMAD7 ubiquitination degradation promotes autophagy-induced EMT and chemoresistance in glioblastoma. Clin. Cancer Res. 2019, 25, 3602–3616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, C.; Shen, F.; Du, J.; Fang, X.; Li, X.; Su, J.; Wang, X.; Huang, X.; Liu, Z. Upregulation of CASC2 sensitized glioma to temozolomide cytotoxicity through autophagy inhibition by sponging miR-193a-5p and regulating mTOR expression. Biomed. Pharmacother. 2018, 97, 844–850. [Google Scholar] [CrossRef]
- Ryabaya, O.O.; Inshakov, A.N.; Egorova, A.V.; Emelyanova, M.A.; Nasedkina, T.V.; Zasedatelev, A.S.; Khochenkov, D.A.; Stepanova, E.V. Autophagy inhibitors chloroquine and LY294002 enhance temozolomide cytotoxicity on cutaneous melanoma cell lines in vitro. Anti-Cancer Drugs 2017, 28, 307–315. [Google Scholar] [CrossRef]
- Chen, G.; Zhu, W.; Shi, D.; Lv, L.; Zhang, C.; Liu, P.; Hu, W. MicroRNA-181a sensitizes human malignant glioma U87MG cells to radiation by targeting Bcl-2. Oncol. Rep. 2010, 23, 997–1003. [Google Scholar]
- Li, H.; Chen, L.; Li, J.-J.; Zhou, Q.; Huang, A.; Liu, W.-W.; Wang, K.; Gao, L.; Qi, S.-T.; Lu, Y.-T. miR-519a enhances chemosensitivity and promotes autophagy in glioblastoma by targeting STAT3/Bcl2 signaling pathway. J. Hematol. Oncol. 2018, 11, 70. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Li, A.; Xu, Y.; Xin, Y. Momelotinib sensitizes glioblastoma cells to temozolomide by enhancement of autophagy via JAK2/STAT3 inhibition. Oncol. Rep. 2019, 41, 1883–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, D.; Li, C.; Yang, C.; Zou, Y.; Feng, B.; Li, L.; Liu, W.; Geng, Y.; Luo, Q.; Chen, Z. Polyphyllin VII Promotes Apoptosis and Autophagic Cell Death via ROS-Inhibited AKT Activity, and Sensitizes Glioma Cells to Temozolomide. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Honorato, J.R.; Hauser-Davis, R.A.; Saggioro, E.M.; Correia, F.V.; Sales-Junior, S.F.; Soares, L.O.; Lima, L.d.R.; Moura-Neto, V.; Lopes, G.P.d.F.; Spohr, T.C.d.S. Role of Sonic hedgehog signaling in cell cycle, oxidative stress, and autophagy of temozolomide resistant glioblastoma. J. Cell. Physiol. 2020, 235, 3798–3814. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, M.I.; Vosoughi, A.R.; Albokashy, M.; Butterfield, Y. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020, 287, 1005–1034. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhang, P.; Li, N.; Kiang, K.M.Y.; Cheng, S.Y.; Wong, V.K.-W.; Leung, G.K.-K. Lovastatin enhances cytotoxicity of temozolomide via impairing autophagic flux in glioblastoma cells. BioMed Res. Int. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Wu, Y.; Mayer, K.; von Rosenstiel, C.; Schecker, J.; Baur, S.; Würstle, S.; Liesche-Starnecker, F.; Gempt, J.; Schlegel, J. Lipid peroxidation plays an important role in chemotherapeutic effects of temozolomide and the development of therapy resistance in human glioblastoma. Transl. Oncol. 2020, 13, 100748. [Google Scholar] [CrossRef]
- Yun, E.-J.; Kim, S.; Hsieh, J.-T.; Baek, S.T. Wnt/β-catenin signaling pathway induces autophagy-mediated temozolomide-resistance in human glioblastoma. Cell Death Dis. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Terzioglu-Usak, S.; Nalli, A.; Elibol, B.; Ozek, E.; Hatiboglu, M.A. Anvirzel(TM)regulates cell death through inhibiting GSK-3 activity in human U87 glioma cells. Neurol. Res. 2020, 42, 68–75. [Google Scholar] [CrossRef]
