
Adipose Tissue Wasting as a Determinant of Pancreatic Cancer-Related Cachexia

{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
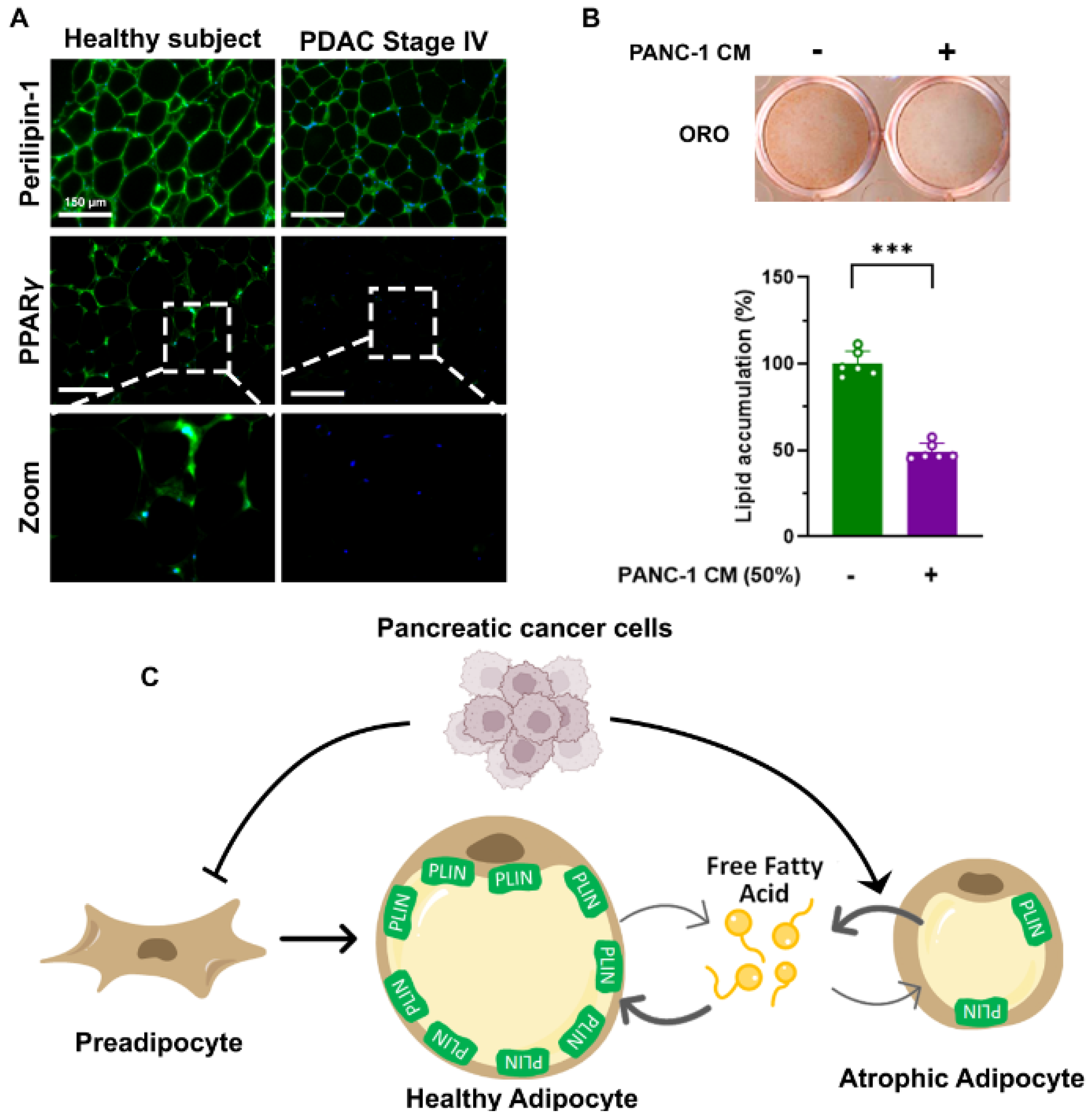
2. Adipose Tissue Wasting as a Critical Determinant for Cachexia and Poor Prognosis in Pancreatic Cancer Patients
3. Decreased Intake and Promoted Adipose Lipid Turnover to Exacerbate Adipose Tissue Loss in Pancreatic Cancer Patients
4. Pancreatic Cancer-Related Adipose Tissue Remodeling to Promote Adipocyte Lipid Turnover and Fibrosis
5. Pancreatic Cancer-Associated Cachectic Mediators and Potential Mechanisms
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw. Open 2021, 4, e214708. [Google Scholar] [CrossRef] [PubMed]
- Dorman, K.; Heinemann, V.; Kobold, S.; von Bergwelt-Baildon, M.; Boeck, S. Novel systemic treatment approaches for metastatic pancreatic cancer. Expert Opin. Investig. Drugs 2022, 31, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Feo, C.F.; Deiana, G.; Ninniri, C.; Cherchi, G.; Crivelli, P.; Fancellu, A.; Ginesu, G.C.; Porcu, A. Vascular resection for locally advanced pancreatic ductal adenocarcinoma: Analysis of long-term outcomes from a single-centre series. World J. Surg. Oncol. 2021, 19, 126. [Google Scholar] [CrossRef] [PubMed]
- Franck, C.; Muller, C.; Rosania, R.; Croner, R.S.; Pech, M.; Venerito, M. Advanced Pancreatic Ductal Adenocarcinoma: Moving Forward. Cancers 2020, 12, 1955. [Google Scholar] [CrossRef]
- Poulia, K.A.; Sarantis, P.; Antoniadou, D.; Koustas, E.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Pancreatic Cancer and Cachexia-Metabolic Mechanisms and Novel Insights. Nutrients 2020, 12, 1543. [Google Scholar] [CrossRef]
- Bachmann, J.; Buchler, M.W.; Friess, H.; Martignoni, M.E. Cachexia in patients with chronic pancreatitis and pancreatic cancer: Impact on survival and outcome. Nutr. Cancer 2013, 65, 827–833. [Google Scholar] [CrossRef]
- Teoule, P.; Rasbach, E.; Oweira, H.; Otto, M.; Rahbari, N.N.; Reissfelder, C.; Ruckert, F.; Birgin, E. Obesity and Pancreatic Cancer: A Matched-Pair Survival Analysis. J. Clin. Med. 2020, 9, 3526. [Google Scholar] [CrossRef]
- Hendifar, A.E.; Chang, J.I.; Huang, B.Z.; Tuli, R.; Wu, B.U. Cachexia, and not obesity, prior to pancreatic cancer diagnosis worsens survival and is negated by chemotherapy. J. Gastrointest. Oncol. 2018, 9, 17–23. [Google Scholar] [CrossRef]
- Martin, L.; Senesse, P.; Gioulbasanis, I.; Antoun, S.; Bozzetti, F.; Deans, C.; Strasser, F.; Thoresen, L.; Jagoe, R.T.; Chasen, M.; et al. Diagnostic criteria for the classification of cancer-associated weight loss. J. Clin. Oncol. 2015, 33, 90–99. [Google Scholar] [CrossRef]
- Gannavarapu, B.S.; Lau, S.K.M.; Carter, K.; Cannon, N.A.; Gao, A.; Ahn, C.; Meyer, J.J.; Sher, D.J.; Jatoi, A.; Infante, R.; et al. Prevalence and Survival Impact of Pretreatment Cancer-Associated Weight Loss: A Tool for Guiding Early Palliative Care. J. Oncol. Pract. 2018, 14, e238–e250. [Google Scholar] [CrossRef]
