
Isoform-Specific Roles of Mutant p63 in Human Diseases


Abstract
:Simple Summary
Abstract
1. Introduction
2. P63 in the Development of Stratified Epithelial Tissues
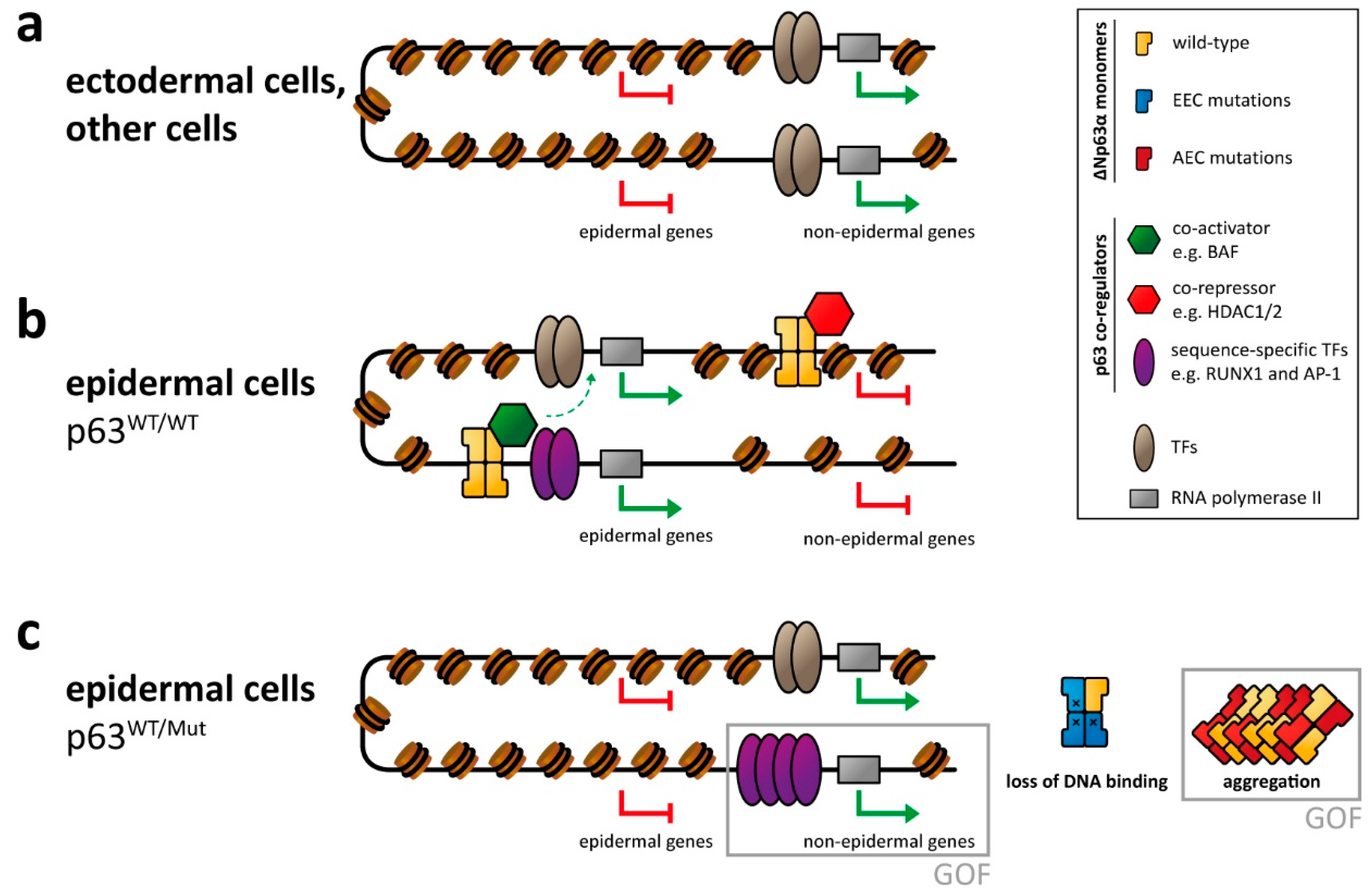
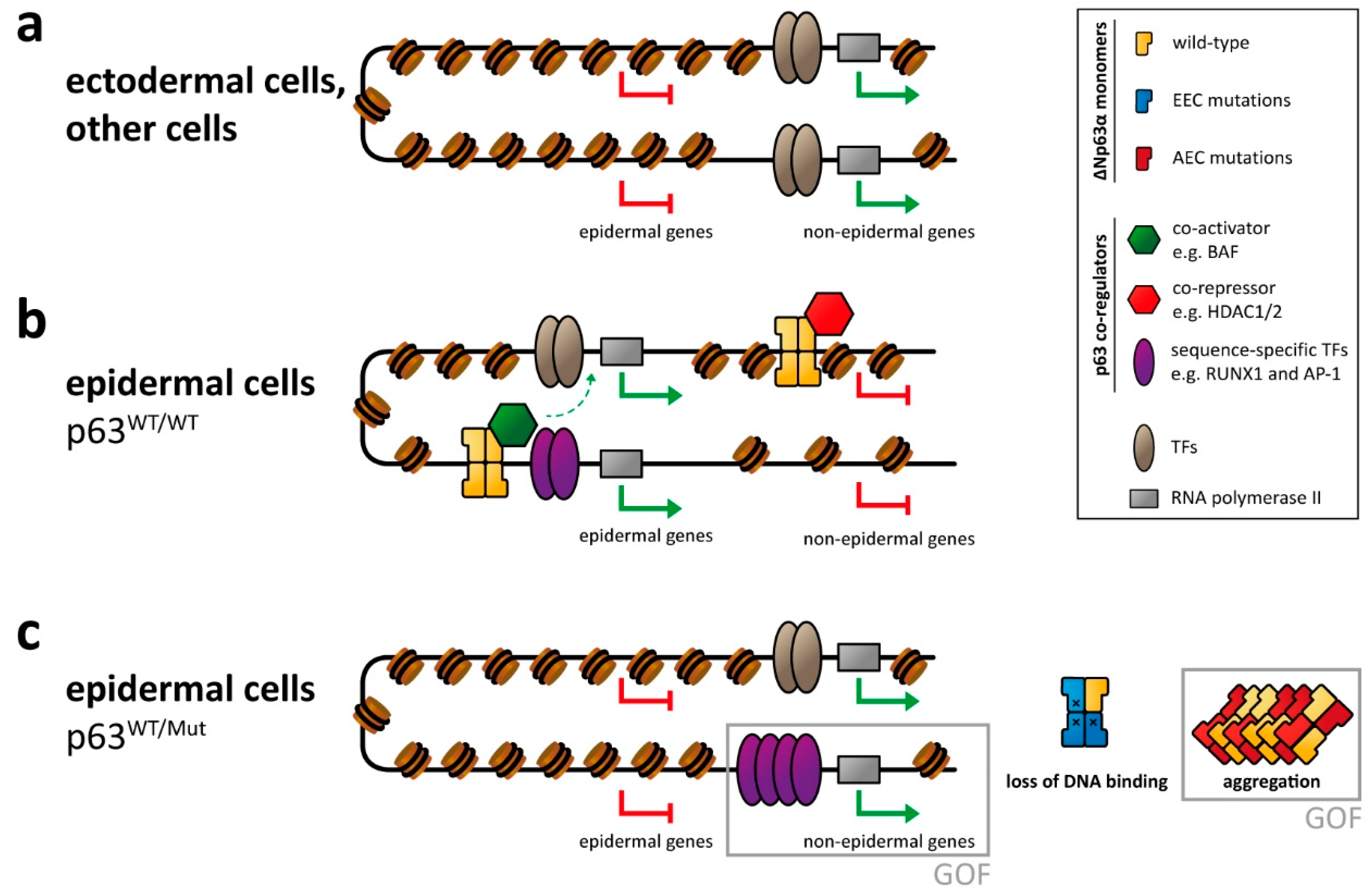
2.1. Binding of p63 to Enhancer Elements
2.2. Cooperation with Other Transcription Factors
2.3. Role of p63 for Chromatin Remodeling
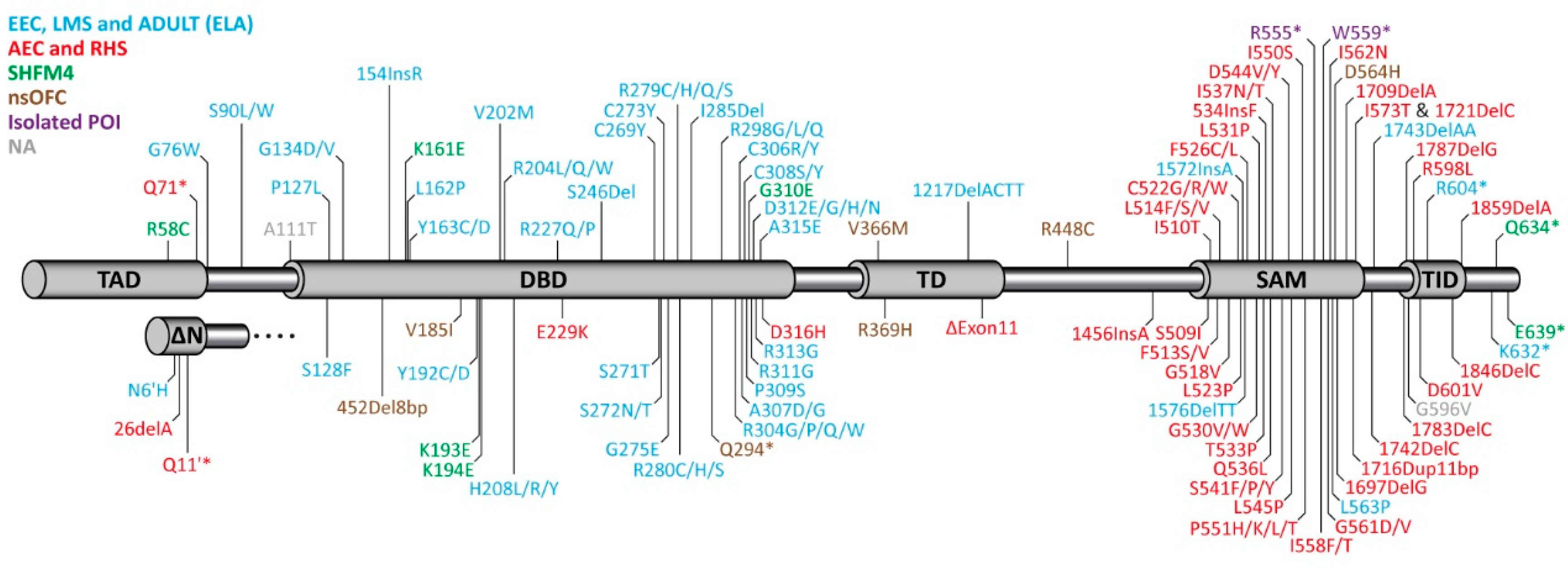
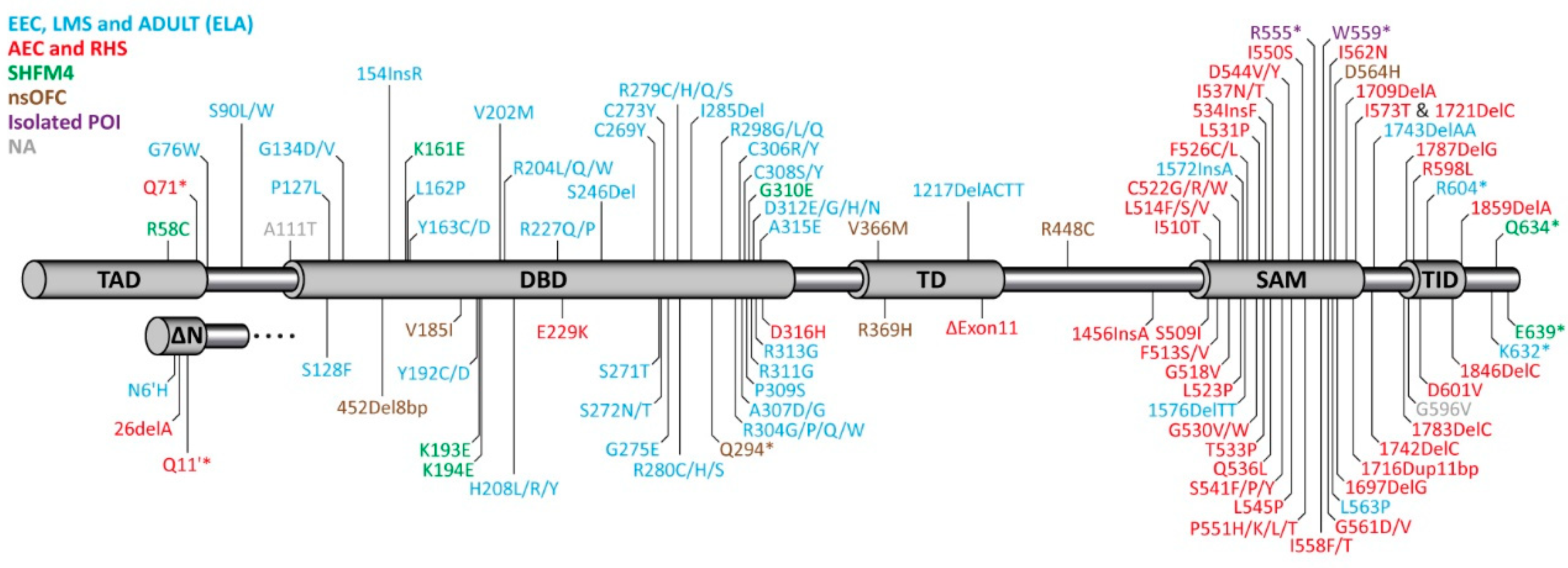
3. Syndromes Caused by Mutations in p63 in Human Patients
4. Molecular Disease Mechanism
4.1. EEC Syndrome
4.2. AEC Syndrome
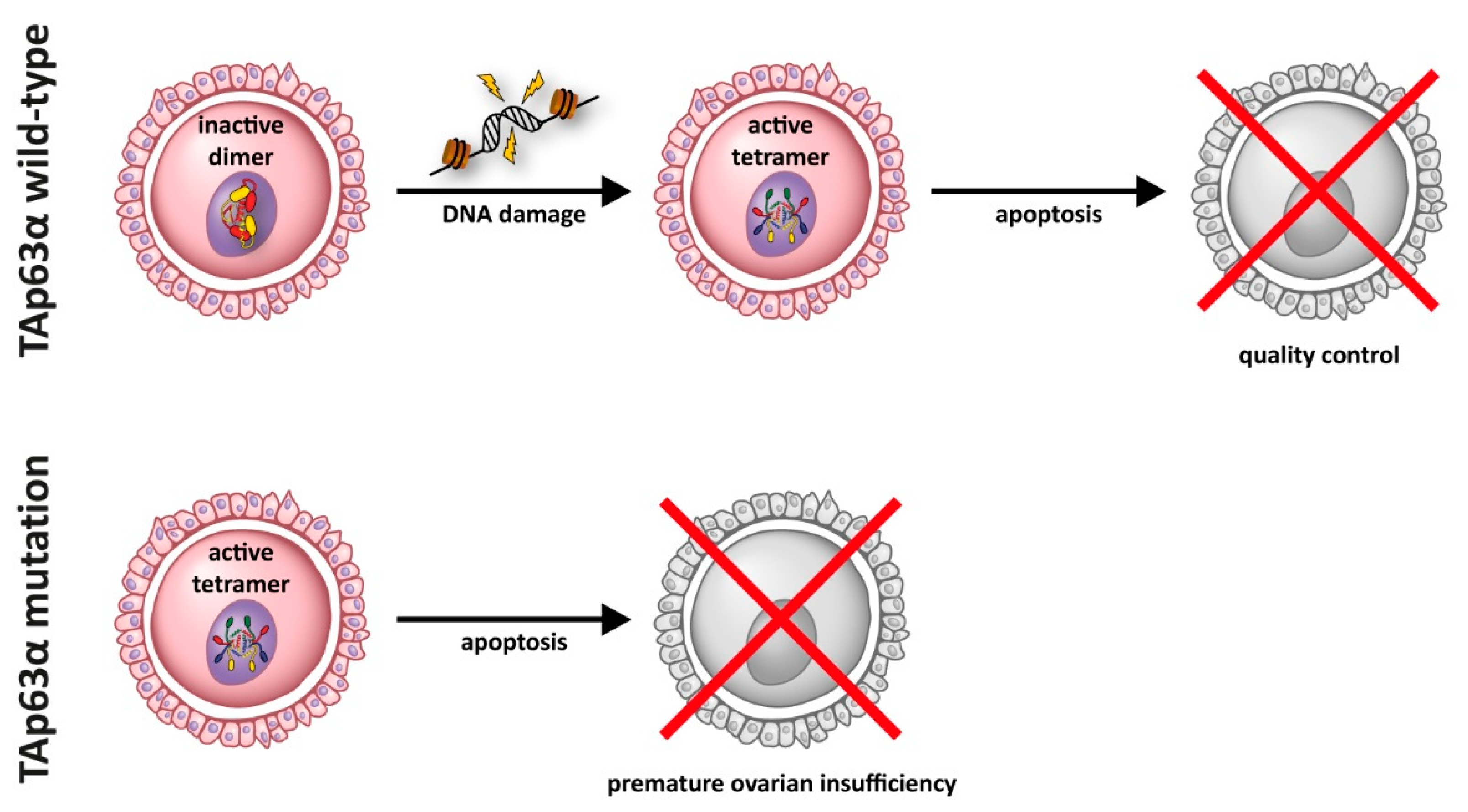
5. Impact of p63 Mutations on Female Fertility
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieging, K.T.; Attardi, L.D. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol. 2012, 22, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- Kenzelmann Broz, D.; Spano Mello, S.; Bieging, K.T.; Jiang, D.; Dusek, R.L.; Brady, C.A.; Sidow, A.; Attardi, L.D. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013, 27, 1016–1031. [Google Scholar] [CrossRef] [Green Version]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dotsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.M.; Dumont, X.; et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997, 90, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Walker, N.; Bronson, R.; Kaghad, M.; Oosterwegel, M.; Bonnin, J.; Vagner, C.; Bonnet, H.; Dikkes, P.; Sharpe, A.; et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000, 404, 99–103. [Google Scholar] [CrossRef]
- Armstrong, J.F.; Kaufman, M.H.; Harrison, D.J.; Clarke, A.R. High-frequency developmental abnormalities in p53-deficient mice. Curr. Biol. 1995, 5, 931–936. [Google Scholar] [CrossRef] [Green Version]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Holembowski, L.; Kramer, D.; Riedel, D.; Sordella, R.; Nemajerova, A.; Dobbelstein, M.; Moll, U.M. TAp73 is essential for germ cell adhesion and maturation in testis. J. Cell Biol. 2014, 204, 1173–1190. [Google Scholar] [CrossRef] [Green Version]
- Inoue, S.; Tomasini, R.; Rufini, A.; Elia, A.J.; Agostini, M.; Amelio, I.; Cescon, D.; Dinsdale, D.; Zhou, L.; Harris, I.S.; et al. TAp73 is required for spermatogenesis and the maintenance of male fertility. Proc. Natl. Acad. Sci. USA 2014, 111, 1843–1848. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C.B.; Mays, D.J.; Beeler, J.S.; Rosenbluth, J.M.; Boyd, K.L.; Santos Guasch, G.L.; Shaver, T.M.; Tang, L.J.; Liu, Q.; Shyr, Y.; et al. p73 is Required for multiciliogenesis and regulates the Foxj1-associated gene network. Cell Rep. 2016, 14, 2289–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Mills, A.A.; Zheng, B.; Wang, X.J.; Vogel, H.; Roop, D.R.; Bradley, A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999, 398, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Senoo, M.; Pinto, F.; Crum, C.P.; McKeon, F. p63 Is essential for the proliferative potential of stem cells in stratified epithelia. Cell 2007, 129, 523–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, E.K.; Yang, A.; Kettenbach, A.; Bamberger, C.; Michaelis, A.H.; Zhu, Z.; Elvin, J.A.; Bronson, R.T.; Crum, C.P.; McKeon, F. p63 protects the female germ line during meiotic arrest. Nature 2006, 444, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Livera, G.; Petre-Lazar, B.; Guerquin, M.J.; Trautmann, E.; Coffigny, H.; Habert, R. p63 null mutation protects mouse oocytes from radio-induced apoptosis. Reproduction 2008, 135, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, B.; Hofmann, K.; Boulton, S.; Gartner, A. The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr. Biol. 2001, 11, 1722–1727. [Google Scholar] [CrossRef] [Green Version]
- Derry, W.B.; Putzke, A.P.; Rothman, J.H. Caenorhabditis elegans p53: Role in apoptosis, meiosis, and stress resistance. Science 2001, 294, 591–595. [Google Scholar] [CrossRef] [Green Version]
- Ollmann, M.; Young, L.M.; Di Como, C.J.; Karim, F.; Belvin, M.; Robertson, S.; Whittaker, K.; Demsky, M.; Fisher, W.W.; Buchman, A.; et al. Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell 2000, 101, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Brodsky, M.H.; Nordstrom, W.; Tsang, G.; Kwan, E.; Rubin, G.M.; Abrams, J.M. Drosophila p53 binds a damage response element at the reaper locus. Cell 2000, 101, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Heering, J.; Jonker, H.R.; Lohr, F.; Schwalbe, H.; Dotsch, V. Structural investigations of the p53/p73 homologs from the tunicate species Ciona intestinalis reveal the sequence requirements for the formation of a tetramerization domain. Protein Sci. 2016, 25, 410–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyi, V.A.; Ak, P.; Markert, E.; Wang, H.; Hu, W.; Puzio-Kuter, A.; Levine, A.J. The origins and evolution of the p53 family of genes. Cold Spring Harb. Perspect. Biol. 2010, 2, a001198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, H.D.; Lohr, F.; Vogel, V.; Mantele, W.; Dotsch, V. Structural evolution of C-terminal domains in the p53 family. EMBO J. 2007, 26, 3463–3473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebel, J.; Tuppi, M.; Sanger, N.; Schumacher, B.; Dotsch, V. DNA Damaged Induced Cell Death in Oocytes. Molecules 2020, 25, 5714. [Google Scholar] [CrossRef]
