
New Insights into the Role of Insulin and Hypothalamic-Pituitary-Adrenal (HPA) Axis in the Metabolic Syndrome


Abstract
:1. Introduction
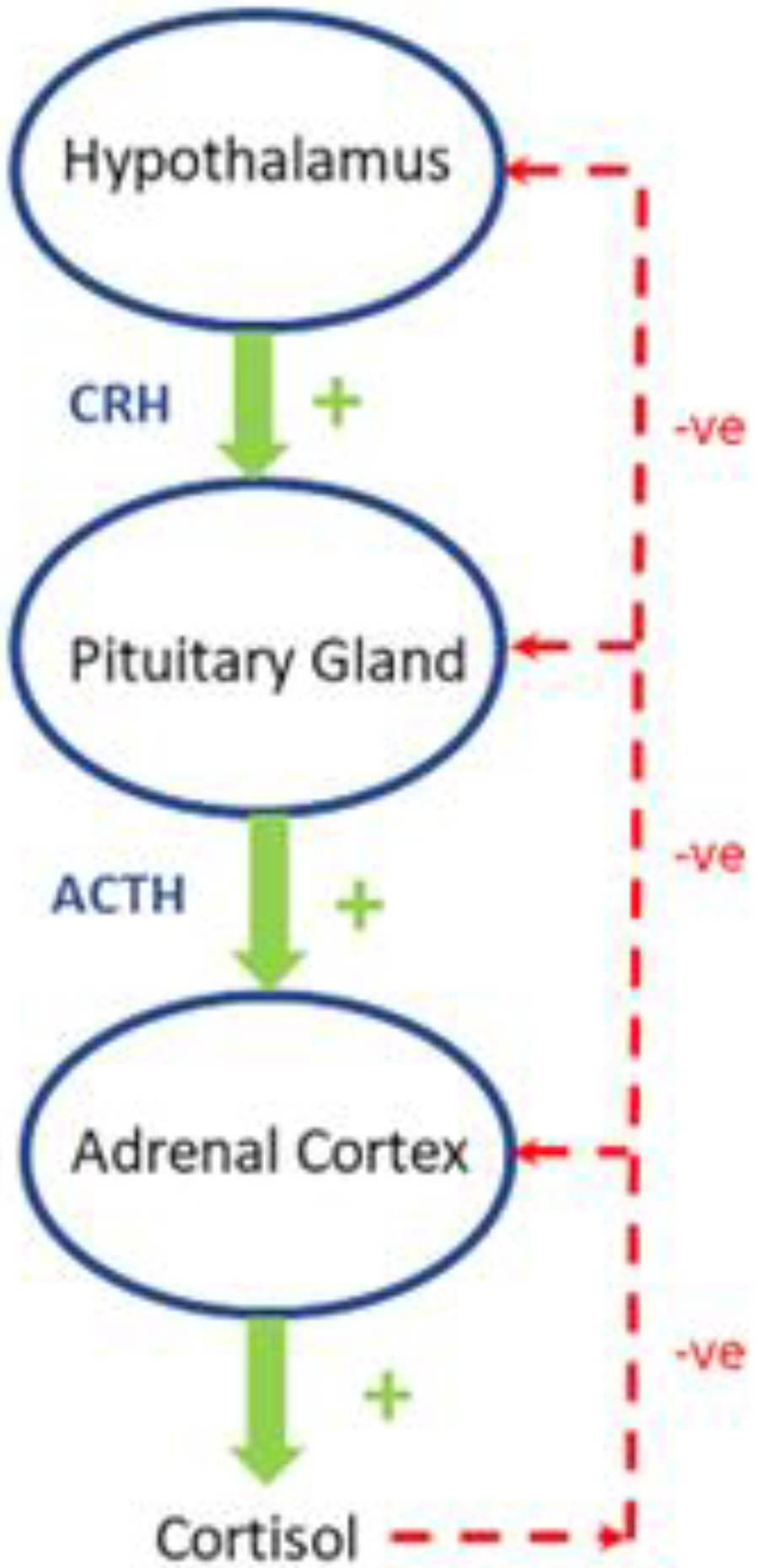
2. Regulation of the Hypothalamic-Pituitary-Adrenal (HPA)-Axis in Healthy Subjects
3. How Does Cortisol Affect Intermediary Metabolism?
4. How Does Insulin Affect Intermediary Metabolism?
5. The Bidirectional Interactions between Insulin and the HPA Axis
6. Effects of Glucocorticoids on Insulin Secretion
7. Glucocorticoids, Insulin Resistance and Metabolism
8. The Biologically Defended Level of Glycemia in Health and Disease
9. Hyperinsulinemia Induces a State of “Functional Hypercortisolism”
10. Evidence for Insulin Regulation of the HPA Activity
11. HPA Activity in Obesity and the Metabolic Syndrome
12. Direct Effects of Insulin on the Adrenals
13. Effects of Insulin on Local Cortisol Metabolism
14. Can Hyperinsulinemia-Induced Activation of the HPA Axis Be Modified?
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reaven, G. Insulin resistance, type 2 diabetes mellitus, and cardiovascular disease: The end of the beginning. Circulation 2005, 112, 3030–3032. [Google Scholar] [CrossRef] [PubMed]
- Shanik, M.H.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin resistance and hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31 (Suppl. 2), S262–S268. [Google Scholar] [CrossRef] [Green Version]
- Corkey, B.E. Diabetes: Have we got it all wrong? Insulin hypersecretion and food additives: Cause of obesity and diabetes? Diabetes Care 2012, 35, 2432–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzadeh, Z.; Faber, C.L.; Schwartz, M.W. Central Nervous System Control of Glucose Homeostasis: A Therapeutic Target for Type 2 Diabetes? Annu. Rev. Pharmacol. Toxicol. 2022, 62, 55–84. [Google Scholar] [CrossRef] [PubMed]
- Trico, D.; Natali, A.; Arslanian, S.; Mari, A.; Ferrannini, E. Identification, pathophysiology, and clinical implications of primary insulin hypersecretion in nondiabetic adults and adolescents. JCI Insight 2018, 3, e124912. [Google Scholar] [CrossRef] [Green Version]
- Brons, C.; Jensen, C.B.; Storgaard, H.; Hiscock, N.J.; White, A.; Appel, J.S.; Jacobsen, S.; Nilsson, E.; Larsen, C.M.; Astrup, A.; et al. Impact of short-term high-fat feeding on glucose and insulin metabolism in young healthy men. J. Physiol. 2009, 587, 2387–2397. [Google Scholar] [CrossRef]
- Ferrannini, E.; Natali, A.; Bell, P.; Cavallo-Perin, P.; Lalic, N.; Mingrone, G. Insulin resistance and hypersecretion in obesity. European Group for the Study of Insulin Resistance (EGIR). J. Clin. Investig. 1997, 100, 1166–1173. [Google Scholar] [CrossRef]
- Loves, S.; van Groningen, L.; Filius, M.; Mekking, M.; Brandon, T.; Tack, C.J.; Hermus, A.; de Boer, H. High-Dose, Diazoxide-Mediated Insulin Suppression Boosts Weight Loss Induced by Lifestyle Intervention. J. Clin. Endocrinol. Metab. 2018, 103, 4014–4022. [Google Scholar] [CrossRef] [Green Version]
