
Clinically Relevant Dose of Pemafibrate, a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator (SPPARMα), Lowers Serum Triglyceride Levels by Targeting Hepatic PPARα in Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice and Treatment
2.2. Histological Analysis
2.3. Biochemical Analysis
2.4. Analysis of mRNA Expression
2.5. Immunoblot Analysis
2.6. Statistical Analysis
3. Results
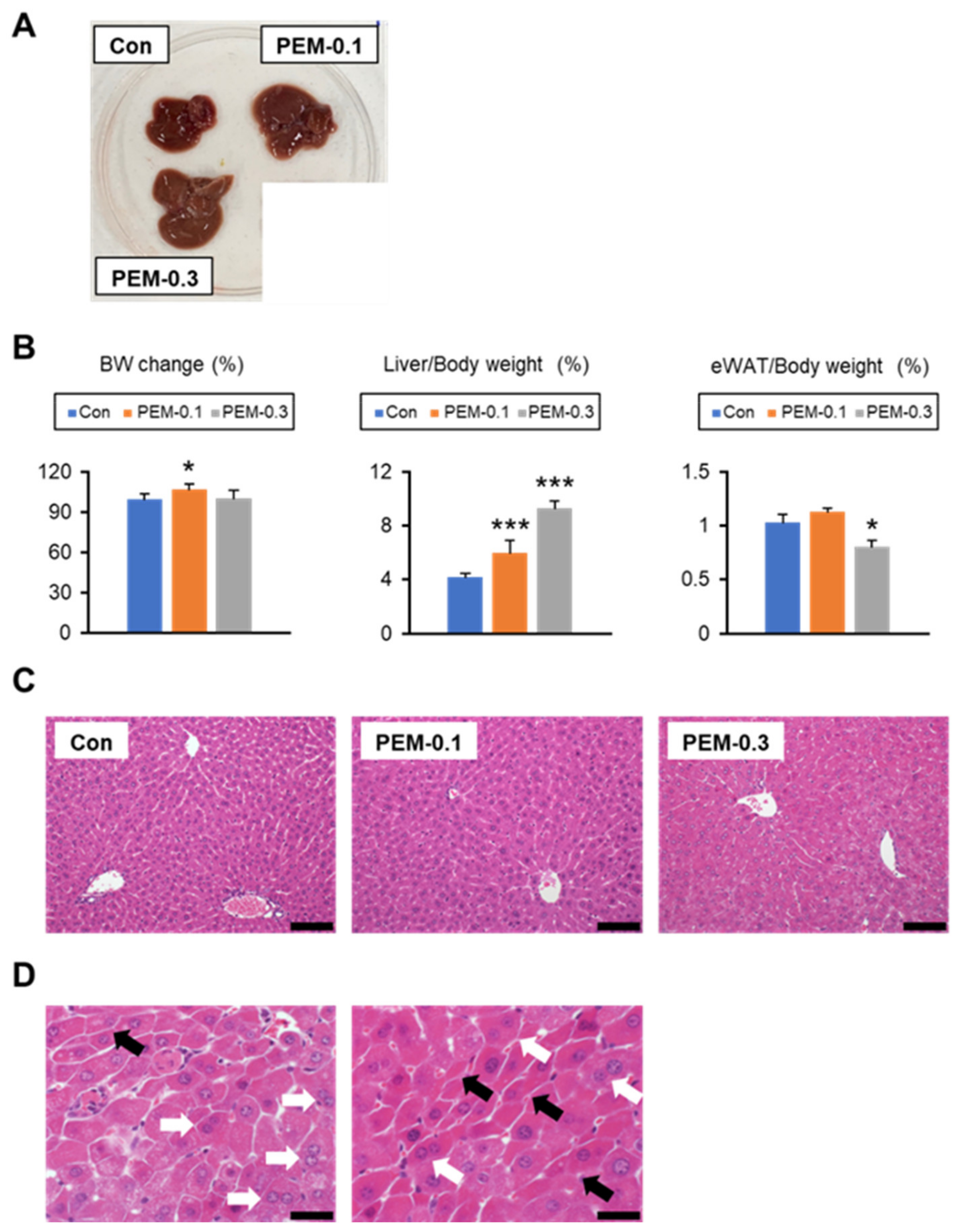
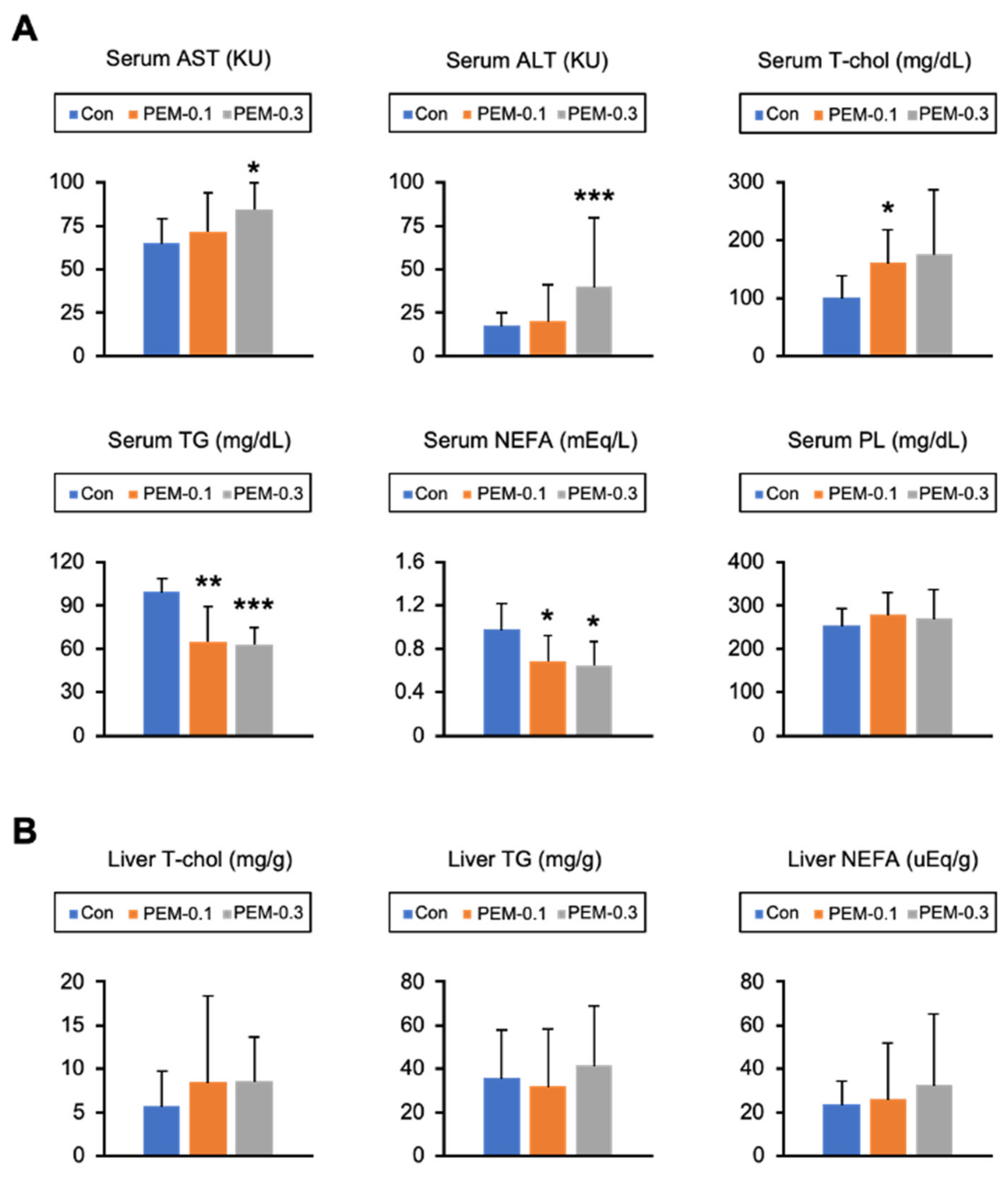
3.1. Lack of Increased Serum AST and ALT at Clinically Relevant Dose of PEM Treatment in Mice
3.2. Clinically Relevant Dose of PEM Reduces Serum TG and NEFA in Mice
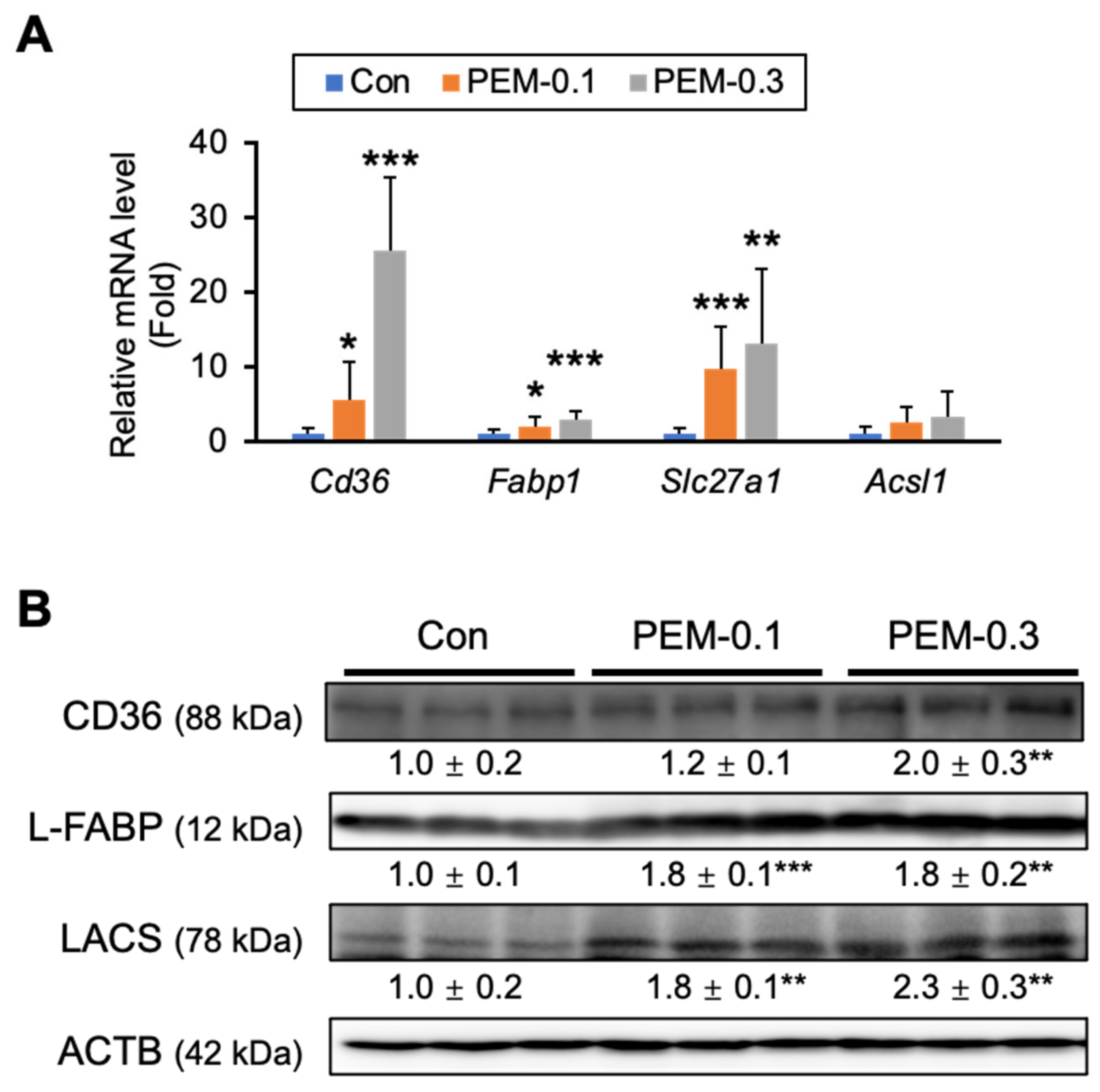
3.3. Clinically Relevant Dose of PEM Increases FA Utilization in the Liver
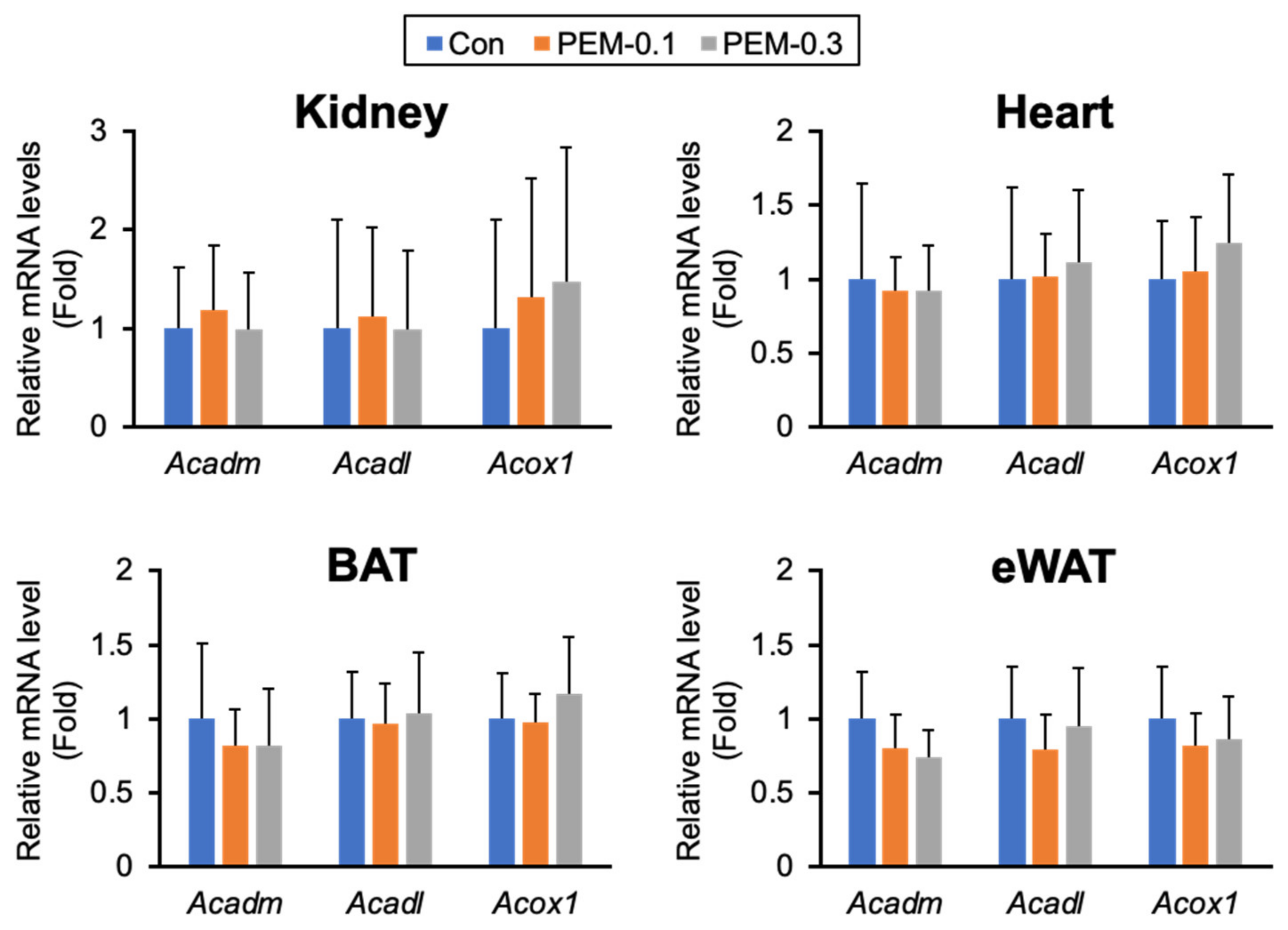
3.4. Clinically Relevant Dose of PEM Enhances Mitochondrial and Peroxisomal FA β-Oxidation
3.5. Effect of PEM on FA/TG Synthesis and VLDL Secretion
3.6. Clinically Relevant Dose of PEM Does Not Affect FA/TG Metabolism in Adipose Tissue
3.7. Clinically Relevant Dose of PEM Activates Hepatic PPARα Only and Increases FGF21 without Enhancing Oxidative Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Goldberg, I.J. Triglyceride: One molecule at the center of health and disease. Biochim. Biophys. Acta 2012, 1821, 719–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, N.; Takahashi, S.; Matsubara, T.; Jiang, C.; Sakamoto, W.; Chanturiya, T.; Teng, R.; Gavrilova, O.; Gonzalez, F.J. Adipocyte-specific Disruption of Fat-specific Protein 27 Causes Hepatosteatosis and Insulin Resistance in High-fat Diet-fed Mice. J. Biol. Chem. 2015, 290, 3092–3105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, C.J.; Lopez, A.D. Mortality by cause for eight regions of the world: Global Burden of Disease Study. Lancet 1997, 349, 1269–1276. [Google Scholar] [CrossRef]
- Tenenbaum, A.; Fisman, E.Z.; Motro, M.; Adler, Y. Atherogenic dyslipidemia in metabolic syndrome and type 2 diabetes: Therapeutic options beyond statins. Cardiovasc. Diabetol. 2006, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Roberto, C.A.; Swinburn, B.; Hawkes, C.; Huang, T.T.