- Keeney, S.; Giroux, C.N.; Kleckner, N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 1997, 88, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Romanienko, P.J.; Camerini-Otero, R.D. The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol. Cell 2000, 6, 975–987. [Google Scholar] [CrossRef]
- Claeys Bouuaert, C.; Tischfield, S.E.; Pu, S.; Mimitou, E.P.; Arias-Palomo, E.; Berger, J.M.; Keeney, S. Structural and functional characterization of the Spo11 core complex. Nat. Struct. Mol. Biol. 2021, 28, 92–102. [Google Scholar] [CrossRef]
- Kerr, J.B.; Hutt, K.J.; Michalak, E.M.; Cook, M.; Vandenberg, C.J.; Liew, S.H.; Bouillet, P.; Mills, A.; Scott, C.L.; Findlay, J.K.; et al. DNA damage-induced primordial follicle oocyte apoptosis and loss of fertility require TAp63-mediated induction of Puma and Noxa. Mol. Cell 2012, 48, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Deutsch, G.B.; Zielonka, E.M.; Coutandin, D.; Weber, T.A.; Schafer, B.; Hannewald, J.; Luh, L.M.; Durst, F.G.; Ibrahim, M.; Hoffmann, J.; et al. DNA damage in oocytes induces a switch of the quality control factor TAp63alpha from dimer to tetramer. Cell 2011, 144, 566–576. [Google Scholar] [CrossRef] [Green Version]
- Coutandin, D.; Osterburg, C.; Srivastav, R.K.; Sumyk, M.; Kehrloesser, S.; Gebel, J.; Tuppi, M.; Hannewald, J.; Schafer, B.; Salah, E.; et al. Quality control in oocytes by p63 is based on a spring-loaded activation mechanism on the molecular and cellular level. eLife 2016, 5. [Google Scholar] [CrossRef]
- Bolcun-Filas, E.; Rinaldi, V.D.; White, M.E.; Schimenti, J.C. Reversal of female infertility by Chk2 ablation reveals the oocyte DNA damage checkpoint pathway. Science 2014, 343, 533–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuppi, M.; Kehrloesser, S.; Coutandin, D.W.; Rossi, V.; Luh, L.M.; Strubel, A.; Hotte, K.; Hoffmeister, M.; Schafer, B.; De Oliveira, T.; et al. Oocyte DNA damage quality control requires consecutive interplay of CHK2 and CK1 to activate p63. Nat. Struct. Mol. Biol. 2018, 25, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Gebel, J.; Tuppi, M.; Chaikuad, A.; Hotte, K.; Schroder, M.; Schulz, L.; Lohr, F.; Gutfreund, N.; Finke, F.; Henrich, E.; et al. p63 uses a switch-like mechanism to set the threshold for induction of apoptosis. Nat. Chem. Biol. 2020, 16, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Woodard, T.L.; Bolcun-Filas, E. Prolonging reproductive life after cancer: The need for fertoprotective therapies. Trends Cancer 2016, 2, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Spears, N.; Lopes, F.; Stefansdottir, A.; Rossi, V.; De Felici, M.; Anderson, R.A.; Klinger, F.G. Ovarian damage from chemotherapy and current approaches to its protection. Hum. Reprod. Update 2019, 25, 673–693. [Google Scholar] [CrossRef]
- Hao, X.; Anastacio, A.; Liu, K.; Rodriguez-Wallberg, K.A. Ovarian follicle depletion induced by chemotherapy and the investigational stages of potential fertility-protective treatments—A review. Int. J. Mol. Sci. 2019, 20, 4720. [Google Scholar] [CrossRef] [Green Version]
- Jeruss, J.S.; Woodruff, T.K. Preservation of fertility in patients with cancer. N. Engl. J. Med. 2009, 360, 902–911. [Google Scholar] [CrossRef] [Green Version]
- Johnston, R.J.; Wallace, W.H. Normal ovarian function and assessment of ovarian reserve in the survivor of childhood cancer. Pediatr. Blood Cancer 2009, 53, 296–302. [Google Scholar] [CrossRef]
- Maltaris, T.; Weigel, M.; Mueller, A.; Schmidt, M.; Seufert, R.; Fischl, F.; Koelbl, H.; Dittrich, R. Cancer and fertility preservation: Fertility preservation in breast cancer patients. Breast Cancer Res. 2008, 10, 206. [Google Scholar] [CrossRef]
- Lena, A.M.; Rossi, V.; Osterburg, S.; Smirnov, A.; Osterburg, C.; Tuppi, M.; Cappello, A.; Amelio, I.; Dotsch, V.; De Felici, M.; et al. The p63 C-terminus is essential for murine oocyte integrity. Nat. Commun. 2021, 12, 383. [Google Scholar] [CrossRef]