- Dankner, R.; Chetrit, A.; Shanik, M.H.; Raz, I.; Roth, J. Basal-state hyperinsulinemia in healthy normoglycemic adults is predictive of type 2 diabetes over a 24-year follow-up: A preliminary report. Diabetes Care 2009, 32, 1464–1466. [Google Scholar] [CrossRef] [Green Version]
- Rizza, R.A.; Mandarino, L.J.; Genest, J.; Baker, B.A.; Gerich, J.E. Production of insulin resistance by hyperinsulinaemia in man. Diabetologia 1985, 28, 70–75. [Google Scholar] [CrossRef]
- Corkey, B.E. Banting lecture 2011: Hyperinsulinemia: Cause or consequence? Diabetes 2012, 61, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, J. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. Int. J. Mol. Sci. 2021, 22, 7797. [Google Scholar] [CrossRef] [PubMed]
- Nolan, C.J.; Ruderman, N.B.; Kahn, S.E.; Pedersen, O.; Prentki, M. Insulin resistance as a physiological defense against metabolic stress: Implications for the management of subsets of type 2 diabetes. Diabetes 2015, 64, 673–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schofield, C.J.; Sutherland, C. Disordered insulin secretion in the development of insulin resistance and Type 2 diabetes. Diabet. Med. 2012, 29, 972–979. [Google Scholar] [CrossRef]
- Abraham, S.B.; Rubino, D.; Sinaii, N.; Ramsey, S.; Nieman, L.K. Cortisol, obesity, and the metabolic syndrome: A cross-sectional study of obese subjects and review of the literature. Obesity 2013, 21, E105–E117. [Google Scholar] [CrossRef] [Green Version]
- Rosmond, R. Role of stress in the pathogenesis of the metabolic syndrome. Psychoneuroendocrinology 2005, 30, 1–10. [Google Scholar] [CrossRef]
- Anagnostis, P.; Athyros, V.G.; Tziomalos, K.; Karagiannis, A.; Mikhailidis, D.P. Clinical review: The pathogenetic role of cortisol in the metabolic syndrome: A hypothesis. J. Clin. Endocrinol. Metab. 2009, 94, 2692–2701. [Google Scholar] [CrossRef] [Green Version]
- Stulnig, T.M.; Waldhausl, W. 11beta-Hydroxysteroid dehydrogenase Type 1 in obesity and Type 2 diabetes. Diabetologia 2004, 47, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Duclos, M.; Marquez Pereira, P.; Barat, P.; Gatta, B.; Roger, P. Increased cortisol bioavailability, abdominal obesity, and the metabolic syndrome in obese women. Obes. Res. 2005, 13, 1157–1166. [Google Scholar] [CrossRef]
- Iida, S.; Nakamura, Y.; Fujii, H.; Nishimura, J.; Tsugawa, M.; Gomi, M.; Fukata, J.; Tarui, S.; Moriwaki, K.; Kitani, T. A patient with hypocortisolism and Cushing’s syndrome-like manifestations: Cortisol hyperreactive syndrome. J. Clin. Endocrinol. Metab. 1990, 70, 729–737. [Google Scholar] [CrossRef]
- Friedman, T.C.; Mastorakos, G.; Newman, T.D.; Mullen, N.M.; Horton, E.G.; Costello, R.; Papadopoulos, N.M.; Chrousos, G.P. Carbohydrate and lipid metabolism in endogenous hypercortisolism: Shared features with metabolic syndrome X and NIDDM. Endocr. J. 1996, 43, 645–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaupere, C.; Liboz, A.; Feve, B.; Blondeau, B.; Guillemain, G. Molecular Mechanisms of Glucocorticoid-Induced Insulin Resistance. Int. J. Mol. Sci. 2021, 22, 623. [Google Scholar] [CrossRef] [PubMed]