- Kordes, M.; Larsson, L.; Engstrand, L.; Lohr, J.M. Pancreatic cancer cachexia: Three dimensions of a complex syndrome. Br. J. Cancer 2021, 124, 1623–1636. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.R.; Yaffee, P.M.; Jamil, L.H.; Lo, S.K.; Nissen, N.; Pandol, S.J.; Tuli, R.; Hendifar, A.E. Pancreatic cancer cachexia: A review of mechanisms and therapeutics. Front. Physiol. 2014, 5, 88. [Google Scholar] [CrossRef] [PubMed]
- Argiles, J.M.; Lopez-Soriano, F.J.; Stemmler, B.; Busquets, S. Therapeutic strategies against cancer cachexia. Eur. J. Transl. Myol. 2019, 29, 7960. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; Feizi, A.; Uhlen, M.; Nielsen, J. A Systematic Investigation of the Malignant Functions and Diagnostic Potential of the Cancer Secretome. Cell Rep. 2019, 26, 2622–2635.e2625. [Google Scholar] [CrossRef] [PubMed]
- Naumann, P.; Eberlein, J.; Farnia, B.; Liermann, J.; Hackert, T.; Debus, J.; Combs, S.E. Cachectic Body Composition and Inflammatory Markers Portend a Poor Prognosis in Patients with Locally Advanced Pancreatic Cancer Treated with Chemoradiation. Cancers 2019, 11, 1655. [Google Scholar] [CrossRef] [PubMed]
- Nemer, L.; Krishna, S.G.; Shah, Z.K.; Conwell, D.L.; Cruz-Monserrate, Z.; Dillhoff, M.; Guttridge, D.C.; Hinton, A.; Manilchuk, A.; Pawlik, T.M.; et al. Predictors of Pancreatic Cancer-Associated Weight Loss and Nutritional Interventions. Pancreas 2017, 46, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Kays, J.K.; Shahda, S.; Stanley, M.; Bell, T.M.; O’Neill, B.H.; Kohli, M.D.; Couch, M.E.; Koniaris, L.G.; Zimmers, T.A. Three cachexia phenotypes and the impact of fat-only loss on survival in FOLFIRINOX therapy for pancreatic cancer. J. Cachexia Sarcopenia Muscle 2018, 9, 673–684. [Google Scholar] [CrossRef]
- Bachmann, J.; Ketterer, K.; Marsch, C.; Fechtner, K.; Krakowski-Roosen, H.; Buchler, M.W.; Friess, H.; Martignoni, M.E. Pancreatic cancer related cachexia: Influence on metabolism and correlation to weight loss and pulmonary function. BMC Cancer 2009, 9, 255. [Google Scholar] [CrossRef]
- Wigmore, S.J.; Plester, C.E.; Richardson, R.A.; Fearon, K.C. Changes in nutritional status associated with unresectable pancreatic cancer. Br. J. Cancer 1997, 75, 106–109. [Google Scholar] [CrossRef]
- Tan, B.H.; Birdsell, L.A.; Martin, L.; Baracos, V.E.; Fearon, K.C. Sarcopenia in an overweight or obese patient is an adverse prognostic factor in pancreatic cancer. Clin. Cancer Res. 2009, 15, 6973–6979. [Google Scholar] [CrossRef] [Green Version]