- Serber, Z.; Lai, H.C.; Yang, A.; Ou, H.D.; Sigal, M.S.; Kelly, A.E.; Darimont, B.D.; Duijf, P.H.; Van Bokhoven, H.; McKeon, F.; et al. A C-terminal inhibitory domain controls the activity of p63 by an intramolecular mechanism. Mol. Cell Biol. 2002, 22, 8601–8611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, R.A.; Smalley, K.; Magraw, C.; Serna, V.A.; Kurita, T.; Raghavan, S.; Sinha, S. DeltaNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development 2012, 139, 772–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candi, E.; Rufini, A.; Terrinoni, A.; Dinsdale, D.; Ranalli, M.; Paradisi, A.; De Laurenzi, V.; Spagnoli, L.G.; Catani, M.V.; Ramadan, S.; et al. Differential roles of p63 isoforms in epidermal development: Selective genetic complementation in p63 null mice. Cell Death Differ. 2006, 13, 1037–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauskopf, K.; Gebel, J.; Kazemi, S.; Tuppi, M.; Lohr, F.; Schafer, B.; Koch, J.; Guntert, P.; Dotsch, V.; Kehrloesser, S. Regulation of the activity in the p53 family depends on the organization of the transactivation domain. Structure 2018, 26, 1091–1100.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krois, A.S.; Ferreon, J.C.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Recognition of the disordered p53 transactivation domain by the transcriptional adapter zinc finger domains of CREB-binding protein. Proc. Natl. Acad. Sci. USA 2016, 113, E1853–E1862. [Google Scholar] [CrossRef] [Green Version]
- Burge, S.; Teufel, D.P.; Townsley, F.M.; Freund, S.M.; Bycroft, M.; Fersht, A.R. Molecular basis of the interactions between the p73 N terminus and p300: Effects on transactivation and modulation by phosphorylation. Proc. Natl. Acad. Sci. USA 2009, 106, 3142–3147. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Zhu, Z.; Kettenbach, A.; Kapranov, P.; McKeon, F.; Gingeras, T.R.; Struhl, K. Genome-wide mapping indicates that p73 and p63 co-occupy target sites and have similar dna-binding profiles in vivo. PLoS ONE 2010, 5, e11572. [Google Scholar] [CrossRef]
- Carroll, D.K.; Carroll, J.S.; Leong, C.O.; Cheng, F.; Brown, M.; Mills, A.A.; Brugge, J.S.; Ellisen, L.W. p63 regulates an adhesion programme and cell survival in epithelial cells. Nat. Cell Biol. 2006, 8, 551–561. [Google Scholar] [CrossRef]
- Truong, A.B.; Kretz, M.; Ridky, T.W.; Kimmel, R.; Khavari, P.A. p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev. 2006, 20, 3185–3197. [Google Scholar] [CrossRef] [Green Version]
- Soares, E.; Zhou, H. Master regulatory role of p63 in epidermal development and disease. Cell Mol. Life Sci. 2018, 75, 1179–1190. [Google Scholar] [CrossRef] [Green Version]
- Kouwenhoven, E.N.; Oti, M.; Niehues, H.; van Heeringen, S.J.; Schalkwijk, J.; Stunnenberg, H.G.; van Bokhoven, H.; Zhou, H. Transcription factor p63 bookmarks and regulates dynamic enhancers during epidermal differentiation. EMBO Rep. 2015, 16, 863–878. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Tanis, S.E.J.; Smits, J.P.H.; Kouwenhoven, E.N.; Oti, M.; van den Bogaard, E.H.; Logie, C.; Stunnenberg, H.G.; van Bokhoven, H.; Mulder, K.W.; et al. Mutant p63 affects epidermal cell identity through rewiring the enhancer landscape. Cell Rep. 2018, 25, 3490–3503.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, E.; Xu, Q.; Li, Q.; Qu, J.; Zheng, Y.; Raeven, H.H.M.; Brandao, K.O.; Petit, I.; van den Akker, W.M.R.; van Heeringen, S.J.; et al. Single-cell RNA-seq identifies a reversible mesodermal activation in abnormally specified epithelia of p63 EEC syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 17361–17370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzales, K.A.U.; Fuchs, E. Skin and its regenerative powers: An alliance between stem cells and their niche. Dev. Cell 2017, 43, 387–401. [Google Scholar] [CrossRef] [Green Version]
- Sada, A.; Jacob, F.; Leung, E.; Wang, S.; White, B.S.; Shalloway, D.; Tumbar, T. Defining the cellular lineage hierarchy in the interfollicular epidermis of adult skin. Nat. Cell Biol. 2016, 18, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Tickle, C. Patterning systems—From one end of the limb to the other. Dev. Cell 2003, 4, 449–458. [Google Scholar] [CrossRef] [Green Version]
- Niswander, L. Interplay between the molecular signals that control vertebrate limb development. Int. J. Dev. Biol. 2002, 46, 877–881. [Google Scholar]
- Coutandin, D.; Lohr, F.; Niesen, F.H.; Ikeya, T.; Weber, T.A.; Schafer, B.; Zielonka, E.M.; Bullock, A.N.; Yang, A.; Guntert, P.; et al. Conformational stability and activity of p73 require a second helix in the tetramerization domain. Cell Death Differ. 2009, 16, 1582–1589. [Google Scholar] [CrossRef] [Green Version]
- Joerger, A.C.; Rajagopalan, S.; Natan, E.; Veprintsev, D.B.; Robinson, C.V.; Fersht, A.R. Structural evolution of p53, p63, and p73: Implication for heterotetramer formation. Proc. Natl. Acad. Sci. USA 2009, 106, 17705–17710. [Google Scholar] [CrossRef] [Green Version]