-K.; Costa, S.A.; Ashe, M.; Zwicker, L.; Cawley, J.H.; Brownell, K.D. Patchy progress on obesity prevention: Emerging examples, entrenched barriers, and new thinking. Lancet 2015, 385, 2400–2409. [Google Scholar] [CrossRef]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Investig. 2017, 127, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.; McCallin, T.; Martinez, J.; Chacko, S.; Yusuf, S. Hyperlipidemia. Pediatr. Rev. 2020, 41, 393–402. [Google Scholar] [CrossRef]
- Botta, M.; Audano, M.; Sahebkar, A.; Sirtori, C.R.; Mitro, N.; Ruscica, M. PPAR Agonists and Metabolic Syndrome: An Established Role? Int. J. Mol. Sci. 2018, 19, 1197. [Google Scholar] [CrossRef] [Green Version]
- Blair, H.A. Pemafibrate: First Global Approval. Drugs 2017, 77, 1805–1810. [Google Scholar] [CrossRef]
- Salakhutdinov, N.F.; Laev, S.S. Triglyceride-lowering agents. Bioorganic Med. Chem. 2014, 22, 3551–3564. [Google Scholar] [CrossRef]
- Lefebvre, P.; Chinetti, G.; Fruchart, J.-C.; Staels, B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J. Clin. Investig. 2006, 116, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Honda, A.; Ikegami, T.; Nakamuta, M.; Miyazaki, T.; Iwamoto, J.; Hirayama, T.; Saito, Y.; Takikawa, H.; Imawari, M.; Matsuzaki, Y. Anticholestatic effects of bezafibrate in patients with primary biliary cirrhosis treated with ursodeoxycholic acid. Hepatology 2013, 57, 1931–1941. [Google Scholar] [CrossRef]
- Nakajima, T.; Tanaka, N.; Kanbe, H.; Hara, A.; Kamijo, Y.; Zhang, X.; Gonzalez, F.J.; Aoyama, T. Bezafibrate at Clinically Relevant Doses Decreases Serum/Liver Triglycerides via Down-Regulation of Sterol Regulatory Element-Binding Protein-1c in Mice: A Novel Peroxisome Proliferator-Activated Receptor α-Independent Mechanism. Mol. Pharmacol. 2009, 75, 782–792. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y.; Raza-Iqbal, S.; Tanaka, T.; Murakami, K.; Anai, M.; Osawa, T.; Matsumura, Y.; Sakai, J.; Kodama, T. Gene Expression Profiles Induced by a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator (SPPARMα) Pemafibrate. Int. J. Mol. Sci. 2019, 20, 5682. [Google Scholar] [CrossRef] [Green Version]
- Abbas, A.; Saraf, S.; Ramachandran, S.; Raju, J.; Ramachandran, S. Fibrates and estimated glomerular filtration rate: Observations from an outpatient clinic setting and clinical implications: Figure 1. Postgrad. Med. J. 2012, 88, 503–506. [Google Scholar] [CrossRef]