- Di Sebastiano, K.M.; Yang, L.; Zbuk, K.; Wong, R.K.; Chow, T.; Koff, D.; Moran, G.R.; Mourtzakis, M. Accelerated muscle and adipose tissue loss may predict survival in pancreatic cancer patients: The relationship with diabetes and anaemia. Br. J. Nutr. 2013, 109, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Nakano, O.; Kawai, H.; Kobayashi, T.; Kohisa, J.; Ikarashi, S.; Hayashi, K.; Yokoyama, J.; Terai, S. Rapid decline in visceral adipose tissue over 1 month is associated with poor prognosis in patients with unresectable pancreatic cancer. Cancer Med. 2021, 10, 4291–4301. [Google Scholar] [CrossRef] [PubMed]
- Mintziras, I.; Miligkos, M.; Wachter, S.; Manoharan, J.; Maurer, E.; Bartsch, D.K. Sarcopenia and sarcopenic obesity are significantly associated with poorer overall survival in patients with pancreatic cancer: Systematic review and meta-analysis. Int. J. Surg. 2018, 59, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Shintakuya, R.; Uemura, K.; Murakami, Y.; Kondo, N.; Nakagawa, N.; Urabe, K.; Okano, K.; Awai, K.; Higaki, T.; Sueda, T. Sarcopenia is closely associated with pancreatic exocrine insufficiency in patients with pancreatic disease. Pancreatology 2017, 17, 70–75. [Google Scholar] [CrossRef]
- Sandini, M.; Patino, M.; Ferrone, C.R.; Alvarez-Perez, C.A.; Honselmann, K.C.; Paiella, S.; Catania, M.; Riva, L.; Tedesco, G.; Casolino, R.; et al. Association Between Changes in Body Composition and Neoadjuvant Treatment for Pancreatic Cancer. JAMA Surg. 2018, 153, 809–815. [Google Scholar] [CrossRef]
- Gresham, G.; Placencio-Hickok, V.R.; Lauzon, M.; Nguyen, T.; Kim, H.; Mehta, S.; Paski, S.; Pandol, S.J.; Osipov, A.; Gong, J.; et al. Feasibility and efficacy of enteral tube feeding on weight stability, lean body mass, and patient-reported outcomes in pancreatic cancer cachexia. J. Cachexia Sarcopenia Muscle 2021, 12, 1959–1968. [Google Scholar] [CrossRef]
- Sobczak, A.I.S.; Blindauer, C.A.; Stewart, A.J. Changes in Plasma Free Fatty Acids Associated with Type-2 Diabetes. Nutrients 2019, 11, 2022. [Google Scholar] [CrossRef]
- Xu, P.C.; You, M.; Yu, S.Y.; Luan, Y.; Eldani, M.; Caffrey, T.C.; Grandgenett, P.M.; O’Connell, K.A.; Shukla, S.K.; Kattamuri, C.; et al. Visceral adipose tissue remodeling in pancreatic ductal adenocarcinoma cachexia: The role of activin A signaling. Sci. Rep. 2022, 12, 1659. [Google Scholar] [CrossRef]
- Sah, R.P.; Sharma, A.; Nagpal, S.; Patlolla, S.H.; Sharma, A.; Kandlakunta, H.; Anani, V.; Angom, R.S.; Kamboj, A.K.; Ahmed, N.; et al. Phases of Metabolic and Soft Tissue Changes in Months Preceding a Diagnosis of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2019, 156, 1742–1752. [Google Scholar] [CrossRef]
- Dzierzek, P.; Kurnol, K.; Hap, W.; Frejlich, E.; Diakun, A.; Karwowski, A.; Kotulski, K.; Rudno-Rudzinska, J.; Kielan, W. Assessment of changes in body composition measured with bioelectrical impedance in patients operated for pancreatic, gastric and colorectal cancer. Pol. Przegl. Chir. 2020, 92, 8–11. [Google Scholar] [CrossRef]