- Gebel, J.; Luh, L.M.; Coutandin, D.; Osterburg, C.; Lohr, F.; Schafer, B.; Frombach, A.S.; Sumyk, M.; Buchner, L.; Krojer, T.; et al. Mechanism of TAp73 inhibition by DeltaNp63 and structural basis of p63/p73 hetero-tetramerization. Cell Death Differ. 2016, 23, 1930–1940. [Google Scholar] [CrossRef] [Green Version]
- Beeler, J.S.; Marshall, C.B.; Gonzalez-Ericsson, P.I.; Shaver, T.M.; Santos Guasch, G.L.; Lea, S.T.; Johnson, K.N.; Jin, H.; Venters, B.J.; Sanders, M.E.; et al. p73 regulates epidermal wound healing and induced keratinocyte programming. PLoS ONE 2019, 14, e0218458. [Google Scholar] [CrossRef] [PubMed]
- Ferone, G.; Mollo, M.R.; Missero, C. Epidermal cell junctions and their regulation by p63 in health and disease. Cell Tissue Res. 2015, 360, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Ferone, G.; Mollo, M.R.; Thomason, H.A.; Antonini, D.; Zhou, H.Q.; Ambrosio, R.; De Rosa, L.; Salvatore, D.; Getsios, S.; van Bokhoven, H.; et al. p63 control of desmosome gene expression and adhesion is compromised in AEC syndrome. Hum. Mol. Genet. 2013, 22, 531–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, B.C.; Lefort, K.; Mandinova, A.; Antonini, D.; Devgan, V.; Della Gatta, G.; Koster, M.I.; Zhang, Z.; Wang, J.; di Vignano, A.T.; et al. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Gene Dev. 2006, 20, 1028–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, R.A.; Birkaya, B.; Sinha, S. A functional enhancer of keratin14 is a direct transcriptional target of deltaNp63. J. Investig. Dermatol. 2007, 127, 1175–1186. [Google Scholar] [CrossRef] [Green Version]
- Lopardo, T.; Lo Iacono, N.; Marinari, B.; Giustizieri, M.L.; Cyr, D.G.; Merlo, G.; Crosti, F.; Costanzo, A.; Guerrini, L. Claudin-1 is a p63 target gene with a crucial role in epithelial development. PLoS ONE 2008, 3, e2715. [Google Scholar] [CrossRef]
- Ihrie, R.A.; Marques, M.R.; Nguyen, B.T.; Horner, J.S.; Papazoglu, C.; Bronson, R.T.; Mills, A.A.; Attardi, L.D. Perp is a p63-regulated gene essential for epithelial integrity. Cell 2005, 120, 843–856. [Google Scholar] [CrossRef] [Green Version]
- Kouwenhoven, E.N.; van Bokhoven, H.; Zhou, H. Gene regulatory mechanisms orchestrated by p63 in epithelial development and related disorders. Biochim. Biophys. Acta 2015, 1849, 590–600. [Google Scholar] [CrossRef]
- Sethi, I.; Gluck, C.; Zhou, H.; Buck, M.J.; Sinha, S. Evolutionary re-wiring of p63 and the epigenomic regulatory landscape in keratinocytes and its potential implications on species-specific gene expression and phenotypes. Nucl. Acids Res. 2017, 45, 8208–8224. [Google Scholar] [CrossRef] [Green Version]
- Kouwenhoven, E.N.; van Heeringen, S.J.; Tena, J.J.; Oti, M.; Dutilh, B.E.; Alonso, M.E.; de la Calle-Mustienes, E.; Smeenk, L.; Rinne, T.; Parsaulian, L.; et al. Genome-wide profiling of p63 DNA-binding sites identifies an element that regulates gene expression during limb development in the 7q21 SHFM1 locus. PLoS Genet. 2010, 6, e1001065. [Google Scholar] [CrossRef] [Green Version]
- McDade, S.S.; Henry, A.E.; Pivato, G.P.; Kozarewa, I.; Mitsopoulos, C.; Fenwick, K.; Assiotis, I.; Hakas, J.; Zvelebil, M.; Orr, N.; et al. Genome-wide analysis of p63 binding sites identifies AP-2 factors as co-regulators of epidermal differentiation. Nucl. Acids Res. 2012, 40, 7190–7206. [Google Scholar] [CrossRef] [PubMed]
- Rapisarda, V.; Malashchuk, I.; Asamaowei, I.E.; Poterlowicz, K.; Fessing, M.Y.; Sharov, A.A.; Karakesisoglou, I.; Botchkarev, V.A.; Mardaryev, A. p63 transcription factor regulates nuclear shape and Expression of nuclear envelope-associated genes in epidermal keratinocytes. J. Investig. Dermatol. 2017, 137, 2157–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, M.R.; He, L.; Forster, N.; Ory, B.; Ellisen, L.W. Physical association of HDAC1 and HDAC2 with p63 mediates transcriptional repression and tumor maintenance in squamous cell carcinoma. Cancer Res. 2011, 71, 4373–4379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, J.; Yi, G.; Zhou, H. p63 cooperates with CTCF to modulate chromatin architecture in skin keratinocytes. Epigenet. Chrom. 2019, 12, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBoeuf, M.; Terrell, A.; Trivedi, S.; Sinha, S.; Epstein, J.A.; Olson, E.N.; Morrisey, E.E.; Millar, S.E. Hdac1 and Hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells. Dev. Cell 2010, 19, 807–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westfall, M.D.; Mays, D.J.; Sniezek, J.C.; Pietenpol, J.A. The Delta Np63 alpha phosphoprotein binds the p21 and 14-3-3 sigma promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome-derived mutations. Mol. Cell Biol. 2003, 23, 2264–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.Y.; Wang, D.M.; Burgmaier, J.E.; Teng, Y.D.; Romano, R.A.; Sinha, S.; Yi, R. Single cell and open chromatin analysis reveals molecular origin of epidermal cells of the skin. Dev. Cell 2018, 47, 133. [Google Scholar] [CrossRef] [Green Version]