- Scaroni, C.; Zilio, M.; Foti, M.; Boscaro, M. Glucose Metabolism Abnormalities in Cushing Syndrome: From Molecular Basis to Clinical Management. Endocr. Rev. 2017, 38, 189–219. [Google Scholar] [CrossRef]
- Traish, A.M.; Guay, A.T.; Zitzmann, M. 5alpha-Reductase inhibitors alter steroid metabolism and may contribute to insulin resistance, diabetes, metabolic syndrome and vascular disease: A medical hypothesis. Horm. Mol. Biol. Clin. Investig. 2014, 20, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.M.; Krozowski, Z.S. 11 beta-Hydroxysteroid dehydrogenase. Vitam. Horm. 1999, 57, 249–324. [Google Scholar]
- Tomlinson, J.W.; Stewart, P.M. Cortisol metabolism and the role of 11beta-hydroxysteroid dehydrogenase. Best Pract. Res. Clin. Endocrinol. Metab. 2001, 15, 61–78. [Google Scholar] [CrossRef]
- Napolitano, A.; Voice, M.W.; Edwards, C.R.; Seckl, J.R.; Chapman, K.E. 11Beta-hydroxysteroid dehydrogenase 1 in adipocytes: Expression is differentiation-dependent and hormonally regulated. J. Steroid Biochem. Mol. Biol. 1998, 64, 251–260. [Google Scholar] [CrossRef]
- Janssen, J.; Lamberts, S. Diabetes associated with glucocorticoid excess. In Diabetes Secondary to Endocrine and Pancreatic Disorders; Ghigo, E., Porta, M., Eds.; Karger: Basel, Switzerland, 2014; pp. 22–33. [Google Scholar]
- Dallman, M.F.; Akana, S.F.; Strack, A.M.; Hanson, E.S.; Sebastian, R.J. The neural network that regulates energy balance is responsive to glucocorticoids and insulin and also regulates HPA axis responsivity at a site proximal to CRF neurons. Ann. N. Y. Acad. Sci. 1995, 771, 730–742. [Google Scholar] [CrossRef]
- Goodman, H.M. Basic Medical Endocrinology, 4th ed.; Elsevier: San Diego, CA, USA, 2009. [Google Scholar]
- Arlt, W.; Stewart, P.M. Adrenal corticosteroid biosynthesis, metabolism, and action. Endocrinol. Metab. Clin. N. Am. 2005, 34, 293–313. [Google Scholar] [CrossRef]
- Christiansen, J.J.; Djurhuus, C.B.; Gravholt, C.H.; Iversen, P.; Christiansen, J.S.; Schmitz, O.; Weeke, J.; Jorgensen, J.O.; Moller, N. Effects of cortisol on carbohydrate, lipid, and protein metabolism: Studies of acute cortisol withdrawal in adrenocortical failure. J. Clin. Endocrinol. Metab. 2007, 92, 3553–3559. [Google Scholar] [CrossRef] [Green Version]
- Geer, E.B.; Islam, J.; Buettner, C. Mechanisms of glucocorticoid-induced insulin resistance: Focus on adipose tissue function and lipid metabolism. Endocrinol. Metab. Clin. N. Am. 2014, 43, 75–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cauter, E.; Blackman, J.D.; Roland, D.; Spire, J.P.; Refetoff, S.; Polonsky, K.S. Modulation of glucose regulation and insulin secretion by circadian rhythmicity and sleep. J. Clin. Investig. 1991, 88, 934–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, R.C.; Walker, B.R. Glucocorticoids and insulin resistance: Old hormones, new targets. Clin. Sci. 1999, 96, 513–523. [Google Scholar] [CrossRef]