- Thaxton, C.S.; Rink, J.S.; Naha, P.C.; Cormode, D.P. Lipoproteins and lipoprotein mimetics for imaging and drug delivery. Adv. Drug Deliv. Rev. 2016, 106, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, S.; de la Puente-Secades, S.; Schurgers, L.; Jankowski, J. Lipids and lipoproteins in cardiovascular diseases: A classification. Trends Endocrinol. Metab. 2022, 33, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Macasek, J.; Vecka, M.; Zak, A.; Urbanek, M.; Krechler, T.; Petruzelka, L.; Stankova, B.; Zeman, M. Plasma fatty acid composition in patients with pancreatic cancer: Correlations to clinical parameters. Nutr. Cancer 2012, 64, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Kobayashi, T.; Chayahara, N.; Imamura, Y.; Toyoda, M.; Kiyota, N.; Mukohara, T.; Nishiumi, S.; Azuma, T.; Yoshida, M.; et al. Metabolomics evaluation of serum markers for cachexia and their intra-day variation in patients with advanced pancreatic cancer. PLoS ONE 2014, 9, e113259. [Google Scholar] [CrossRef]
- Zuijdgeest-van Leeuwen, S.D.; van der Heijden, M.S.; Rietveld, T.; van den Berg, J.W.; Tilanus, H.W.; Burgers, J.A.; Wilson, J.H.; Dagnelie, P.C. Fatty acid composition of plasma lipids in patients with pancreatic, lung and oesophageal cancer in comparison with healthy subjects. Clin. Nutr. 2002, 21, 225–230. [Google Scholar] [CrossRef]
- Hodson, L.; Skeaff, C.M.; Fielding, B.A. Fatty acid composition of adipose tissue and blood in humans and its use as a biomarker of dietary intake. Prog. Lipid Res. 2008, 47, 348–380. [Google Scholar] [CrossRef]
- Kaur, N.; Chugh, V.; Gupta, A.K. Essential fatty acids as functional components of foods—A review. J. Food Sci. Technol. 2014, 51, 2289–2303. [Google Scholar] [CrossRef]
- Foster, M.T.; Pagliassotti, M.J. Metabolic alterations following visceral fat removal and expansion: Beyond anatomic location. Adipocyte 2012, 1, 192–199. [Google Scholar] [CrossRef]
- Sztalryd, C.; Brasaemle, D.L. The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1221–1232. [Google Scholar] [CrossRef]
- Tansey, J.T.; Sztalryd, C.; Gruia-Gray, J.; Roush, D.L.; Zee, J.V.; Gavrilova, O.; Reitman, M.L.; Deng, C.X.; Li, C.; Kimmel, A.R.; et al. Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc. Natl. Acad. Sci. USA 2001, 98, 6494–6499. [Google Scholar] [CrossRef] [Green Version]
- Corella, D.; Qi, L.; Sorli, J.V.; Godoy, D.; Portoles, O.; Coltell, O.; Greenberg, A.S.; Ordovas, J.M. Obese subjects carrying the 11482G>A polymorphism at the perilipin locus are resistant to weight loss after dietary energy restriction. J. Clin. Endocrinol. Metab. 2005, 90, 5121–5126. [Google Scholar] [CrossRef] [PubMed]
- Michalkova, L.; Hornik, S.; Sykora, J.; Habartova, L.; Setnicka, V.; Bunganic, B. Early Detection of Pancreatic Cancer in Type 2 Diabetes Mellitus Patients Based on (1)H NMR Metabolomics. J. Proteome Res. 2021, 20, 1744–1753. [Google Scholar] [CrossRef] [PubMed]
- Cotter, D.G.; Schugar, R.C.; Crawford, P.A. Ketone body metabolism and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1060–H1076. [Google Scholar] [CrossRef]
