
Glutamine Synthetase as a Therapeutic Target for Cancer Treatment


Abstract
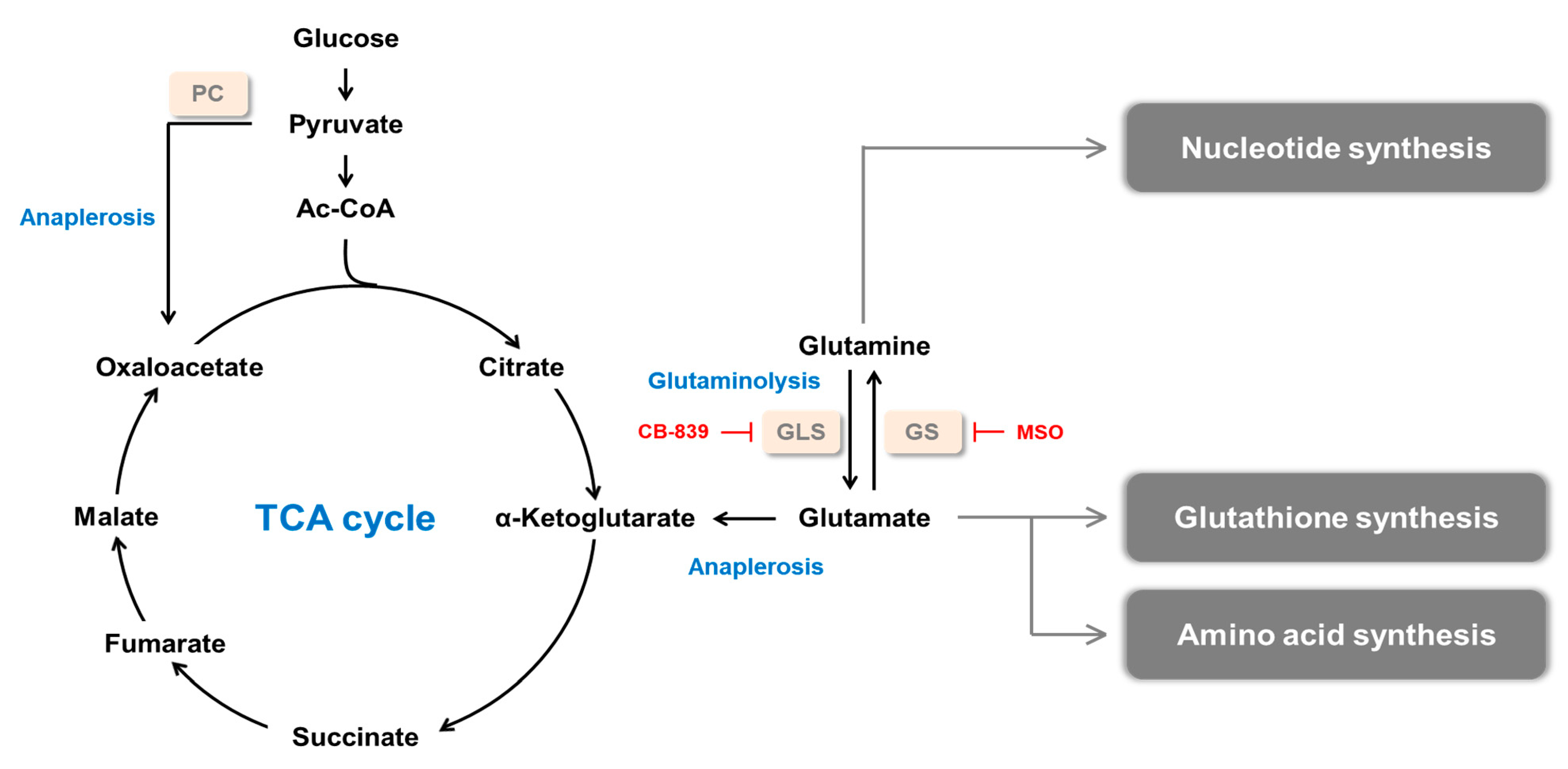
:1. Introduction
2. Glutamine Synthetase and Cancer
2.1. Dysregulation of Glutamine Synthetase in Tumor
2.1.1. Glioma
2.1.2. Liver Cancer
2.1.3. Breast Cancer
2.1.4. Ovarian Cancer
2.1.5. Lung Cancer
2.1.6. Pancreatic Cancer
2.1.7. Other Cancers
2.2. Dysregulation of Glutamine Synthetase in the Tumor Microenvironment
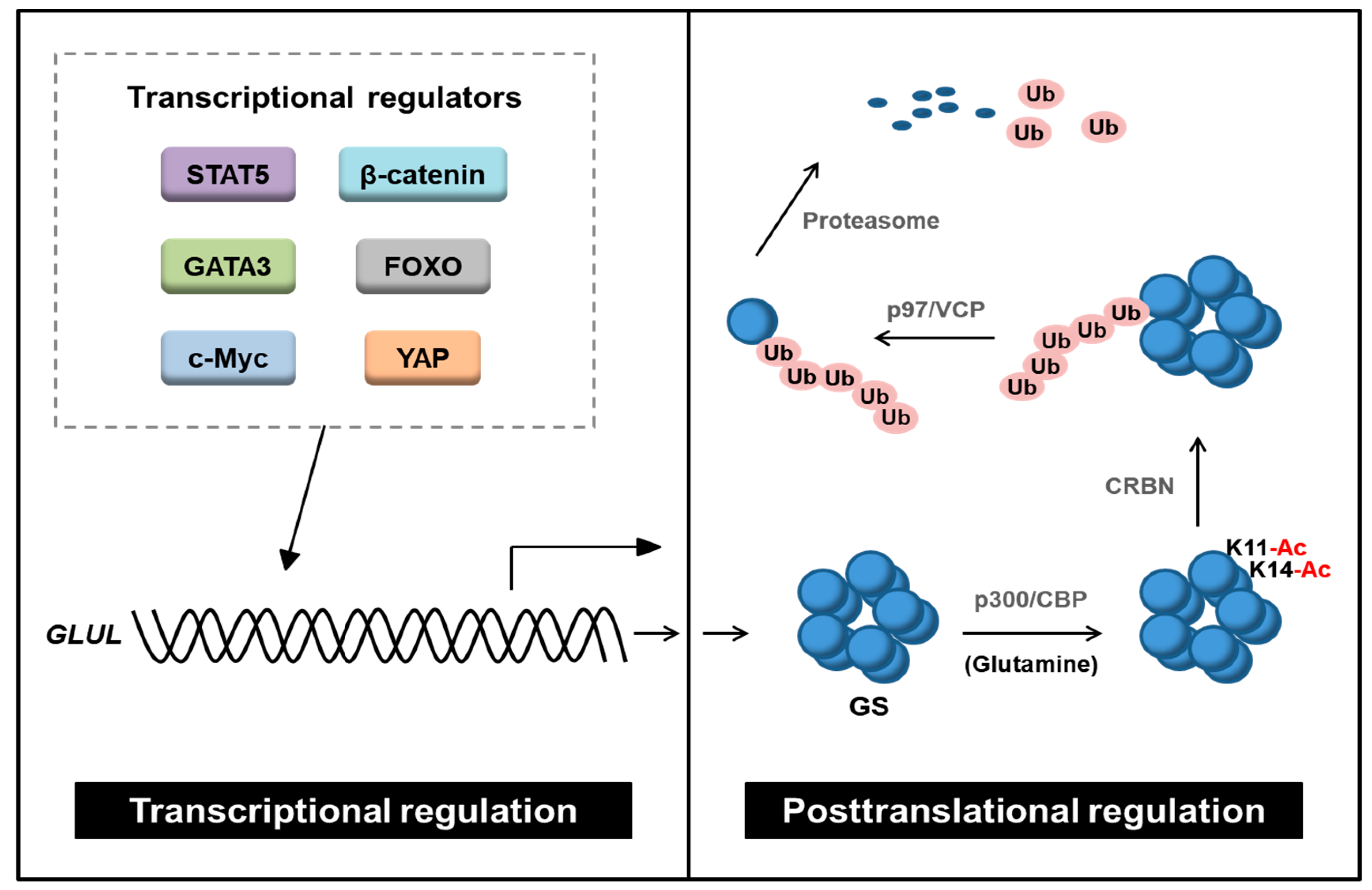
3. Regulation of GS
3.1. Transcriptional Regulation of GS
3.1.1. c-Myc
3.1.2. β-Catenin
3.1.3. GATA Binding Protein 3 (GATA3)
3.1.4. Members of the Class O of Forkhead Box Transcription Factors (FOXO)
3.1.5. YAP1
3.1.6. Signal Transducer and Activator of Transcription 5 (STAT5)
3.2. Posttranslational Modifications of GS
4. GS Inhibitors
5. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATP | adenosine triphosphate |
| ALL | acute lymphoblastic leukemia |
| ASNase | L-asparaginase |
| BCAA | branched-chain amino acid |
| CAF | cancer-associated fibroblast |
| CRBN | cereblon |
| DFS | disease-free survival |
| TDG | thymine-DNA glycosylase |
| FNH | focal nodular hyperplasia |
| FOXO | members of the class O of forkhead box transcription factors |
| GPCRs | G protein-coupled receptors |
| GC | gastric cancer |
| GATA3 | GATA Binding Protein 3 |
| GBM | glioblastoma |
| GLUL | glutamate-ammonia ligase |
| GLS | glutaminase |
| GS | glutamine synthetase |
| GSH | glutathione |
| HA | hepatic adenoma |
| HBV | hepatitis B virus |
| HB | hepatoblastoma |
| HCC | hepatocellular carcinoma |
| HOT | human orthotopic tumor |
| MM | multiple myeloma |
| MSC | mesenchymal stromal cell |
| HIF1α | hypoxia inducible factor 1 α |
| MSO | methionine sulfoximine |
| NPC | nasopharyngeal carcinoma |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NSCLC | non-small cell lung cancer |
| NOF | normal ovarian fibroblast |
| OD | oligodendroglioma |
| OVC | ovarian cancer |
| OS | overall survival |
| PDAC | pancreatic ductal carcinoma |
| PTM | posttranslational modification |
| PC | pyruvate carboxylase |
| STAT5 | signal transducer and activator of transcription 5 |
| TCA | tricarboxylic acid |
| TME | tumor microenvironment |
| VCP | valosin-containing protein |
| YAP | yes-associated protein |
| ZNRF1 | zinc and ring finger 1 |
| α-KG | α-ketoglutarate |
References
- Cruzat, V.; Macedo Rogero, M.; Noel Keane, K.; Curi, R.; Newsholme, P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watford, M. Glutamine and glutamate metabolism across the liver sinusoid. J. Nutr. 2000, 130, 983S–987S. [Google Scholar] [CrossRef] [PubMed]
- Janicki, R.H.; Goldstein, L. Glutamine synthetase and renal ammonia metabolism. Am. J. Physiol. 1969, 216, 1107–1110. [Google Scholar] [CrossRef]
- Smith, R.J.; Larson, S.; Stred, S.E.; Durschlag, R.P. Regulation of glutamine synthetase and glutaminase activities in cultured skeletal muscle cells. J. Cell. Physiol. 1984, 120, 197–203. [Google Scholar] [CrossRef]
- Suarez, I.; Bodega, G.; Fernandez, B. Glutamine synthetase in brain: Effect of ammonia. Neurochem. Int. 2002, 41, 123–142. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [Green Version]