- Cavazza, A.; Miccio, A.; Romano, O.; Petiti, L.; Malagoli Tagliazucchi, G.; Peano, C.; Severgnini, M.; Rizzi, E.; De Bellis, G.; Bicciato, S.; et al. Dynamic Transcriptional and Epigenetic Regulation of Human Epidermal Keratinocyte Differentiation. Stem Cell Rep. 2016, 6, 618–632. [Google Scholar] [CrossRef] [Green Version]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [Green Version]
- Pasquali, L.; Gaulton, K.J.; Rodriguez-Segui, S.A.; Mularoni, L.; Miguel-Escalada, I.; Akerman, I.; Tena, J.J.; Moran, I.; Gomez-Marin, C.; van de Bunt, M.; et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat. Genet. 2014, 46, 136–143. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, L.; Datta, D.; Serrat, J.; Morey, L.; Solanas, G.; Avgustinova, A.; Blanco, E.; Pons, J.I.; Matallanas, D.; Von Kriegsheim, A.; et al. Dnmt3a and Dnmt3b associate with enhancers to regulate human epidermal stem cell homeostasis. Cell Stem Cell 2016, 19, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Wang, Y.; Torkelson, J.L.; Shankar, G.; Pattison, J.M.; Zhen, H.H.; Fang, F.; Duren, Z.; Xin, J.; Gaddam, S.; et al. TFAP2C- and p63-dependent networks sequentially rearrange chromatin landscapes to drive human epidermal lineage commitment. Cell Stem Cell 2019, 24, 271–284.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.; Zhu, Z.; Kapranov, P.; McKeon, F.; Church, G.M.; Gingeras, T.R.; Struhl, K. Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol. Cell 2006, 24, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Sethi, I.; Sinha, S.; Buck, M.J. Role of chromatin and transcriptional co-regulators in mediating p63-genome interactions in keratinocytes. BMC Genom. 2014, 15, 1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Mistry, D.S.; Sen, G.L. Highly rapid and efficient conversion of human fibroblasts to keratinocyte-like cells. J. Investig. Dermatol. 2014, 134, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Russo, C.; Osterburg, C.; Sirico, A.; Antonini, D.; Ambrosio, R.; Wurz, J.M.; Rinnenthal, J.; Ferniani, M.; Kehrloesser, S.; Schafer, B.; et al. Protein aggregation of the p63 transcription factor underlies severe skin fragility in AEC syndrome. Proc. Natl. Acad. Sci. USA 2018, 115, E906–E915. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, H.; Xue, Y.; Lin, Y.; Zhang, X.; Xi, L.; Patel, S.; Cai, H.; Luo, J.; Zhang, M.; Zhang, M.; et al. WNT7A and PAX6 define corneal epithelium homeostasis and pathogenesis. Nature 2014, 511, 358–361. [Google Scholar] [CrossRef]
- Di Iorio, E.; Kaye, S.B.; Ponzin, D.; Barbaro, V.; Ferrari, S.; Bohm, E.; Nardiello, P.; Castaldo, G.; McGrath, J.A.; Willoughby, C.E. Limbal stem cell deficiency and ocular phenotype in ectrodactyly-ectodermal dysplasia-clefting syndrome caused by p63 mutations. Ophthalmology 2012, 119, 74–83. [Google Scholar] [CrossRef]
- Secker, G.A.; Daniels, J.T. Corneal epithelial stem cells: Deficiency and regulation. Stem Cell Rev. 2008, 4, 159–168. [Google Scholar] [CrossRef]
- Fessing, M.Y.; Mardaryev, A.N.; Gdula, M.R.; Sharov, A.A.; Sharova, T.Y.; Rapisarda, V.; Gordon, K.B.; Smorodchenko, A.D.; Poterlowicz, K.; Ferone, G.; et al. p63 regulates Satb1 to control tissue-specific chromatin remodeling during development of the epidermis. J. Cell Biol. 2011, 194, 825–839. [Google Scholar] [CrossRef] [Green Version]
- Mardaryev, A.N.; Liu, B.; Rapisarda, V.; Poterlowicz, K.; Malashchuk, I.; Rudolf, J.; Sharov, A.A.; Jahoda, C.A.; Fessing, M.Y.; Benitah, S.A.; et al. Cbx4 maintains the epithelial lineage identity and cell proliferation in the developing stratified epithelium. J. Cell Biol. 2016, 212, 77–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardaryev, A.N.; Gdula, M.R.; Yarker, J.L.; Emelianov, V.U.; Poterlowicz, K.; Sharov, A.A.; Sharova, T.Y.; Scarpa, J.A.; Joffe, B.; Solovei, I.; et al. p63 and Brg1 control developmentally regulated higher-order chromatin remodelling at the epidermal differentiation complex locus in epidermal progenitor cells. Development 2014, 141, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyes, W.M.; Pecoraro, M.; Aranda, V.; Vernersson-Lindahl, E.; Li, W.; Vogel, H.; Guo, X.; Garcia, E.L.; Michurina, T.V.; Enikolopov, G.; et al. DeltaNp63alpha is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell 2011, 8, 164–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straub, W.E.; Weber, T.A.; Schafer, B.; Candi, E.; Durst, F.; Ou, H.D.; Rajalingam, K.; Melino, G.; Dotsch, V. The C-terminus of p63 contains multiple regulatory elements with different functions. Cell Death Dis. 2010, 1, e5. [Google Scholar] [CrossRef]