- Kriel, J.; Müller-Nedebock, K.; Maarman, G.; Mbizana, S.; Ojuka, E.; Klumperman, B.; Loos, B. Coordinated autophagy modulation overcomes glioblastoma chemoresistance through disruption of mitochondrial bioenergetics. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Liang, N.; Jia, L.; Liu, Y.; Liang, B.; Kong, D.; Yan, M.; Ma, S.; Liu, X. ATM pathway is essential for ionizing radiation-induced autophagy. Cell. Signal. 2013, 25, 2530–2539. [Google Scholar] [CrossRef]
- Ito, H.; Daido, S.; Kanzawa, T.; Kondo, S.; Kondo, Y. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int. J. Oncol. 2005, 26, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Daido, S.; Yamamoto, A.; Fujiwara, K.; Sawaya, R.; Kondo, S.; Kondo, Y. Inhibition of the DNA-dependent protein kinase catalytic subunit radiosensitizes malignant glioma cells by inducing autophagy. Cancer Res. 2005, 65, 4368–4375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apel, A.; Herr, I.; Schwarz, H.; Rodemann, H.P.; Mayer, A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008, 68, 1485–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, A.; Kanehisa, A.; Martins, I.; Senovilla, L.; Chargari, C.; Dugue, D.; Marino, G.; Kepp, O.; Michaud, M.; Perfettini, J. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. 2014, 21, 92–99. [Google Scholar] [CrossRef]
- Lomonaco, S.L.; Finniss, S.; Xiang, C.; DeCarvalho, A.; Umansky, F.; Kalkanis, S.N.; Mikkelsen, T.; Brodie, C. The induction of autophagy by γ-radiation contributes to the radioresistance of glioma stem cells. Int. J. Cancer 2009, 125, 717–722. [Google Scholar] [CrossRef]
- Paglin, S.; Hollister, T.; Delohery, T.; Hackett, N.; McMahill, M.; Sphicas, E.; Domingo, D.; Yahalom, J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001, 61, 439–444. [Google Scholar]
- Rikiishi, H. Novel insights into the interplay between apoptosis and autophagy. Int. J. Cell Biol. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Gewirtz, D.A. When cytoprotective autophagy isn’t… and even when it is. Autophagy 2014, 10, 391–392. [Google Scholar] [CrossRef] [Green Version]
- Panganiban, R.-A.M.; Snow, A.L.; Day, R.M. Mechanisms of radiation toxicity in transformed and non-transformed cells. Int. J. Mol. Sci. 2013, 14, 15931–15958. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-J.; Long, L.-M.; Yang, N.; Zhang, Q.-Q.; Ji, W.-J.; Zhao, J.-H.; Qin, Z.-H.; Wang, Z.; Chen, G.; Liang, Z.-Q. NVP-BEZ235, a novel dual PI3K/mTOR inhibitor, enhances the radiosensitivity of human glioma stem cells in vitro. Acta Pharmacol. Sin. 2013, 34, 681–690. [Google Scholar] [CrossRef] [Green Version]
- Singer, E.; Judkins, J.; Salomonis, N.; Matlaf, L.; Soteropoulos, P.; McAllister, S.; Soroceanu, L. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis. 2015, 6, e1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.; Kim, C.K.; Alvarez, A.A.; Pangeni, R.P.; Wan, X.; Song, X.; Shi, T.; Yang, Y.; Sastry, N.; Horbinski, C.M. MST4 phosphorylation of ATG4B regulates autophagic activity, tumorigenicity, and radioresistance in glioblastoma. Cancer Cell 2017, 32, 840–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kegelman, T.P.; Wu, B.; Das, S.K.; Talukdar, S.; Beckta, J.M.; Hu, B.; Emdad, L.; Valerie, K.; Sarkar, D.; Furnari, F.B. Inhibition of radiation-induced glioblastoma invasion by genetic and pharmacological targeting of MDA-9/Syntenin. Proc. Natl. Acad. Sci. USA 2017, 114, 370–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koukourakis, M.I.; Mitrakas, A.G.; Giatromanolaki, A. Therapeutic interactions of autophagy with radiation and temozolomide in glioblastoma: Evidence and issues to resolve. Br. J. Cancer 2016, 114, 485–496. [Google Scholar] [CrossRef]