- Krajewski, W.W.; Collins, R.; Holmberg-Schiavone, L.; Jones, T.A.; Karlberg, T.; Mowbray, S.L. Crystal structures of mammalian glutamine synthetases illustrate substrate-induced conformational changes and provide opportunities for drug and herbicide design. J. Mol. Biol. 2008, 375, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qie, S.; Yoshida, A.; Parnham, S.; Oleinik, N.; Beeson, G.C.; Beeson, C.C.; Ogretmen, B.; Bass, A.J.; Wong, K.K.; Rustgi, A.K.; et al. Targeting glutamine-addiction and overcoming CDK4/6 inhibitor resistance in human esophageal squamous cell carcinoma. Nat. Commun. 2019, 10, 1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, R.M.; Dytfeld, D.; Reyes, L.; Robinson, R.M.; Smith, B.; Manevich, Y.; Jakubowiak, A.; Komarnicki, M.; Przybylowicz-Chalecka, A.; Szczepaniak, T.; et al. Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget 2017, 8, 35863–35876. [Google Scholar] [CrossRef] [Green Version]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef] [Green Version]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.O.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [Green Version]
- Kung, H.N.; Marks, J.R.; Chi, J.T. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 2011, 7, e1002229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, T.; Sudderth, J.; Yang, C.; Mullen, A.R.; Jin, E.S.; Mates, J.M.; DeBerardinis, R.J. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8674–8679. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Z.; Qi, M.; Li, J.; Okamoto, H.; Xu, D.S.; Iyer, R.R.; Lu, J.; Yang, C.; Weil, R.J.; Vortmeyer, A.; et al. Proteomic identification of glutamine synthetase as a differential marker for oligodendrogliomas and astrocytomas. J. Neurosurg. 2011, 115, 789–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, M.; Taurino, G.; Bianchi, M.G.; Ottaviani, L.; Andreoli, R.; Ciociola, T.; Lagrasta, C.A.M.; Tardito, S.; Bussolati, O. Oligodendroglioma Cells Lack Glutamine Synthetase and Are Auxotrophic for Glutamine, but Do not Depend on Glutamine Anaplerosis for Growth. Int. J. Mol. Sci. 2018, 19, 1099. [Google Scholar] [CrossRef] [Green Version]
- Wasfy, R.E.; Shams Eldeen, A.A. Roles of Combined Glypican-3 and Glutamine Synthetase in Differential Diagnosis of Hepatocellular Lesions. Asian Pac. J. Cancer Prev. 2015, 16, 4769–4775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.; Wang, H.; Lang, Z.; Wang, T.; Long, M.; Wang, B. Expression level of glutamine synthetase is increased in hepatocellular carcinoma and liver tissue with cirrhosis and chronic hepatitis B. Hepatol. Int. 2011, 5, 698–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moudi, B.; Heidari, Z.; Mahmoudzadeh-Sagheb, H.; Alavian, S.M.; Lankarani, K.B.; Farrokh, P.; Randel Nyengaard, J. Concomitant use of heat-shock protein 70, glutamine synthetase and glypican-3 is useful in diagnosis of HBV-related hepatocellular carcinoma with higher specificity and sensitivity. Eur. J. Histochem. 2018, 62, 2859. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.Y.; Yu, M.W.; Lin, S.M.; Lee, S.D.; Chen, C.L.; Chen, D.S.; Chen, P.J. Genome-wide association analysis identifies a GLUL haplotype for familial hepatitis B virus-related hepatocellular carcinoma. Cancer 2017, 123, 3966–3976. [Google Scholar] [CrossRef]