- Davidson, M.H.; Armani, A.; McKenney, J.M.; Jacobson, T. Safety Considerations with Fibrate Therapy. Am. J. Cardiol. 2007, 99, S3–S18. [Google Scholar] [CrossRef]
- Jacobson, T.A. Myopathy with statin–fibrate combination therapy: Clinical considerations. Nat. Rev. Endocrinol. 2009, 5, 507–518. [Google Scholar] [CrossRef]
- Tanaka, N.; Honda, A. Pemafibrate for primary biliary cholangitis with dyslipidemia: A proposal of a new treatment from Japan. Hepatol. Res. 2022, 52, 495–496. [Google Scholar] [CrossRef]
- Yamashita, S.; Masuda, D.; Matsuzawa, Y. Pemafibrate, a New Selective PPARα Modulator: Drug Concept and Its Clinical Applications for Dyslipidemia and Metabolic Diseases. Curr. Atheroscler. Rep. 2020, 22, 5. [Google Scholar] [CrossRef] [Green Version]
- Hounslow, N.; Mair, S.; Suganami, H.; Nakamura, M. Pemafibrate Has High Bioavailability and is Principally Excreted via the Liver. Atheroscler. Suppl. 2018, 32, 157. [Google Scholar] [CrossRef]
- Yokote, K.; Yamashita, S.; Arai, H.; Araki, E.; Suganami, H.; Ishibashi, S. Long-Term Efficacy and Safety of Pemafibrate, a Novel Selective Peroxisome Proliferator-Activated Receptor-α Modulator (SPPARMα), in Dyslipidemic Patients with Renal Impairment. Int. J. Mol. Sci. 2019, 20, 706. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, S.-I.; Shimizu, M.; Kamiya, Y.; Uehara, S.; Suemizu, H.; Yamazaki, H. Increased plasma concentrations of an antidyslipidemic drug pemafibrate co-administered with rifampicin or cyclosporine A in cynomolgus monkeys genotyped for the organic anion transporting polypeptide 1B1. Drug Metab. Pharmacokinet. 2020, 35, 354–360. [Google Scholar] [CrossRef]
- Raza-Iqbal, S.; Tanaka, T.; Anai, M.; Inagaki, T.; Matsumura, Y.; Ikeda, K.; Taguchi, A.; Gonzalez, F.J.; Sakai, J.; Kodama, T. Transcriptome Analysis of K-877 (a Novel Selective PPARα Modulator (SPPARMα))-Regulated Genes in Primary Human Hepatocytes and the Mouse Liver. J. Atheroscler. Thromb. 2015, 22, 754–772. [Google Scholar] [CrossRef] [Green Version]