- Jun, F.; Liu, Z.-G.; Liu, X.-M.; Chen, F.-R.; Shi, H.-L.; Pangjesse, C.-S.; NG, H.-K.; Chen, Z.-P. Glioblastoma stem cells resistant to temozolomide-induced autophagy. Chin. Med. J. 2009, 122, 1255–1259. [Google Scholar]
- Ciechomska, I.A.; Przanowski, P.; Jackl, J.; Wojtas, B.; Kaminska, B. BIX01294, an inhibitor of histone methyltransferase, induces autophagy-dependent differentiation of glioma stem-like cells. Sci. Rep. 2016, 6, 38723. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, S.; Comincini, S. Autophagy and ionizing radiation in tumors: The “survive or not survive” dilemma. J. Cell. Physiol. 2013, 228, 1–8. [Google Scholar] [CrossRef]
- Jo, G.H.; Bögler, O.; Chwae, Y.-J.; Yoo, H.; Lee, S.H.; Park, J.B.; Kim, Y.-J.; Kim, J.H.; Gwak, H.-S. Radiation-induced autophagy contributes to cell death and induces apoptosis partly in malignant glioma cells. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2015, 47, 221. [Google Scholar] [CrossRef]
- Mendiburu-Eliçabe, M.; Gil-Ranedo, J.; Izquierdo, M. Efficacy of rapamycin against glioblastoma cancer stem cells. Clin. Transl. Oncol. 2014, 16, 495–502. [Google Scholar] [CrossRef]
- Cerniglia, G.J.; Karar, J.; Tyagi, S.; Christofidou-Solomidou, M.; Rengan, R.; Koumenis, C.; Maity, A. Inhibition of autophagy as a strategy to augment radiosensitization by the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Mol. Pharmacol. 2012, 82, 1230–1240. [Google Scholar] [CrossRef] [Green Version]
- Del Alcazar, C.R.G.; Hardebeck, M.C.; Mukherjee, B.; Tomimatsu, N.; Gao, X.; Yan, J.; Xie, X.-J.; Bachoo, R.; Li, L.; Habib, A.A. Inhibition of DNA double-strand break repair by the dual PI3K/mTOR inhibitor NVP-BEZ235 as a strategy for radiosensitization of glioblastoma. Clin. Cancer Res. 2014, 20, 1235–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int. J. Cancer 2011, 129, 2720–2731. [Google Scholar] [CrossRef] [PubMed]
- Laks, D.R.; Oses-Prieto, J.A.; Alvarado, A.G.; Nakashima, J.; Chand, S.; Azzam, D.B.; Gholkar, A.A.; Sperry, J.; Ludwig, K.; Condro, M.C. A molecular cascade modulates MAP1B and confers resistance to mTOR inhibition in human glioblastoma. Neuro-Oncology 2018, 20, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wang, B.; Zheng, R.; Zhang, J.; Huang, C.; Zheng, R.; Huang, Z.; Qiu, W.; Liu, M.; Yang, K. Linc-RA1 inhibits autophagy and promotes radioresistance by preventing H2Bub1/USP44 combination in glioma cells. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Zheng, W.; Chen, Q.; Wang, C.; Yao, D.; Zhu, L.; Pan, Y.; Zhang, J.; Bai, Y.; Shao, C. Inhibition of Cathepsin D (CTSD) enhances radiosensitivity of glioblastoma cells by attenuating autophagy. Mol. Carcinog. 2020, 59, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Mitrakas, A.G.; Kalamida, D.; Giatromanolaki, A.; Pouliliou, S.; Tsolou, A.; Kyranas, R.; Koukourakis, M.I. Autophagic flux response and glioblastoma sensitivity to radiation. Cancer Biol. Med. 2018, 15, 260. [Google Scholar]