- Swanson, B.J.; Yearsley, M.M.; Marsh, W.; Frankel, W.L. A triple stain of reticulin, glypican-3, and glutamine synthetase: A useful aid in the diagnosis of liver lesions. Arch. Pathol. Lab. Med. 2015, 139, 537–542. [Google Scholar] [CrossRef]
- Bioulac-Sage, P.; Laumonier, H.; Rullier, A.; Cubel, G.; Laurent, C.; Zucman-Rossi, J.; Balabaud, C. Over-expression of glutamine synthetase in focal nodular hyperplasia: A novel easy diagnostic tool in surgical pathology. Liver Int. 2009, 29, 459–465. [Google Scholar] [CrossRef]
- Cadoret, A.; Ovejero, C.; Terris, B.; Souil, E.; Levy, L.; Lamers, W.H.; Kitajewski, J.; Kahn, A.; Perret, C. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002, 21, 8293–8301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, Q.; Molina, L.; Li, J.; Adebayo Michael, A.O.; Russell, J.O.; Preziosi, M.E.; Singh, S.; Poddar, M.; Matz-Soja, M.; Ranganathan, S.; et al. beta-Catenin and Yes-Associated Protein 1 Cooperate in Hepatoblastoma Pathogenesis. Am. J. Pathol. 2019, 189, 1091–1104. [Google Scholar] [CrossRef] [PubMed]
- Dal Bello, B.; Rosa, L.; Campanini, N.; Tinelli, C.; Torello Viera, F.; D’Ambrosio, G.; Rossi, S.; Silini, E.M. Glutamine synthetase immunostaining correlates with pathologic features of hepatocellular carcinoma and better survival after radiofrequency thermal ablation. Clin. Cancer Res. 2010, 16, 2157–2166. [Google Scholar] [CrossRef] [Green Version]
- Hale, G.; Liu, X.; Hu, J.; Xu, Z.; Che, L.; Solomon, D.; Tsokos, C.; Shafizadeh, N.; Chen, X.; Gill, R.; et al. Correlation of exon 3 beta-catenin mutations with glutamine synthetase staining patterns in hepatocellular adenoma and hepatocellular carcinoma. Mod. Pathol. 2016, 29, 1370–1380. [Google Scholar] [CrossRef] [Green Version]
- Sohn, B.H.; Park, I.Y.; Shin, J.H.; Yim, S.Y.; Lee, J.S. Glutamine synthetase mediates sorafenib sensitivity in beta-catenin-active hepatocellular carcinoma cells. Exp. Mol. Med. 2018, 50, e421. [Google Scholar] [CrossRef] [Green Version]
- Adebayo Michael, A.O.; Ko, S.; Tao, J.; Moghe, A.; Yang, H.; Xu, M.; Russell, J.O.; Pradhan-Sundd, T.; Liu, S.; Singh, S.; et al. Inhibiting Glutamine-Dependent mTORC1 Activation Ameliorates Liver Cancers Driven by beta-Catenin Mutations. Cell Metab. 2019, 29, 1135–1150. [Google Scholar] [CrossRef]
- Huang, W.J.; Tsai, J.H.; Jeng, Y.M. Complementary roles of beta-catenin and glutamine synthetase immunostaining in diagnosis of chemotherapy-treated and untreated hepatoblastoma. J. Formos. Med. Assoc. 2017, 116, 549–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, A.G.; Hwang, K.L.; Brown, K.K.; Evason, K.; Beltz, S.; Tsomides, A.; O’Connor, K.; Galli, G.G.; Yimlamai, D.; Chhangawala, S.; et al. Yap reprograms glutamine metabolism to increase nucleotide biosynthesis and enable liver growth. Nat. Cell Biol. 2016, 18, 886–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.L. The Hippo Pathway: Biology and Pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Wang, Y.; Zhang, Z.; Lu, J.; Wu, Z.; Shan, Q.; Sun, C.; Wu, D.; Li, M.; Sheng, N.; et al. High expression of glutamate-ammonia ligase is associated with unfavorable prognosis in patients with ovarian cancer. J. Cell. Biochem. 2018, 119, 6008–6015. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef]