- Sammons, M.A.; Zhu, J.J.; Drake, A.M.; Berger, S.L. TP53 engagement with the genome occurs in distinct local chromatin environments via pioneer factor activity. Genome Res. 2015, 25, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Rubin, A.J.; Qu, K.; Zhang, J.; Giresi, P.G.; Chang, H.Y.; Khavari, P.A. A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF and p63. Genome Biol. 2015, 16, 284. [Google Scholar] [CrossRef] [Green Version]
- Santos-Pereira, J.M.; Gallardo-Fuentes, L.; Neto, A.; Acemel, R.D.; Tena, J.J. Pioneer and repressive functions of p63 during zebrafish embryonic ectoderm specification. Nat. Commun. 2019, 10, 3049. [Google Scholar] [CrossRef]
- Celli, J.; Duijf, P.; Hamel, B.C.; Bamshad, M.; Kramer, B.; Smits, A.P.; Newbury-Ecob, R.; Hennekam, R.C.; Van Buggenhout, G.; van Haeringen, A.; et al. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell 1999, 99, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Duijf, P.H.; van Bokhoven, H.; Brunner, H.G. Pathogenesis of split-hand/split-foot malformation. Hum. Mol. Genet. 2003, 12, R51–R60. [Google Scholar] [CrossRef] [Green Version]
- McGrath, J.A.; Duijf, P.H.; Doetsch, V.; Irvine, A.D.; de Waal, R.; Vanmolkot, K.R.; Wessagowit, V.; Kelly, A.; Atherton, D.J.; Griffiths, W.A.; et al. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum. Mol. Genet. 2001, 10, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Rinne, T.; Bolat, E.; Meijer, R.; Scheffer, H.; van Bokhoven, H. Spectrum of p63 mutations in a selected patient cohort affected with ankyloblepharon-ectodermal defects-cleft lip/palate syndrome (AEC). Am. J. Med. Genet. 2009, 149, 1948–1951. [Google Scholar] [CrossRef] [PubMed]
- Rinne, T.; Hamel, B.; van Bokhoven, H.; Brunner, H.G. Pattern of p63 mutations and their phenotypes—Update. Am. J. Med. Genet. 2006, 140, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Vernersson Lindahl, E.; Garcia, E.L.; Mills, A.A. An allelic series of Trp63 mutations defines TAp63 as a modifier of EEC syndrome. Am. J. Med. Genet. 2013, 161, 1961–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prontera, P.; Garelli, E.; Isidori, I.; Mencarelli, A.; Carando, A.; Silengo, M.C.; Donti, E. Cleft palate and ADULT phenotype in a patient with a novel TP63 mutation suggests lumping of EEC/LM/ADULT syndromes into a unique entity: ELA syndrome. Am. J. Med. Genet. 2011, 155, 2746–2749. [Google Scholar] [CrossRef]
- Maillard, A.; Alby, C.; Gabison, E.; Doan, S.; Caux, F.; Bodemer, C.; Hadj-Rabia, S. P63-related disorders: Dermatological characteristics in 22 patients. Exp. Dermatol 2019, 28, 1190–1195. [Google Scholar] [CrossRef]
- van Bokhoven, H.; Hamel, B.C.; Bamshad, M.; Sangiorgi, E.; Gurrieri, F.; Duijf, P.H.; Vanmolkot, K.R.; van Beusekom, E.; van Beersum, S.E.; Celli, J.; et al. p63 Gene mutations in eec syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. Am. J. Hum. Genet. 2001, 69, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Leoyklang, P.; Siriwan, P.; Shotelersuk, V. A mutation of the p63 gene in non-syndromic cleft lip. J. Med. Genet. 2006, 43, e28. [Google Scholar] [CrossRef] [Green Version]
- Chitayat, D.; Babul, R.; Silver, M.M.; Jay, V.; Teshima, I.E.; Babyn, P.; Becker, L.E. Terminal deletion of the long arm of chromosome 3 [46,XX,del(3)(q27-->qter)]. Am. J. Med. Genet. 1996, 61, 45–48. [Google Scholar] [CrossRef]
- Khandelwal, K.D.; van den Boogaard, M.H.; Mehrem, S.L.; Gebel, J.; Fagerberg, C.; van Beusekom, E.; van Binsbergen, E.; Topaloglu, O.; Steehouwer, M.; Gilissen, C.; et al. Deletions and loss-of-function variants in TP63 associated with orofacial clefting. Eur. J. Hum. Genet. 2019, 27, 1101–1112. [Google Scholar] [CrossRef]
- Guazzarotti, L.; Caprio, C.; Rinne, T.K.; Bosoni, M.; Pattarino, G.; Mauri, S.; Tadini, G.L.; van Bokhoven, H.; Zuccotti, G.V. Limb-mammary syndrome (LMS) associated with internal female genitalia dysgenesia: A new genotype/phenotype correlation? Am. J. Med. Genet. 2008, 146, 2001–2004. [Google Scholar] [CrossRef]
- Mathorne, S.W.; Ravn, P.; Hansen, D.; Beck-Nielsen, S.S.; Gjorup, H.; Sorensen, K.P.; Fagerberg, C.R. Novel phenotype of syndromic premature ovarian insufficiency associated with TP63 molecular defect. Clin. Genet. 2020, 97, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Holder-Espinasse, M.; Martin-Coignard, D.; Escande, F.; Manouvrier-Hanu, S. A new mutation in TP63 is associated with age-related pathology. Eur. J. Hum. Genet. 