- Leal, A.M.; Moreira, A.C. Food and the circadian activity of the hypothalamic-pituitary-adrenal axis. Braz. J. Med. Biol. Res. 1997, 30, 1391–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tataranni, P.A.; Larson, D.E.; Snitker, S.; Young, J.B.; Flatt, J.P.; Ravussin, E. Effects of glucocorticoids on energy metabolism and food intake in humans. Am. J. Physiol. 1996, 271, E317–E325. [Google Scholar] [CrossRef]
- Burt, M.G.; Gibney, J.; Ho, K.K. Characterization of the metabolic phenotypes of Cushing’s syndrome and growth hormone deficiency: A study of body composition and energy metabolism. Clin. Endocrinol. 2006, 64, 436–443. [Google Scholar] [CrossRef]
- Torres, S.J.; Nowson, C.A. Relationship between stress, eating behavior, and obesity. Nutrition 2007, 23, 887–894. [Google Scholar] [CrossRef] [Green Version]
- Norton, L.; Shannon, C.; Gastaldelli, A.; DeFronzo, R.A. Insulin: The master regulator of glucose metabolism. Metabolism 2022, 129, 155142. [Google Scholar] [CrossRef]
- Hatting, M.; Tavares, C.D.J.; Sharabi, K.; Rines, A.K.; Puigserver, P. Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci. 2018, 1411, 21–35. [Google Scholar] [CrossRef]
- Rahman, M.S.; Hossain, K.S.; Das, S.; Kundu, S.; Adegoke, E.O.; Rahman, M.A.; Hannan, M.A.; Uddin, M.J.; Pang, M.G. Role of Insulin in Health and Disease: An Update. Int. J. Mol. Sci. 2021, 22, 6403. [Google Scholar] [CrossRef]
- Ganong, W.F. Circumventricular organs: Definition and role in the regulation of endocrine and autonomic function. Clin. Exp. Pharmacol. Physiol. 2000, 27, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Baura, G.D.; Foster, D.M.; Porte, D., Jr.; Kahn, S.E.; Bergman, R.N.; Cobelli, C.; Schwartz, M.W. Saturable transport of insulin from plasma into the central nervous system of dogs In Vivo. A mechanism for regulated insulin delivery to the brain. J. Clin. Investig. 1993, 92, 1824–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baura, G.D.; Foster, D.M.; Kaiyala, K.; Porte, D., Jr.; Kahn, S.E.; Schwartz, M.W. Insulin transport from plasma into the central nervous system is inhibited by dexamethasone in dogs. Diabetes 1996, 45, 86–90. [Google Scholar] [CrossRef]
- Gray, S.M.; Meijer, R.I.; Barrett, E.J. Insulin regulates brain function, but how does it get there? Diabetes 2014, 63, 3992–3997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselbalch, S.G.; Knudsen, G.M.; Videbaek, C.; Pinborg, L.H.; Schmidt, J.F.; Holm, S.; Paulson, O.B. No effect of insulin on glucose blood-brain barrier transport and cerebral metabolism in humans. Diabetes 1999, 48, 1915–1921. [Google Scholar] [CrossRef]
- Seaquist, E.R.; Damberg, G.S.; Tkac, I.; Gruetter, R. The effect of insulin on In Vivo cerebral glucose concentrations and rates of glucose transport/metabolism in humans. Diabetes 2001, 50, 2203–2209. [Google Scholar] [CrossRef] [Green Version]
- McEwen, B.S.; Reagan, L.P. Glucose transporter expression in the central nervous system: Relationship to synaptic function. Eur. J. Pharmacol. 2004, 490, 13–24. [Google Scholar] [CrossRef]