- Furusawa, A.; Miyamoto, M.; Takano, M.; Tsuda, H.; Song, Y.S.; Aoki, D.; Miyasaka, N.; Inazawa, J.; Inoue, J. Ovarian cancer therapeutic potential of glutamine depletion based on GS expression. Carcinogenesis 2018, 39, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Yuneva, M.O.; Fan, T.W.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Mates, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Peng, W.; Wu, T.; Deng, P.; Zhao, Y.L. Increased glutamine anabolism sensitizes non-small cell lung cancer to gefitinib treatment. Cell Death Discov. 2018, 4, 24. [Google Scholar] [CrossRef]
- Muthu, M.; Kumar, R.; Syed Khaja, A.S.; Gilthorpe, J.D.; Persson, J.L.; Nordstrom, A. GLUL Ablation Can Confer Drug Resistance to Cancer Cells via a Malate-Aspartate Shuttle-Mediated Mechanism. Cancers 2019, 11, 1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bott, A.J.; Shen, J.; Tonelli, C.; Zhan, L.; Sivaram, N.; Jiang, Y.P.; Yu, X.; Bhatt, V.; Chiles, E.; Zhong, H.; et al. Glutamine Anabolism Plays a Critical Role in Pancreatic Cancer by Coupling Carbon and Nitrogen Metabolism. Cell Rep. 2019, 29, 1287–1298. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Fan, S.; Lu, J.; Zhang, Z.; Wu, D.; Wu, Z.; Zheng, Y. GLUL Promotes Cell Proliferation in Breast Cancer. J. Cell. Biochem. 2017, 118, 2018–2025. [Google Scholar] [CrossRef]
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Brunelli, L.; Caiola, E.; Marabese, M.; Broggini, M.; Pastorelli, R. Capturing the metabolomic diversity of KRAS mutants in non-small-cell lung cancer cells. Oncotarget 2014, 5, 4722–4731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellers, K.; Fox, M.P.; Bousamra, M., 2nd; Slone, S.P.; Higashi, R.M.; Miller, D.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R.; et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Investig. 2015, 125, 687–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O’Brien, J.P.; Pierce, K.A.; et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Annese, T.; Tamma, R.; Ruggieri, S.; Ribatti, D. Angiogenesis in Pancreatic Cancer: Pre-Clinical and Clinical Studies. Cancers 2019, 11, 381. [Google Scholar] [CrossRef] [Green Version]
- Biancur, D.E.; Paulo, J.A.; Malachowska, B.; Quiles Del Rey, M.; Sousa, C.M.; Wang, X.; Sohn, A.S.W.; Chu, G.C.; Gygi, S.P.; Harper, J.W.; et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 2017, 8, 15965. [Google Scholar] [CrossRef] [PubMed]