2007, 15, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Browne, G.; Cipollone, R.; Lena, A.M.; Serra, V.; Zhou, H.; van Bokhoven, H.; Dotsch, V.; Merico, D.; Mantovani, R.; Terrinoni, A.; et al. Differential altered stability and transcriptional activity of DeltaNp63 mutants in distinct ectodermal dysplasias. J. Cell Sci. 2011, 124, 2200–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della Gatta, G.; Bansal, M.; Ambesi-Impiombato, A.; Antonini, D.; Missero, C.; di Bernardo, D. Direct targets of the TRP63 transcription factor revealed by a combination of gene expression profiling and reverse engineering. Genome Res. 2008, 18, 939–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; van den Bogaard, E.H.; Kouwenhoven, E.N.; Bykov, V.J.; Rinne, T.; Zhang, Q.; Tjabringa, G.S.; Gilissen, C.; van Heeringen, S.J.; Schalkwijk, J.; et al. APR-246/PRIMA-1(MET) rescues epidermal differentiation in skin keratinocytes derived from EEC syndrome patients with p63 mutations. Proc. Natl. Acad. Sci. USA 2013, 110, 2157–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Iacono, N.; Mantero, S.; Chiarelli, A.; Garcia, E.; Mills, A.A.; Morasso, M.I.; Costanzo, A.; Levi, G.; Guerrini, L.; Merlo, G.R. Regulation of Dlx5 and Dlx6 gene expression by p63 is involved in EEC and SHFM congenital limb defects. Development 2008, 135, 1377–1388. [Google Scholar] [CrossRef] [Green Version]
- Merlo, G.R.; Zerega, B.; Paleari, L.; Trombino, S.; Mantero, S.; Levi, G. Multiple functions of Dlx genes. Int. J. Dev. Biol. 2000, 44, 619–626. [Google Scholar]
- Merlo, G.R.; Paleari, L.; Mantero, S.; Genova, F.; Beverdam, A.; Palmisano, G.L.; Barbieri, O.; Levi, G. Mouse model of split hand/foot malformation type I. Genesis 2002, 33, 97–101. [Google Scholar] [CrossRef]
- Acampora, D.; Merlo, G.R.; Paleari, L.; Zerega, B.; Postiglione, M.P.; Mantero, S.; Bober, E.; Barbieri, O.; Simeone, A.; Levi, G. Craniofacial, vestibular and bone defects in mice lacking the Distal-less-related gene Dlx5. Development 1999, 126, 3795–3809. [Google Scholar]
- Bakkers, J.; Hild, M.; Kramer, C.; Furutani-Seiki, M.; Hammerschmidt, M. Zebrafish DeltaNp63 is a direct target of Bmp signaling and encodes a transcriptional repressor blocking neural specification in the ventral ectoderm. Dev. Cell 2002, 2, 617–627. [Google Scholar] [CrossRef] [Green Version]
- Ferone, G.; Thomason, H.A.; Antonini, D.; De Rosa, L.; Hu, B.; Gemei, M.; Zhou, H.; Ambrosio, R.; Rice, D.P.; Acampora, D.; et al. Mutant p63 causes defective expansion of ectodermal progenitor cells and impaired FGF signalling in AEC syndrome. EMBO Mol. Med. 2012, 4, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Zarnegar, B.J.; Webster, D.E.; Lopez-Pajares, V.; Vander Stoep Hunt, B.; Qu, K.; Yan, K.J.; Berk, D.R.; Sen, G.L.; Khavari, P.A. Genomic profiling of a human organotypic model of AEC syndrome reveals ZNF750 as an essential downstream target of mutant TP63. Am. J. Hum. Genet. 2012, 91, 435–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, G.L.; Boxer, L.D.; Webster, D.E.; Bussat, R.T.; Qu, K.; Zarnegar, B.J.; Johnston, D.; Siprashvili, Z.; Khavari, P.A. ZNF750 is a p63 target gene that induces KLF4 to drive terminal epidermal differentiation. Dev. Cell 2012, 22, 669–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorch, J.H.; Klessner, J.; Park, J.K.; Getsios, S.; Wu, Y.L.; Stack, M.S.; Green, K.J. Epidermal growth factor receptor inhibition promotes desmosome assembly and strengthens intercellular adhesion in squamous cell carcinoma cells. J. Biol. Chem. 2004, 279, 37191–37200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, T.F.; Getsios, S.; Caldelari, R.; Godsel, L.M.; Kowalczyk, A.P.; Muller, E.J.; Green, K.J. Mechanisms of plakoglobin-dependent adhesion—Desmosome-specific functions in assembly and regulation by epidermal growth factor receptor. J. Biol. Chem. 2005, 280, 40355–40363. [Google Scholar] [CrossRef] [Green Version]
- Sathyamurthy, A.; Freund, S.M.; Johnson, C.M.; Allen, M.D.; Bycroft, M. Structural basis of p63alpha SAM domain mutants involved in AEC syndrome. FEBS J. 2011, 278, 2680–2688. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Tomasini, R.; McKeon, F.D.; Mak, T.W.; Melino, G. The p53 family: Guardians of maternal reproduction. Nat. Rev. 2011, 12, 259–265. [Google Scholar] [CrossRef]
- Gebel, J.; Tuppi, M.; Krauskopf, K.; Coutandin, D.; Pitzius, S.; Kehrloesser, S.; Osterburg, C.; Dotsch, V. Control mechanisms in germ cells mediated by p53 family proteins. J. Cell Sci. 2017, 130, 2663–2671. [Google Scholar] [CrossRef] [Green Version]
- Tucker, E.J.; Grover, S.R.; Robevska, G.; van den Bergen, J.; Hanna, C.; Sinclair, A.H. Identification of variants in pleiotropic genes causing “isolated” premature ovarian insufficiency: Implications for medical practice. Eur. J. Hum. Genet. 2018, 26, 1319–1328. [Google Scholar] [CrossRef] [Green Version]
- Tucker, E.J.; Jaillard, S.; Grover, S.R.; van den Bergen, J.; Robevska, G.; Bell, K.M.; Sadedin, S.; Hanna, C.; Dulon, J.; Touraine, P.; et al. TP63-truncating variants cause isolated premature ovarian insufficiency. Hum. Mutat. 2019, 40, 886–892. [Google Scholar] [CrossRef]