- Hennuyer, N.; Duplan, I.; Paquet, C.; Vanhoutte, J.; Woitrain, E.; Touche, V.; Colin, S.; Vallez, E.; Lestavel, S.; Lefebvre, P.; et al. The novel selective PPARα modulator (SPPARMα) pemafibrate improves dyslipidemia, enhances reverse cholesterol transport and decreases inflammation and atherosclerosis. Atherosclerosis 2016, 249, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Takahashi, S.; Fang, Z.-Z.; Matsubara, T.; Krausz, K.W.; Qu, A.; Gonzalez, F.J. Role of white adipose lipolysis in the development of NASH induced by methionine- and choline-deficient diet. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2014, 1841, 1596–1607. [Google Scholar] [CrossRef] [Green Version]
- Hara, A.; Radin, N.S. Lipid extraction of tissues with a low-toxicity solvent. Anal. Biochem. 1978, 90, 420–426. [Google Scholar] [CrossRef] [Green Version]
- Carr, T.P.; Andresen, C.J.; Rudel, L.L. Enzymatic determination of triglyceride, free cholesterol, and total cholesterol in tissue lipid extracts. Clin. Biochem. 1993, 26, 39–42. [Google Scholar] [CrossRef]
- Tanaka, N.; Takahashi, S.; Zhang, Y.; Krausz, K.W.; Smith, P.B.; Patterson, A.D.; Gonzalez, F.J. Role of fibroblast growth factor 21 in the early stage of NASH induced by methionine- and choline-deficient diet. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2015, 1852, 1242–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, F.; Diao, P.; Wang, X.; Hu, X.; Kimura, T.; Nakamuta, M.; Nakamura, I.; Shirotori, S.; Sato, Y.; Moriya, K.; et al. Dietary Restriction Suppresses Steatosis-Associated Hepatic Tumorigenesis in Hepatitis C Virus Core Gene Transgenic Mice. Liver Cancer 2020, 9, 529–548. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Peters, J.M.; Iritani, N.; Nakajima, T.; Furihata, K.; Hashimoto, T.; Gonzalez, F.J. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J. Biol. Chem. 1998, 273, 5678–5684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, J.; Jahangir, Z.; Al-Qarni, A.A. Microsomal Triglyceride Transfer Protein: From Lipid Metabolism to Metabolic Diseases. Adv. Exp. Med. Biol. 2020, 1276, 37–52. [Google Scholar] [CrossRef]
- Higuchi, N.; Kato, M.; Tanaka, M.; Miyazaki, M.; Takao, S.; Kohjima, M.; Kotoh, K.; Enjoji, M.; Nakamuta, M.; Takayanagi, R. Effects of insulin resistance and hepatic lipid accumulation on hepatic mRNA expression levels of apoB, MTP and L-FABP in non-alcoholic fatty liver disease. Exp. Ther. Med. 2011, 2, 1077–1081. [Google Scholar] [CrossRef] [Green Version]