- Tardito, S.; Uggeri, J.; Bozzetti, C.; Bianchi, M.G.; Rotoli, B.M.; Franchi-Gazzola, R.; Gazzola, G.C.; Gatti, R.; Bussolati, O. The inhibition of glutamine synthetase sensitizes human sarcoma cells to l-asparaginase. Cancer Chemother. Pharmacol. 2007, 60, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Issaq, S.H.; Mendoza, A.; Fox, S.D.; Helman, L.J. Glutamine synthetase is necessary for sarcoma adaptation to glutamine deprivation and tumor growth. Oncogenesis 2019, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Huang, Q.; Xu, J.; Huang, J.; Wang, J.; Zhong, W.; Chen, W.; Lin, X.; Lin, X. Targeting of glutamine transporter ASCT2 and glutamine synthetase suppresses gastric cancer cell growth. J. Cancer Res. Clin. Oncol. 2018, 144, 821–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, S.; Li, Z.; Xiao, L.; Hu, W.; Zhang, L.; Xie, B.; Zhou, Q.; He, J.; Qiu, Y.; Wen, M.; et al. Glutamine Synthetase Promotes Radiation Resistance via Facilitating Nucleotide Metabolism and Subsequent DNA Damage Repair. Cell Rep. 2019, 28, 1136–1143.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Fu, S.; Hu, W.; Qiu, Y.; Zhang, L.; Tan, R.; Sun, L.Q. Glutamine synthetase facilitates cancer cells to recover from irradiation-induced G2/M arrest. Cancer Biol. Ther. 2020, 21, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Krall, A.S.; Christofk, H.R. Rethinking glutamine addiction. Nat. Cell Biol. 2015, 17, 1515–1517. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Menga, A.; Lebrun, A.; Hooper, D.C.; Butterfield, D.A.; Mazzone, M.; Castegna, A. Blockade of Glutamine Synthetase Enhances Inflammatory Response in Microglial Cells. Antioxid. Redox Signal. 2017, 26, 351–363. [Google Scholar] [CrossRef]
- Choi, J.; Stradmann-Bellinghausen, B.; Yakubov, E.; Savaskan, N.E.; Regnier-Vigouroux, A. Glioblastoma cells induce differential glutamatergic gene expressions in human tumor-associated microglia/macrophages and monocyte-derived macrophages. Cancer Biol. Ther. 2015, 16, 1205–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, E.M.; Menga, A.; Martin-Perez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquiere, B.; McVicar, D.W.; et al. Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef] [Green Version]
- Ehsanipour, E.A.; Sheng, X.; Behan, J.W.; Wang, X.; Butturini, A.; Avramis, V.I.; Mittelman, S.D. Adipocytes cause leukemia cell resistance to L-asparaginase via release of glutamine. Cancer Res. 2013, 73, 2998–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolzoni, M.; Chiu, M.; Accardi, F.; Vescovini, R.; Airoldi, I.; Storti, P.; Todoerti, K.; Agnelli, L.; Missale, G.; Andreoli, R.; et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: A new attractive target. Blood 2016, 128, 667–679. [Google Scholar] [CrossRef]
- Soncini, D.; Minetto, P.; Martinuzzi, C.; Becherini, P.; Fenu, V.; Guolo, F.; Todoerti, K.; Calice, G.; Contini, P.; Miglino, M.; et al. Amino acid depletion triggered by L-asparaginase sensitizes MM cells to carfilzomib by inducing mitochondria ROS-mediated cell death. Blood Adv. 2020, 4, 4312–4326. [Google Scholar] [CrossRef]
- Xiang, Y.; Stine, Z.E.; Xia, J.; Lu, Y.; O’Connor, R.S.; Altman, B.J.; Hsieh, A.L.; Gouw, A.M.; Thomas, A.G.; Gao, P.; et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J. Clin. Investig. 2015, 125, 2293–2306. [Google Scholar] [CrossRef] [Green Version]