- Porte, D., Jr.; Baskin, D.G.; Schwartz, M.W. Insulin signaling in the central nervous system: A critical role in metabolic homeostasis and disease from C. elegans to humans. Diabetes 2005, 54, 1264–1276. [Google Scholar] [CrossRef] [Green Version]
- Obici, S.; Zhang, B.B.; Karkanias, G.; Rossetti, L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat. Med. 2002, 8, 1376–1382. [Google Scholar] [CrossRef]
- Inoue, H.; Ogawa, W.; Asakawa, A.; Okamoto, Y.; Nishizawa, A.; Matsumoto, M.; Teshigawara, K.; Matsuki, Y.; Watanabe, E.; Hiramatsu, R.; et al. Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab. 2006, 3, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Inoue, H.; Ogawa, W.; Ozaki, M.; Haga, S.; Matsumoto, M.; Furukawa, K.; Hashimoto, N.; Kido, Y.; Mori, T.; Sakaue, H.; et al. Role of STAT-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism In Vivo. Nat. Med. 2004, 10, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, C.; Lyall, H.; Gould, G.W. Hypothalamic GLUT 4 expression: A glucose- and insulin-sensing mechanism? Mol. Cell Endocrinol. 1995, 107, 67–70. [Google Scholar] [CrossRef]
- Lucignani, G.; Namba, H.; Nehlig, A.; Porrino, L.J.; Kennedy, C.; Sokoloff, L. Effects of insulin on local cerebral glucose utilization in the rat. J. Cereb. Blood Flow Metab. 1987, 7, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strack, A.M.; Sebastian, R.J.; Schwartz, M.W.; Dallman, M.F. Glucocorticoids and insulin: Reciprocal signals for energy balance. Am. J. Physiol. 1995, 268, R142–R149. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. The source of cerebral insulin. Eur. J. Pharmacol. 2004, 490, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Kamba, A.; Daimon, M.; Murakami, H.; Otaka, H.; Matsuki, K.; Sato, E.; Tanabe, J.; Takayasu, S.; Matsuhashi, Y.; Yanagimachi, M.; et al. Association between Higher Serum Cortisol Levels and Decreased Insulin Secretion in a General Population. PLoS ONE 2016, 11, e0166077. [Google Scholar] [CrossRef] [Green Version]
- Mazziotti, G.; Gazzaruso, C.; Giustina, A. Diabetes in Cushing syndrome: Basic and clinical aspects. Trends Endocrinol. Metab. 2011, 22, 499–506. [Google Scholar] [CrossRef]
- Lambillotte, C.; Gilon, P.; Henquin, J.C. Direct glucocorticoid inhibition of insulin secretion. An in vitro study of dexamethasone effects in mouse islets. J. Clin. Investig. 1997, 99, 414–423. [Google Scholar] [CrossRef] [Green Version]
- van Raalte, D.H.; Ouwens, D.M.; Diamant, M. Novel insights into glucocorticoid-mediated diabetogenic effects: Towards expansion of therapeutic options? Eur. J. Clin. Investig. 2009, 39, 81–93. [Google Scholar] [CrossRef]
- Hansen, K.B.; Vilsboll, T.; Bagger, J.I.; Holst, J.J.; Knop, F.K. Reduced glucose tolerance and insulin resistance induced by steroid treatment, relative physical inactivity, and high-calorie diet impairs the incretin effect in healthy subjects. J. Clin. Endocrinol. Metab. 2010, 95, 3309–3317. [Google Scholar] [CrossRef] [Green Version]