- Bestetti, I.; Castronovo, C.; Sironi, A.; Caslini, C.; Sala, C.; Rossetti, R.; Crippa, M.; Ferrari, I.; Pistocchi, A.; Toniolo, D.; et al. High-resolution array-CGH analysis on 46,XX patients affected by early onset primary ovarian insufficiency discloses new genes involved in ovarian function. Hum. Reprod. 2019, 34, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Keyes, W.M.; Vogel, H.; Koster, M.I.; Guo, X.; Qi, Y.; Petherbridge, K.M.; Roop, D.R.; Bradley, A.; Mills, A.A. p63 heterozygous mutant mice are not prone to spontaneous or chemically induced tumors. Proc. Natl. Acad. Sci. USA 2006, 103, 8435–8440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, M.R.; Wilson, C.; Ory, B.; Rothenberg, S.M.; Faquin, W.; Mills, A.A.; Ellisen, L.W. FGFR2 signaling underlies p63 oncogenic function in squamous cell carcinoma. J. Clin. Investig. 2013, 123, 3525–3538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocco, J.W.; Leong, C.O.; Kuperwasser, N.; DeYoung, M.P.; Ellisen, L.W. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 2006, 9, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, R.; Wei, Y.; Hwang, J.; Hang, X.; Andres Blanco, M.; Choudhury, A.; Tiede, B.; Romano, R.A.; DeCoste, C.; Mercatali, L.; et al. DeltaNp63 promotes stem cell activity in mammary gland development and basal-like breast cancer by enhancing Fzd7 expression and Wnt signalling. Nat. Cell Biol. 2014, 16, 1004–1015. [Google Scholar] [CrossRef]
- Yalcin-Ozuysal, O.; Fiche, M.; Guitierrez, M.; Wagner, K.U.; Raffoul, W.; Brisken, C. Antagonistic roles of Notch and p63 in controlling mammary epithelial cell fates. Cell Death Differ. 2010, 17, 1600–1612. [Google Scholar] [CrossRef] [Green Version]
- Su, X.H.; Chakravarti, D.; Cho, M.S.; Liu, L.Z.; Gi, Y.J.; Lin, Y.L.; Leung, M.L.; El-Naggar, A.; Creighton, C.J.; Suraokar, M.B.; et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 2010, 467, 986–990. [Google Scholar] [CrossRef] [Green Version]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [Green Version]
- Pitzius, S.; Osterburg, C.; Gebel, J.; Tascher, G.; Schafer, B.; Zhou, H.; Munch, C.; Dotsch, V. TA*p63 and GTAp63 achieve tighter transcriptional regulation in quality control by converting an inhibitory element into an additional transactivation domain. Cell Death Dis. 2019, 10, 686. [Google Scholar] [CrossRef] [Green Version]
- Gatti, V.; Bongiorno-Borbone, L.; Fierro, C.; Annicchiarico-Petruzzelli, M.; Melino, G.; Peschiaroli, A. p63 at the Crossroads between Stemness and Metastasis in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 2683. [Google Scholar] [CrossRef] [Green Version]
- Forster, N.; Saladi, S.V.; van Bragt, M.; Sfondouris, M.E.; Jones, F.E.; Li, Z.; Ellisen, L.W. Basal cell signaling by p63 controls luminal progenitor function and lactation via NRG1. Dev. Cell 2014, 28, 147–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| EEC | LMS | ADULT | AEC | RHS | SHFM4 | nsOFC | ||
|---|---|---|---|---|---|---|---|---|
| Limb Defects | Ectrodactyly | +++ | +++ | +++ | - | - | +++ | - |
| Syndactyly | +++ | +++ | +++ | + | + | +++ | - | |
| Orofacial Clefting | Cleft Lip | +++ | - | - | +++ | +++ | - | +++ |
| Cleft Palate | +++ | +++ | - | +++ | +++ | - | +++ | |
| Ectodermal Dysplasia | Skin | +++ | + | +++ (#) | +++ (*) | +++ | - | - |
| Hair | +++ | + | +++ | +++ | +++ | - | - | |
| Nails | +++ | +++ | +++ | +++ | +++ | - | - | |
| Teeth | +++ | +++ | +++ | +++ | +++ | - | - | |
| Lacrimal Duct | +++ | +++ | +++ | +++ | +++ | - | - | |
| Mammary Gland/Nipple | + | +++ | +++ | + | + | - | - | |
| Hypohydrosis | + | +++ | + | + | +++ | - | - | |
| Ankyloblepharon | - | - | - | +++ | + | - | - | |
| Hearing Impairment | + | + | - | +++ | + | - | - | |
| Genitourinary/Kidney | + | +++ (1) | - | +++ | + (2) | - | - | |
| Limbal Stem Cell Deficiency | +++ | - | - | - | - | - | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osterburg, C.; Osterburg, S.; Zhou, H.; Missero, C.; Dötsch, V. Isoform-Specific Roles of Mutant p63 in Human Diseases. Cancers 2021, 13, 536. https://doi.org/10.3390/cancers13030536
Osterburg C, Osterburg S, Zhou H, Missero C, Dötsch V. Isoform-Specific Roles of Mutant p63 in Human Diseases. Cancers. 2021; 13(3):536. https://doi.org/10.3390/cancers13030536
Chicago/Turabian StyleOsterburg, Christian, Susanne Osterburg, Huiqing Zhou, Caterina Missero, and Volker Dötsch. 2021. "Isoform-Specific Roles of Mutant p63 in Human Diseases" Cancers 13, no. 3: 536. https://doi.org/10.3390/cancers13030536
APA StyleOsterburg, C., Osterburg, S., Zhou, H., Missero, C., & Dötsch, V. (2021). Isoform-Specific Roles of Mutant p63 in Human Diseases. Cancers, 13(3), 536. https://doi.org/10.3390/cancers13030536