- Chiu, M.; Toscani, D.; Marchica, V.; Taurino, G.; Costa, F.; Bianchi, M.G.; Andreoli, R.; Franceschi, V.; Storti, P.; Burroughs-Garcia, J.; et al. Myeloma Cells Deplete Bone Marrow Glutamine and Inhibit Osteoblast Differentiation Limiting Asparagine Availability. Cancers 2020, 12, 3267. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.A.; Neeley, C.K.; Baker, N.A.; Washabaugh, A.R.; Flesher, C.G.; Nelson, B.S.; Frankel, T.L.; Lumeng, C.N.; Lyssiotis, C.A.; Wynn, M.L.; et al. Adipocytes promote pancreatic cancer cell proliferation via glutamine transfer. Biochem. Biophys. Rep. 2016, 7, 144–149. [Google Scholar] [CrossRef] [Green Version]
- Eelen, G.; Dubois, C.; Cantelmo, A.R.; Goveia, J.; Brüning, U.; DeRan, M.; Jarugumilli, G.; van Rijssel, J.; Saladino, G.; Comitani, F.; et al. Role of glutamine synthetase in angiogenesis beyond glutamine synthesis. Nature 2018, 561, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Bott, A.J.; Peng, I.C.; Fan, Y.; Faubert, B.; Zhao, L.; Li, J.; Neidler, S.; Sun, Y.; Jaber, N.; Krokowski, D.; et al. Oncogenic Myc Induces Expression of Glutamine Synthetase through Promoter Demethylation. Cell Metab. 2015, 22, 1068–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeppen, S.; Schneider, D.; Gaunitz, F.; Gebhardt, R.; Kurek, R.; Buchmann, A.; Schwarz, M. Overexpression of glutamine synthetase is associated with beta-catenin-mutations in mouse liver tumors during promotion of hepatocarcinogenesis by phenobarbital. Cancer Res. 2002, 62, 5685–5688. [Google Scholar]
- Audard, V.; Grimber, G.; Elie, C.; Radenen, B.; Audebourg, A.; Letourneur, F.; Soubrane, O.; Vacher-Lavenu, M.C.; Perret, C.; Cavard, C.; et al. Cholestasis is a marker for hepatocellular carcinomas displaying beta-catenin mutations. J. Pathol. 2007, 212, 345–352. [Google Scholar] [CrossRef]
- Austinat, M.; Dunsch, R.; Wittekind, C.; Tannapfel, A.; Gebhardt, R.; Gaunitz, F. Correlation between beta-catenin mutations and expression of Wnt-signaling target genes in hepatocellular carcinoma. Mol. Cancer 2008, 7, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucman-Rossi, J.; Jeannot, E.; Nhieu, J.T.; Scoazec, J.Y.; Guettier, C.; Rebouissou, S.; Bacq, Y.; Leteurtre, E.; Paradis, V.; Michalak, S.; et al. Genotype-phenotype correlation in hepatocellular adenoma: New classification and relationship with HCC. Hepatology 2006, 43, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ao, X.; Ding, W.; Ponnusamy, M.; Wu, W.; Hao, X.; Yu, W.; Wang, Y.; Li, P.; Wang, J. Critical role of FOXO3a in carcinogenesis. Mol. Cancer 2018, 17, 104. [Google Scholar] [CrossRef] [Green Version]
- van der Vos, K.E.; Eliasson, P.; Proikas-Cezanne, T.; Vervoort, S.J.; van Boxtel, R.; Putker, M.; van Zutphen, I.J.; Mauthe, M.; Zellmer, S.; Pals, C.; et al. Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat. Cell Biol. 2012, 14, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.V.; Lee, J.E.; Sweredoski, M.J.; Yang, S.J.; Jeon, S.J.; Harrison, J.S.; Yim, J.H.; Lee, S.G.; Handa, H.; Kuhlman, B.; et al. Glutamine Triggers Acetylation-Dependent Degradation of Glutamine Synthetase via the Thalidomide Receptor Cereblon. Mol. Cell 2016, 61, 809–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.V.; Li, J.; Lu, C.J.; Mamrosh, J.L.; Lu, G.; Cathers, B.E.; Deshaies, R.J. p97/VCP promotes degradation of CRBN substrate glutamine synthetase and neosubstrates. Proc. Natl. Acad. Sci. USA 2017, 114, 3565–3571. [Google Scholar] [CrossRef] [Green Version]