- Shukla, S.K.; Gebregiworgis, T.; Purohit, V.; Chaika, N.V.; Gunda, V.; Radhakrishnan, P.; Mehla, K.; Pipinos, I.I.; Powers, R.; Yu, F.; et al. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab. 2014, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; TeSlaa, T.; Ng, S.; Nofal, M.; Wang, L.; Lan, T.; Zeng, X.; Cowan, A.; McBride, M.; Lu, W.; et al. Ketogenic diet and chemotherapy combine to disrupt pancreatic cancer metabolism and growth. Med 2022, 3, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Westermark, A.M.; Zhang, Y.; Yuan, C.; Li, Z.; Lau, A.N.; Sapp, K.M.; Wolpin, B.M.; Vander Heiden, M.G. Low glycaemic diets alter lipid metabolism to influence tumour growth. Nature 2021, 599, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Cochran, B.J.; Ryder, W.J.; Parmar, A.; Klaeser, K.; Reilhac, A.; Angelis, G.I.; Meikle, S.R.; Barter, P.J.; Rye, K.A. Determining Glucose Metabolism Kinetics Using 18F-FDG Micro-PET/CT. J. Vis. Exp. 2017, 123, e55184. [Google Scholar] [CrossRef]
- Van de Wiele, C.; Van Vlaenderen, M.; D’Hulst, L.; Delcourt, A.; Copin, D.; De Spiegeleer, B.; Maes, A. Metabolic and morphological measurements of subcutaneous and visceral fat and their relationship with disease stage and overall survival in newly diagnosed pancreatic adenocarcinoma: Metabolic and morphological fat measurements in pancreatic adenocarcinoma. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 110–116. [Google Scholar] [CrossRef]
- Okumura, T.; Ohuchida, K.; Sada, M.; Abe, T.; Endo, S.; Koikawa, K.; Iwamoto, C.; Miura, D.; Mizuuchi, Y.; Moriyama, T.; et al. Extra-pancreatic invasion induces lipolytic and fibrotic changes in the adipose microenvironment, with released fatty acids enhancing the invasiveness of pancreatic cancer cells. Oncotarget 2017, 8, 18280–18295. [Google Scholar] [CrossRef]
- Cai, Z.; Liang, Y.; Xing, C.; Wang, H.; Hu, P.; Li, J.; Huang, H.; Wang, W.; Jiang, C. Cancer associated adipocytes exhibit distinct phenotypes and facilitate tumor progression in pancreatic cancer. Oncol. Rep. 2019, 42, 2537–2549. [Google Scholar] [CrossRef] [Green Version]
- Takehara, M.; Sato, Y.; Kimura, T.; Noda, K.; Miyamoto, H.; Fujino, Y.; Miyoshi, J.; Nakamura, F.; Wada, H.; Bando, Y.; et al. Cancer-associated adipocytes promote pancreatic cancer progression through SAA1 expression. Cancer Sci. 2020, 111, 2883–2894. [Google Scholar] [CrossRef] [PubMed]
- Sagar, G.; Sah, R.P.; Javeed, N.; Dutta, S.K.; Smyrk, T.C.; Lau, J.S.; Giorgadze, N.; Tchkonia, T.; Kirkland, J.L.; Chari, S.T.; et al. Pathogenesis of pancreatic cancer exosome-induced lipolysis in adipose tissue. Gut 2016, 65, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Rupert, J.E.; Narasimhan, A.; Jengelley, D.H.A.; Jiang, Y.; Liu, J.; Au, E.; Silverman, L.M.; Sandusky, G.; Bonetto, A.; Cao, S.; et al. Tumor-derived IL-6 and trans-signaling among tumor, fat, and muscle mediate pancreatic cancer cachexia. J. Exp. Med. 2021, 218, e20190450. [Google Scholar] [CrossRef]