- Pereira, I.V.; Stefano, J.T.; Oliveira, C.P. Microsomal triglyceride transfer protein and nonalcoholic fatty liver disease. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 245–251. [Google Scholar] [CrossRef]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Kawanishi, H.; Ohashi, K.; Ogawa, H.; Otaka, N.; Takikawa, T.; Fang, L.; Ozaki, Y.; Takefuji, M.; Murohara, T.; Ouchi, N. A novel selective PPARα modulator, pemafibrate promotes ischemia-induced revascularization through the eNOS-dependent mechanisms. PLoS ONE 2020, 15, e0235362. [Google Scholar] [CrossRef]
- Yoshida, M.; Nakamura, K.; Miyoshi, T.; Yoshida, M.; Kondo, M.; Akazawa, K.; Kimura, T.; Ohtsuka, H.; Ohno, Y.; Miura, D.; et al. Combination therapy with pemafibrate (K-877) and pitavastatin improves vascular endothelial dysfunction in dahl/salt-sensitive rats fed a high-salt and high-fat diet. Cardiovasc. Diabetol. 2020, 19, 149. [Google Scholar] [CrossRef]
- Horikawa, T.; Kawanami, T.; Hamaguchi, Y.; Tanaka, Y.; Kita, S.; Ryorin, R.; Takashi, Y.; Takahashi, H.; Tanabe, M.; Yanase, T.; et al. Pemafibrate, a PPAR alpha agonist, attenuates neointima formation after vascular injury in mice fed normal chow and a high-fat diet. Heliyon 2020, 6, e05431. [Google Scholar] [CrossRef]
- Suto, K.; Fukuda, D.; Shinohara, M.; Ganbaatar, B.; Yagi, S.; Kusunose, K.; Yamada, H.; Soeki, T.; Hirata, K.-I.; Sata, M. Pemafibrate, A Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator, Reduces Plasma Eicosanoid Levels and Ameliorates Endothelial Dysfunction in Diabetic Mice. J. Atheroscler. Thromb. 2021, 28, 1349–1360. [Google Scholar] [CrossRef]
- Fruchart, J.-C.; Hermans, M.P.; Fruchart-Najib, J.; Kodama, T. Selective Peroxisome Proliferator-Activated Receptor Alpha Modulators (SPPARMα) in the Metabolic Syndrome: Is Pemafibrate Light at the End of the Tunnel? Curr. Atheroscler. Rep. 2021, 23, 3. [Google Scholar] [CrossRef]