- Meyer, H.; Weihl, C.C. The VCP/p97 system at a glance: Connecting cellular function to disease pathogenesis. J. Cell Sci. 2014, 127, 3877–3883. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, F.; Araki, T. Proteasomal degradation of glutamine synthetase regulates schwann cell differentiation. J. Neurosci. 2010, 30, 1204–1212. [Google Scholar] [CrossRef]
- Linder, M.E.; Deschenes, R.J. Palmitoylation: Policing protein stability and traffic. Nat. Rev. Mol. Cell Biol. 2007, 8, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Savas, J.N.; Roth, A.F.; Sanders, S.S.; Singaraja, R.R.; Hayden, M.R.; Yates, J.R., 3rd; Davis, N.G. Tracking brain palmitoylation change: Predominance of glial change in a mouse model of Huntington’s disease. Chem. Biol. 2013, 20, 1421–1434. [Google Scholar] [CrossRef] [Green Version]
- Huyghe, D.; Nakamura, Y.; Terunuma, M.; Faideau, M.; Haydon, P.; Pangalos, M.N.; Moss, S.J. Glutamine synthetase stability and subcellular distribution in astrocytes are regulated by gamma-aminobutyric type B receptors. J. Biol. Chem. 2014, 289, 28808–28815. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, D.; Gill, H.S.; Pfluegl, G.M.; Rotstein, S.H. Structure-function relationships of glutamine synthetases. Biochim. Biophys. Acta 2000, 1477, 122–145. [Google Scholar] [CrossRef] [Green Version]
- Berlicki, L. Inhibitors of glutamine synthetase and their potential application in medicine. Mini Rev. Med. Chem. 2008, 8, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Sellinger, O.Z.; Azcurra, J.M.; Ohlsson, W.G. Methionine sulfoximine seizures. 8. The dissociation of the convulsant and glutamine synthetase inhibitory effects. J. Pharmacol. Exp. Ther. 1968, 164, 212–222. [Google Scholar] [PubMed]
- Brusilow, W.S.; Peters, T.J. Therapeutic effects of methionine sulfoximine in multiple diseases include and extend beyond inhibition of glutamine synthetase. Expert Opin. Ther. Targets 2017, 21, 461–469. [Google Scholar] [CrossRef]
- Shang, S.; Hua, F.; Hu, Z.W. The regulation of beta-catenin activity and function in cancer: Therapeutic opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [Green Version]
- Igelmann, S.; Neubauer, H.A.; Ferbeyre, G. STAT3 and STAT5 Activation in Solid Cancers. Cancers 2019, 11, 1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Muller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dansen, T.B.; Burgering, B.M. Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol. 2008, 18, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Hornsveld, M.; Smits, L.M.M.; Meerlo, M.; van Amersfoort, M.; Groot Koerkamp, M.J.A.; van Leenen, D.; Kloet, D.E.A.; Holstege, F.C.P.; Derksen, P.W.B.; Burgering, B.M.T.; et al. FOXO Transcription Factors Both Suppress and Support Breast Cancer Progression. Cancer Res. 2018, 78, 2356–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornsveld, M.; Dansen, T.B.; Derksen, P.W.; Burgering, B.M.T. Re-evaluating the role of FOXOs in cancer. Semin. Cancer Biol. 2018, 50, 90–100. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Cancer Type | GS Expression | Role of GS | Experimental Models | References |