- Zoico, E.; Darra, E.; Rizzatti, V.; Budui, S.; Franceschetti, G.; Mazzali, G.; Rossi, A.P.; Fantin, F.; Menegazzi, M.; Cinti, S.; et al. Adipocytes WNT5a mediated dedifferentiation: A possible target in pancreatic cancer microenvironment. Oncotarget 2016, 7, 20223–20235. [Google Scholar] [CrossRef] [PubMed]
- Wafer, R.; Tandon, P.; Minchin, J.E.N. The Role of Peroxisome Proliferator-Activated Receptor Gamma (PPARG) in Adipogenesis: Applying Knowledge from the Fish Aquaculture Industry to Biomedical Research. Front. Endocrinol. 2017, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Tamori, Y.; Masugi, J.; Nishino, N.; Kasuga, M. Role of peroxisome proliferator-activated receptor-gamma in maintenance of the characteristics of mature 3T3-L1 adipocytes. Diabetes 2002, 51, 2045–2055. [Google Scholar] [CrossRef]
- Ye, R.; Wang, Q.A.; Tao, C.; Vishvanath, L.; Shao, M.; McDonald, J.G.; Gupta, R.K.; Scherer, P.E. Impact of tamoxifen on adipocyte lineage tracing: Inducer of adipogenesis and prolonged nuclear translocation of Cre recombinase. Mol. Metab. 2015, 4, 771–778. [Google Scholar] [CrossRef]
- Petruzzelli, M.; Schweiger, M.; Schreiber, R.; Campos-Olivas, R.; Tsoli, M.; Allen, J.; Swarbrick, M.; Rose-John, S.; Rincon, M.; Robertson, G.; et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 2014, 20, 433–447. [Google Scholar] [CrossRef]
- Luan, Y.; Zhang, Y.; Yu, S.Y.; You, M.; Xu, P.C.; Chung, S.; Kurita, T.; Zhu, J.; Kim, S.Y. Development of ovarian tumour causes significant loss of muscle and adipose tissue: A novel mouse model for cancer cachexia study. J. Cachexia Sarcopenia Muscle 2022, 13, 1289–1301. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, S.; Shangguan, J.; Pan, L.; Zhou, X.; Yaghmai, V.; Velichko, Y.; Hu, C.; Yang, J.; Zhang, Z. MRI Assessment of Associations between Brown Adipose Tissue and Cachexia in Murine Pancreatic Ductal Adenocarcinoma. Intern. Med. Open Access 2019, 9, 301. [Google Scholar] [CrossRef]
- Park, J.; Shin, S.; Liu, L.; Jahan, I.; Ong, S.G.; Xu, P.; Berry, D.C.; Jiang, Y. Progenitor-like characteristics in a subgroup of UCP1+ cells within white adipose tissue. Dev. Cell 2021, 56, 985–999.e984. [Google Scholar] [CrossRef]
- Corvera, S. Cellular Heterogeneity in Adipose Tissues. Annu. Rev. Physiol. 2021, 83, 257–278. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.J.; Kim, Y.J.; Park, B.; Yim, S.; Park, C.; Roh, H.; Moon, Y.; Seong, J.K.; Park, H. YAP-dependent Wnt5a induction in hypertrophic adipocytes restrains adiposity. Cell Death Dis. 2022, 13, 407. [Google Scholar] [CrossRef] [PubMed]