- Xiao, N.; Li, C.; Liao, W.; Yin, J.; Zhang, S.; Zhang, P.; Yuan, L.; Hong, M. FOXG1 mediates the radiosensitivity of glioma cells through regulation of autophagy. Int. J. Radiat. Biol. 2020, 1–32. [Google Scholar] [CrossRef]
- Skaga, E.; Kulesskiy, E.; Fayzullin, A.; Sandberg, C.J.; Potdar, S.; Kyttälä, A.; Langmoen, I.A.; Laakso, A.; Gaál-Paavola, E.; Perola, M. Intertumoral heterogeneity in patient-specific drug sensitivities in treatment-naïve glioblastoma. BMC Cancer 2019, 19, 628. [Google Scholar] [CrossRef] [Green Version]
- Boyd, N.H.; Walker, K.; Fried, J.; Hackney, J.R.; McDonald, P.C.; Benavides, G.A.; Spina, R.; Audia, A.; Scott, S.E.; Libby, C.J. Addition of carbonic anhydrase 9 inhibitor SLC-0111 to temozolomide treatment delays glioblastoma growth in vivo. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Rulseh, A.M.; Keller, J.; Klener, J.; Šroubek, J.; Dbalý, V.; Syrůček, M.; Tovaryš, F.; Vymazal, J. Long-term survival of patients suffering from glioblastoma multiforme treated with tumor-treating fields. World J. Surg. Oncol. 2012, 10, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Weller, M.; Felsberg, J.; Hartmann, C.; Berger, H.; Steinbach, J.P.; Schramm, J.; Westphal, M.; Schackert, G.; Simon, M.; Tonn, J.C. Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: A prospective translational study of the German Glioma Network. J. Clin. Oncol. 2009, 27, 5743–5750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriksson, R.; Asklund, T.; Poulsen, H.S. Impact of therapy on quality of life, neurocognitive function and their correlates in glioblastoma multiforme: A review. J. Neuro-Oncol. 2011, 104, 639–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalva, C.; Cortes, U.; Wager, M.; Tourani, J.-M.; Rivet, P.; Marquant, C.; Martin, S.; Turhan, A.G.; Karayan-Tapon, L. O6-Methylguanine-Methyltransferase (MGMT) promoter methylation status in glioma stem-like cells is correlated to temozolomide sensitivity under differentiation-promoting conditions. Int. J. Mol. Sci. 2012, 13, 6983–6994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suvà, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Zhong, Z.; Luo, C.; Xiao, Y.; Li, L.; Zhang, X.; Yang, L.; Xiao, K.; Ning, Y.; Chen, L. The miR-26a/AP-2α/Nanog signaling axis mediates stem cell self-renewal and temozolomide resistance in glioma. Theranostics 2019, 9, 5497. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Jahangiri, A.; Chen, W.; Nguyen, A.T.; Yagnik, G.; Pereira, M.P.; Jain, S.; Garcia, J.H.; Shah, S.S.; Wadhwa, H. Clonal ZEB1-Driven Mesenchymal Transition Promotes Targetable Oncologic Antiangiogenic Therapy Resistance. Cancer Res. 2020, 80, 1498–1511. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Fujioka, K.; Ikeda, K.; Murayama, Y.; Manome, Y. Temozolomide does not influence the transcription or activity of matrix metalloproteinases 9 and 2 in glioma cell lines. J. Clin. Neurosci. 2017, 41, 144–149. [Google Scholar] [CrossRef]
- Suzuki, H.; Osawa, T.; Fujioka, Y.; Noda, N.N. Structural biology of the core autophagy machinery. Curr. Opin. Struct. Biol. 2017, 43, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xu, R.; Zhang, C.; Xu, Y.; Han, M.; Huang, B.; Chen, A.; Qiu, C.; Thorsen, F.; Prestegarden, L. Trifluoperazine, a novel autophagy inhibitor, increases radiosensitivity in glioblastoma by impairing homologous recombination. J. Exp. Clin. Cancer Res. 2017, 36, 118. [Google Scholar] [CrossRef] [Green Version]