|---|---|---|---|---|
| GBM | High | GS sustains nucleotide biosynthesis and cell growth of GBM in gln starved conditions | Human GBM patients, GBM PDX model | [17,19] |
| OVC | High | GS supports the proliferation of OVC cells GShigh OVC shows low invasiveness | OVC cells | [38,39] |
| Low | GSlow OVC shows high invasiveness | Xenograft mouse model | [39,40] | |
| Breast cancer (luminal) | High | High expression of GS contributes to gln independence GLS inhibitor has no anti-proliferative activity | Luminal type breast cancer cells | [13,18] |
| Breast cancer (basal) | Low | Low expression of GS contributes to gln dependence GLS inhibitor has anti-proliferative activity | Basal type breast cancer cells, Xenograft model of basal like breast cancer | [13,18] |
| Lung cancer | High | Increased GS accumulates gln in cancer cells although gln catabolism is activated | GEMs (Myc-induced lung tumors) | [41] |
| - | GS confers gefitinib resistance | NSCLC cells | [42,43] | |
| PDAC | High | GS contributes to cataplerotic usage of α-KG GLUL ablation suppresses tumor growth | KPC tumor cell organoids, Orthotopic mouse model | [44] |
| TME Cell Type | GS Expression | Role of GS | Experimental Models | References |
|---|---|---|---|---|
| TAMs | High | GS maintains M2 macrophage phenotype by suppressing the accumulation of succinate and HIF1α GS supports vascularization and metastasis of cancer cells | Lewis lung carcinoma implanted GLUL conditional knockout mice | [61] |
| Microglial cells | High | GS modulates inflammatory responses GS ablation in microglia increases inflammatory responses | Microglial-specific GLUL conditional knockout mice, Experimental autoimmune encephalomyelitis | [60] |
| GBM astrocytes | High | Astrocytes synthesize gln via GS and provide gln to GSlow GBM cells, supporting cell proliferation | Co-culture of rat primary cortical astrocytes and GBM cells | [17] |
| OVC CAFs | High | GS supports gln catabolism in OVC cells via crosstalk between CAFs and OVC Co-targeting of stromal GS and cancer GLS significantly suppresses tumor growth | Orthotopic mouse model | [46] |
| ALL adipocytes | High | GS protects ALL cells from L-asparaginase by supplying gln | Co-culture of leukemic cells with adipocytes, Leukemic mouse model | [62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, G.W.; Lee, D.H.; Jeon, Y.H.; Yoo, J.; Kim, S.Y.; Lee, S.W.; Cho, H.Y.; Kwon, S.H. Glutamine Synthetase as a Therapeutic Target for Cancer Treatment. Int. J. Mol. Sci. 2021, 22, 1701. https://doi.org/10.3390/ijms22041701
Kim GW, Lee DH, Jeon YH, Yoo J, Kim SY, Lee SW, Cho HY, Kwon SH. Glutamine Synthetase as a Therapeutic Target for Cancer Treatment. International Journal of Molecular Sciences. 2021; 22(4):1701. https://doi.org/10.3390/ijms22041701
Chicago/Turabian StyleKim, Go Woon, Dong Hoon Lee, Yu Hyun Jeon, Jung Yoo, So Yeon Kim, Sang Wu Lee, Ha Young Cho, and So Hee Kwon. 2021. "Glutamine Synthetase as a Therapeutic Target for Cancer Treatment" International Journal of Molecular Sciences 22, no. 4: 1701. https://doi.org/10.3390/ijms22041701