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Naslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nature 2008, 453, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Li, S.N.; Wu, J.F. TGF-beta/SMAD signaling regulation of mesenchymal stem cells in adipocyte commitment. Stem Cell Res. Ther. 2020, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- de Winter, T.J.J.; Nusse, R. Running Against the Wnt: How Wnt/beta-Catenin Suppresses Adipogenesis. Front. Cell Dev. Biol. 2021, 9, 627429. [Google Scholar] [CrossRef]
- Kraus, N.A.; Ehebauer, F.; Zapp, B.; Rudolphi, B.; Kraus, B.J.; Kraus, D. Quantitative assessment of adipocyte differentiation in cell culture. Adipocyte 2016, 5, 351–358. [Google Scholar] [CrossRef]
- Togashi, Y.; Kogita, A.; Sakamoto, H.; Hayashi, H.; Terashima, M.; de Velasco, M.A.; Sakai, K.; Fujita, Y.; Tomida, S.; Kitano, M.; et al. Activin signal promotes cancer progression and is involved in cachexia in a subset of pancreatic cancer. Cancer Lett. 2015, 356, 819–827. [Google Scholar] [CrossRef]
- Mancinelli, G.; Torres, C.; Krett, N.; Bauer, J.; Castellanos, K.; McKinney, R.; Dawson, D.; Guzman, G.; Hwang, R.; Grimaldo, S.; et al. Role of stromal activin A in human pancreatic cancer and metastasis in mice. Sci. Rep. 2021, 11, 7986. [Google Scholar] [CrossRef]
- Chen, J.L.; Walton, K.L.; Qian, H.; Colgan, T.D.; Hagg, A.; Watt, M.J.; Harrison, C.A.; Gregorevic, P. Differential Effects of IL6 and Activin A in the Development of Cancer-Associated Cachexia. Cancer Res. 2016, 76, 5372–5382. [Google Scholar] [CrossRef] [Green Version]
- Walton, K.L.; Chen, J.L.; Arnold, Q.; Kelly, E.; La, M.; Lu, L.; Lovrecz, G.; Hagg, A.; Colgan, T.D.; Qian, H.; et al. Activin A-Induced Cachectic Wasting Is Attenuated by Systemic Delivery of Its Cognate Propeptide in Male Mice. Endocrinology 2019, 160, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Narasimhan, A.; Silverman, L.M.; Young, A.R.; Shahda, S.; Liu, S.; Wan, J.; Liu, Y.; Koniaris, L.G.; Zimmers, T.A. Sex specificity of pancreatic cancer cachexia phenotypes, mechanisms, and treatment in mice and humans: Role of Activin. J. Cachexia Sarcopenia Muscle 2022, 13, 2146–2161. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Rousseau, F.; Guilhot, F.; Malinge, P.; Magistrelli, G.; Herren, S.; Jones, S.A.; Jones, G.W.; Scheller, J.; Lissilaa, R.; et al. Novel Insights into Interleukin 6 (IL-6) Cis- and Trans-signaling Pathways by Differentially Manipulating the Assembly of the IL-6 Signaling Complex. J. Biol. Chem. 2015, 290, 26943–26953. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lu, C.; Meng, Q.; Halim, A.; Yean, T.J.; Wu, G. Plasma concentration of interleukin-6 was upregulated in cancer cachexia patients and was positively correlated with plasma free fatty acid in female patients. Nutr. Metab. 2019, 16, 80. [Google Scholar] [CrossRef]
- Okada, S.; Okusaka, T.; Ishii, H.; Kyogoku, A.; Yoshimori, M.; Kajimura, N.; Yamaguchi, K.; Kakizoe, T. Elevated serum interleukin-6 levels in patients with pancreatic cancer. Jpn. J. Clin. Oncol. 1998, 28, 12–15. [Google Scholar] [CrossRef]