- Sairyo, M.; Kobayashi, T.; Masuda, D.; Kanno, K.; Zhu, Y.; Okada, T.; Koseki, M.; Ohama, T.; Nishida, M.; Sakata, Y.; et al. A Novel Selective PPARα Modulator (SPPARMα), K-877 (Pemafibrate), Attenuates Postprandial Hypertriglyceridemia in Mice. J. Atheroscler. Thromb. 2018, 25, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Mukaiyama, K.; Morikawa, A.; Kawakami, S.; Ichise, Y.; Kimura, T.; Horiuchi, A. Pemafibrate, a novel selective PPARα modulator, attenuates tamoxifen-induced fatty liver disease. Clin. J. Gastroenterol. 2021, 14, 846–851. [Google Scholar] [CrossRef]
- Yamashita, S.; Masuda, D.; Matsuzawa, Y. Clinical Applications of a Novel Selective PPARα Modulator, Pemafibrate, in Dyslipidemia and Metabolic Diseases. J. Atheroscler. Thromb. 2019, 26, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.E.; Ebling, F.J.; Samms, R.J.; Tsintzas, K. Going Back to the Biology of FGF21: New Insights. Trends Endocrinol. Metab. 2019, 30, 491–504. [Google Scholar] [CrossRef]
- Keinicke, H.; Sun, G.; Mentzel, C.; Fredholm, M.; John, L.M.; Andersen, B.; Raun, K.; Kjaergaard, M. FGF21 regulates hepatic metabolic pathways to improve steatosis and inflammation. Endocr. Connect. 2020, 9, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef] [Green Version]
- Do, H.T.; Tselykh, T.V.; Mäkelä, J.; Ho, T.H.; Olkkonen, V.; Bornhauser, B.C.; Korhonen, L.; Zelcer, N.; Lindholm, D. Fibroblast Growth Factor-21 (FGF21) Regulates Low-density Lipoprotein Receptor (LDLR) Levels in Cells via the E3-ubiquitin Ligase Mylip/Idol and the Canopy2 (Cnpy2)/Mylip-interacting Saposin-like Protein (Msap). J. Biol. Chem. 2012, 287, 12602–12611. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Shao, Y.; Wu, F.; Wang, Y.; Xiong, R.; Zheng, J.; Tian, H.; Wang, B.; Wang, Y.; Zhang, Y.; et al. FGF21 Prevents Angiotensin II-Induced Hypertension and Vascular Dysfunction by Activation of ACE2/Angiotensin-(1–7) Axis in Mice. Cell Metab. 2018, 27, 1323–1337.e5. [Google Scholar] [CrossRef] [Green Version]
- Tomita, Y.; Lee, D.; Miwa, Y.; Jiang, X.; Ohta, M.; Tsubota, K.; Kurihara, T. Pemafibrate Protects Against Retinal Dysfunction in a Murine Model of Diabetic Retinopathy. Int. J. Mol. Sci. 2020, 21, 6243. [Google Scholar] [CrossRef]
- Tanajak, P.; Chattipakorn, S.C.; Chattipakorn, N. Effects of fibroblast growth factor 21 on the heart. J. Endocrinol. 2015, 227, R13–R30. [Google Scholar] [CrossRef] [Green Version]