- Qayed, M.; Cash, T.; Tighiouart, M.; MacDonald, T.J.; Goldsmith, K.C.; Tanos, R.; Kean, L.; Watkins, B.; Suessmuth, Y.; Wetmore, C. A phase I study of sirolimus in combination with metronomic therapy (CHOAnome) in children with recurrent or refractory solid and brain tumors. Pediatr. Blood Cancer 2020, 67, e28134. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Iwado, E.; Kondo, Y.; Aoki, H.; Hayashi, Y.; Georgescu, M.; Sawaya, R.; Hess, K.; Mills, G.; Kawamura, H. Autophagy-inducing agents augment the antitumor effect of telerase-selve oncolytic adenovirus OBP-405 on glioblastoma cells. Gene Ther. 2008, 15, 1233–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.-Y.; Kim, S.Y.; Shin, S.-L.; Choi, Y.-S.; Lee, J.-H.; Tsujimoto, Y.; Lee, J.-H. Reactive astrocytes express bis, a bcl-2-binding protein, after transient forebrain ischemia. Exp. Neurol. 2002, 175, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Guadagno, E.; Borrelli, G.; Pignatiello, S.; Donato, A.; Presta, I.; Arcidiacono, B.; Malara, N.; Solari, D.; Somma, T.; Cappabianca, P. Anti-Apoptotic and Anti-Oxidant Proteins in Glioblastomas: Immunohistochemical Expression of Beclin and DJ-1 and Its Correlation with Prognosis. Int. J. Mol. Sci. 2019, 20, 4066. [Google Scholar] [CrossRef] [Green Version]
- Balvers, R.K.; Lamfers, M.L.; Kloezeman, J.J.; Kleijn, A.; Pont, L.M.B.; Dirven, C.M.; Leenstra, S. ABT-888 enhances cytotoxic effects of temozolomide independent of MGMT status in serum free cultured glioma cells. J. Transl. Med. 2015, 13, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Liu, L.; Wang, P.; Yao, Y.; Xue, Y.; Liu, Y. EMAP-II sensitize U87MG and glioma stem-like cells to temozolomide via induction of autophagy-mediated cell death and G2/M arrest. Cell Cycle 2017, 16, 1085–1092. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.H.; Yu, J.; Brown, K.; Levine, B. Beclin 1 contains a leucine-rich nuclear export signal that is required for its autophagy and tumor suppressor function. Cancer Res. 2001, 61, 3443–3449. [Google Scholar]
- Tassa, A.; Roux, M.P.; Attaix, D.; Bechet, D.M. Class III phosphoinositide 3-kinase–Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem. J. 2003, 376, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martin, P.; Lahuerta, M.; Viana, R.; Knecht, E.; Sanz, P. Regulation of the autophagic PI3KC3 complex by laforin/malin E3-ubiquitin ligase, two proteins involved in Lafora disease. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2020, 1867, 118613. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Li, X.; Lu, Z. Protein kinase activity of the glycolytic enzyme PGK1 regulates autophagy to promote tumorigenesis. Autophagy 2017, 13, 1246–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaverina, N.V.; Kadagidze, Z.G.; Borovjagin, A.V.; Karseladze, A.I.; Kim, C.K.; Lesniak, M.S.; Miska, J.; Zhang, P.; Baryshnikova, M.A.; Xiao, T. Tamoxifen overrides autophagy inhibition in Beclin-1-deficient glioma cells and their resistance to adenovirus-mediated oncolysis via upregulation of PUMA and BAX. Oncogene 2018, 37, 6069–6082. [Google Scholar] [CrossRef] [PubMed]
- Ulasov, I.V.; Mijanovic, O.; Savchuk, S.; Gonzalez-Buendia, E.; Sonabend, A.; Xiao, T.; Timashev, P.; Lesniak, M.S. TMZ regulates GBM stemness via MMP14-DLL4-Notch3 pathway. Int. J. Cancer 2020, 146, 2218–2228. [Google Scholar] [CrossRef]