- Martignoni, M.E.; Kunze, P.; Hildebrandt, W.; Kunzli, B.; Berberat, P.; Giese, T.; Kloters, O.; Hammer, J.; Buchler, M.W.; Giese, N.A.; et al. Role of mononuclear cells and inflammatory cytokines in pancreatic cancer-related cachexia. Clin. Cancer Res. 2005, 11, 5802–5808. [Google Scholar] [CrossRef]
- Ramsey, M.L.; Talbert, E.; Ahn, D.; Bekaii-Saab, T.; Badi, N.; Bloomston, P.M.; Conwell, D.L.; Cruz-Monserrate, Z.; Dillhoff, M.; Farren, M.R.; et al. Circulating interleukin-6 is associated with disease progression, but not cachexia in pancreatic cancer. Pancreatology 2019, 19, 80–87. [Google Scholar] [CrossRef]
- Tsai, V.W.; Zhang, H.P.; Manandhar, R.; Schofield, P.; Christ, D.; Lee-Ng, K.K.M.; Lebhar, H.; Marquis, C.P.; Husaini, Y.; Brown, D.A.; et al. GDF15 mediates adiposity resistance through actions on GFRAL neurons in the hindbrain AP/NTS. Int. J. Obes. 2019, 43, 2370–2380. [Google Scholar] [CrossRef]
- Mullican, S.E.; Lin-Schmidt, X.; Chin, C.N.; Chavez, J.A.; Furman, J.L.; Armstrong, A.A.; Beck, S.C.; South, V.J.; Dinh, T.Q.; Cash-Mason, T.D.; et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med. 2017, 23, 1150–1157. [Google Scholar] [CrossRef]
- Johnen, H.; Lin, S.; Kuffner, T.; Brown, D.A.; Tsai, V.W.; Bauskin, A.R.; Wu, L.; Pankhurst, G.; Jiang, L.; Junankar, S.; et al. Tumor-induced anorexia and weight loss are mediated by the TGF-beta superfamily cytokine MIC-1. Nat. Med. 2007, 13, 1333–1340. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Tian, H.; Qi, J.; Li, M.; Fu, C.; Wu, F.; Wang, Y.; Cheng, D.; Zhao, W.; et al. Macrophage inhibitory cytokine 1 (MIC-1/GDF15) as a novel diagnostic serum biomarker in pancreatic ductal adenocarcinoma. BMC Cancer 2014, 14, 578. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.B.; Kleinert, M.; Richter, E.A.; Clemmensen, C. GDF15 in Appetite and Exercise: Essential Player or Coincidental Bystander? Endocrinology 2022, 163, bqab242. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, S.-Y.; Luan, Y.; Dong, R.; Abazarikia, A.; Kim, S.-Y. Adipose Tissue Wasting as a Determinant of Pancreatic Cancer-Related Cachexia. Cancers 2022, 14, 4754. https://doi.org/10.3390/cancers14194754
Yu S-Y, Luan Y, Dong R, Abazarikia A, Kim S-Y. Adipose Tissue Wasting as a Determinant of Pancreatic Cancer-Related Cachexia. Cancers. 2022; 14(19):4754. https://doi.org/10.3390/cancers14194754
Chicago/Turabian StyleYu, Seok-Yeong, Yi Luan, Rosemary Dong, Amirhossein Abazarikia, and So-Youn Kim. 2022. "Adipose Tissue Wasting as a Determinant of Pancreatic Cancer-Related Cachexia" Cancers 14, no. 19: 4754. https://doi.org/10.3390/cancers14194754
APA StyleYu, S. -Y., Luan, Y., Dong, R., Abazarikia, A., & Kim, S. -Y. (2022). Adipose Tissue Wasting as a Determinant of Pancreatic Cancer-Related Cachexia. Cancers, 14(19), 4754. https://doi.org/10.3390/cancers14194754