- Planavila, A.; Redondo-Angulo, I.; Ribas, F.; Garrabou, G.; Casademont, J.; Giralt, M.; Villarroya, F. Fibroblast growth factor 21 protects the heart from oxidative stress. Cardiovasc. Res. 2015, 106, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, D.; Long, X.-X.; Fang, Q.-C.; Jia, W.-P.; Li, H.-T. The role of FGF21 in the pathogenesis of cardiovascular disease. Chin. Med. J. 2021, 134, 2931–2943. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Wroblewski, V.J.; Koester, A.; Chen, Y.-F.; Clutinger, C.K.; Tigno, X.T.; Hansen, B.C.; Shanafelt, A.B.; Etgen, G.J. The Metabolic State of Diabetic Monkeys Is Regulated by Fibroblast Growth Factor-21. Endocrinology 2007, 148, 774–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diener, J.L.; Mowbray, S.; Huang, W.-J.; Yowe, D.; Xu, J.; Caplan, S.; Misra, A.; Kapur, A.; Shapiro, J.; Ke, X.; et al. FGF21 Normalizes Plasma Glucose in Mouse Models of Type 1 Diabetes and Insulin Receptor Dysfunction. Endocrinology 2021, 162, bqab092. [Google Scholar] [CrossRef] [PubMed]
- Laeger, T.; Baumeier, C.; Wilhelmi, I.; Würfel, J.; Kamitz, A.; Schürmann, A. FGF21 improves glucose homeostasis in an obese diabetes-prone mouse model independent of body fat changes. Diabetologia 2017, 60, 2274–2284. [Google Scholar] [CrossRef] [Green Version]
- Honda, Y.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M.; Takizawa, T.; Saito, S.; Nagashima, Y.; et al. Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator, improves the pathogenesis in a rodent model of nonalcoholic steatohepatitis. Sci. Rep. 2017, 7, 42477. [Google Scholar] [CrossRef]
- Sasaki, Y.; Asahiyama, M.; Tanaka, T.; Yamamoto, S.; Murakami, K.; Kamiya, W.; Matsumura, Y.; Osawa, T.; Anai, M.; Fruchart, J.-C.; et al. Pemafibrate, a selective PPARα modulator, prevents non-alcoholic steatohepatitis development without reducing the hepatic triglyceride content. Sci. Rep. 2020, 10, 7818. [Google Scholar] [CrossRef]
- Araki, M.; Nakagawa, Y.; Oishi, A.; Han, S.-I.; Wang, Y.; Kumagai, K.; Ohno, H.; Mizunoe, Y.; Iwasaki, H.; Sekiya, M.; et al. The Peroxisome Proliferator-Activated Receptor α (PPARα) Agonist Pemafibrate Protects against Diet-Induced Obesity in Mice. Int. J. Mol. Sci. 2018, 19, 2148. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Aoyama, T.; Kimura, S.; Gonzalez, F.J. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol. Ther. 2017, 179, 142–157. [Google Scholar] [CrossRef]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [Green Version]