- Witusik-Perkowska, M.; Zakrzewska, M.; Sikorska, B.; Papierz, W.; Jaskolski, D.J.; Szemraj, J.; Liberski, P.P. Glioblastoma-derived cells in vitro unveil the spectrum of drug resistance capability—Comparative study of tumour chemosensitivity in different culture systems. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef]
- Vázquez, P.; Arroba, A.I.; Cecconi, F.; de la Rosa, E.J.; Boya, P.; De Pablo, F. Atg5 and Ambra1 differentially modulate neurogenesis in neural stem cells. Autophagy 2012, 8, 187–199. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liang, C.-C.; Bian, Z.C.; Zhu, Y.; Guan, J.-L. FIP200 is required for maintenance and differentiation of postnatal neural stem cells. Nat. Neurosci. 2013, 16, 532–542. [Google Scholar] [CrossRef] [Green Version]
- Yazdankhah, M.; Farioli-Vecchioli, S.; Tonchev, A.; Stoykova, A.; Cecconi, F. The autophagy regulators Ambra1 and Beclin 1 are required for adult neurogenesis in the brain subventricular zone. Cell Death Dis. 2014, 5, e1403. [Google Scholar] [CrossRef]
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.E.; Richard-Londt, A.; Brandner, S.; Waddington, S.N.; Schapira, A.H.; Duchen, M.R. Mitochondria and quality control defects in a mouse model of Gaucher disease—Links to Parkinson’s disease. Cell Metab. 2013, 17, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, C.J.; Lenk, G.M.; Meisler, M.H. Defective autophagy in neurons and astrocytes from mice deficient in PI (3, 5) P2. Hum. Mol. Genet. 2009, 18, 4868–4878. [Google Scholar] [CrossRef] [Green Version]
- Tang, G.; Yue, Z.; Talloczy, Z.; Hagemann, T.; Cho, W.; Messing, A.; Sulzer, D.L.; Goldman, J.E. Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum. Mol. Genet. 2008, 17, 1540–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magini, A.; Polchi, A.; Di Meo, D.; Mariucci, G.; Sagini, K.; De Marco, F.; Cassano, T.; Giovagnoli, S.; Dolcetta, D.; Emiliani, C. TFEB activation restores migration ability to Tsc1-deficient adult neural stem/progenitor cells. Hum. Mol. Genet. 2017, 26, 3303–3312. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, I.; Baig, M.H.; Mahfooz, S.; Rahim, M.; Karacam, B.; Elbasan, E.B.; Ulasov, I.; Dong, J.-J.; Hatiboglu, M.A. Deciphering the Role of Autophagy in Treatment of Resistance Mechanisms in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 1318. https://doi.org/10.3390/ijms22031318
Khan I, Baig MH, Mahfooz S, Rahim M, Karacam B, Elbasan EB, Ulasov I, Dong J-J, Hatiboglu MA. Deciphering the Role of Autophagy in Treatment of Resistance Mechanisms in Glioblastoma. International Journal of Molecular Sciences. 2021; 22(3):1318. https://doi.org/10.3390/ijms22031318
Chicago/Turabian StyleKhan, Imran, Mohammad Hassan Baig, Sadaf Mahfooz, Moniba Rahim, Busra Karacam, Elif Burce Elbasan, Ilya Ulasov, Jae-June Dong, and Mustafa Aziz Hatiboglu. 2021. "Deciphering the Role of Autophagy in Treatment of Resistance Mechanisms in Glioblastoma" International Journal of Molecular Sciences 22, no. 3: 1318. https://doi.org/10.3390/ijms22031318
APA StyleKhan, I., Baig, M. H., Mahfooz, S., Rahim, M., Karacam, B., Elbasan, E. B., Ulasov, I., Dong, J.-J., & Hatiboglu, M. A. (2021). Deciphering the Role of Autophagy in Treatment of Resistance Mechanisms in Glioblastoma. International Journal of Molecular Sciences, 22(3), 1318. https://doi.org/10.3390/ijms22031318