- E Foreman, J.; Koga, T.; Kosyk, O.; Kang, B.-H.; Zhu, X.; Cohen, S.M.; Billy, L.J.; Sharma, A.K.; Amin, S.; Gonzalez, F.J.; et al. Species Differences between Mouse and Human PPARα in Modulating the Hepatocarcinogenic Effects of Perinatal Exposure to a High-Affinity Human PPARα Agonist in Mice. Toxicol. Sci. 2021, 183, 81–92. [Google Scholar] [CrossRef]
- Lee, D.; Tomita, Y.; Jeong, H.; Miwa, Y.; Tsubota, K.; Negishi, K.; Kurihara, T. Pemafibrate Prevents Retinal Dysfunction in a Mouse Model of Unilateral Common Carotid Artery Occlusion. Int. J. Mol. Sci. 2021, 22, 9408. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 659–661. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Diao, P.; Zhang, X.; Nakajima, T.; Kimura, T.; Tanaka, N. Clinically Relevant Dose of Pemafibrate, a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator (SPPARMα), Lowers Serum Triglyceride Levels by Targeting Hepatic PPARα in Mice. Biomedicines 2022, 10, 1667. https://doi.org/10.3390/biomedicines10071667
Zhang Z, Diao P, Zhang X, Nakajima T, Kimura T, Tanaka N. Clinically Relevant Dose of Pemafibrate, a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator (SPPARMα), Lowers Serum Triglyceride Levels by Targeting Hepatic PPARα in Mice. Biomedicines. 2022; 10(7):1667. https://doi.org/10.3390/biomedicines10071667
Chicago/Turabian StyleZhang, Zhe, Pan Diao, Xuguang Zhang, Takero Nakajima, Takefumi Kimura, and Naoki Tanaka. 2022. "Clinically Relevant Dose of Pemafibrate, a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator (SPPARMα), Lowers Serum Triglyceride Levels by Targeting Hepatic PPARα in Mice" Biomedicines 10, no. 7: 1667. https://doi.org/10.3390/biomedicines10071667
APA StyleZhang, Z., Diao, P., Zhang, X., Nakajima, T., Kimura, T., & Tanaka, N. (2022). Clinically Relevant Dose of Pemafibrate, a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator (SPPARMα), Lowers Serum Triglyceride Levels by Targeting Hepatic PPARα in Mice. Biomedicines, 10(7), 1667. https://doi.org/10.3390/biomedicines10071667