- Olefsky, J.M. Effect of dexamethasone on insulin binding, glucose transport, and glucose oxidation of isolated rat adipocytes. J. Clin. Investig. 1975, 56, 1499–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horner, H.C.; Munck, A.; Lienhard, G.E. Dexamethasone causes translocation of glucose transporters from the plasma membrane to an intracellular site in human fibroblasts. J. Biol. Chem. 1987, 262, 17696–17702. [Google Scholar] [CrossRef]
- Kahn, C.R.; Goldfine, I.D.; Neville, D.M., Jr.; De Meyts, P. Alterations in insulin binding induced by changes In Vivo in the levels of glucocorticoids and growth hormone. Endocrinology 1978, 103, 1054–1066. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Hines, E., 3rd; Kitabchi, A.E. Hypercortisolism and insulin resistance: Comparative effects of prednisone, hydrocortisone, and dexamethasone on insulin binding of human erythrocytes. J. Clin. Endocrinol. Metab. 1982, 55, 910–915. [Google Scholar] [CrossRef]
- Pivonello, R.; De Leo, M.; Vitale, P.; Cozzolino, A.; Simeoli, C.; De Martino, M.C.; Lombardi, G.; Colao, A. Pathophysiology of diabetes mellitus in Cushing’s syndrome. Neuroendocrinology 2010, 92 (Suppl. 1), 77–81. [Google Scholar] [CrossRef]
- Qi, D.; Rodrigues, B. Glucocorticoids produce whole body insulin resistance with changes in cardiac metabolism. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E654–E667. [Google Scholar] [CrossRef] [Green Version]
- Kola, B.; Christ-Crain, M.; Lolli, F.; Arnaldi, G.; Giacchetti, G.; Boscaro, M.; Grossman, A.B.; Korbonits, M. Changes in adenosine 5′-monophosphate-activated protein kinase as a mechanism of visceral obesity in Cushing’s syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 4969–4973. [Google Scholar] [CrossRef] [Green Version]
- Hauner, H.; Schmid, P.; Pfeiffer, E.F. Glucocorticoids and insulin promote the differentiation of human adipocyte precursor cells into fat cells. J. Clin. Endocrinol. Metab. 1987, 64, 832–835. [Google Scholar] [CrossRef]
- Santiago, J.V.; Clarke, W.L.; Shah, S.D.; Cryer, P.E. Epinephrine, norepinephrine, glucagon, and growth hormone release in association with physiological decrements in the plasma glucose concentration in normal and diabetic man. J. Clin. Endocrinol. Metab. 1980, 51, 877–883. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Hendler, R.; Christensen, N. Stimulation of counterregulatory hormonal responses in diabetic man by a fall in glucose concentration. Diabetes 1980, 29, 125–131. [Google Scholar] [CrossRef]
- Spyer, G.; Hattersley, A.T.; MacDonald, I.A.; Amiel, S.; MacLeod, K.M. Hypoglycaemic counter-regulation at normal blood glucose concentrations in patients with well controlled type-2 diabetes. Lancet 2000, 356, 1970–1974. [Google Scholar] [CrossRef]
- Chakera, A.J.; Hurst, P.S.; Spyer, G.; Ogunnowo-Bada, E.O.; Marsh, W.J.; Riches, C.H.; Yueh, C.Y.; Markkula, S.P.; Dalley, J.W.; Cox, R.D.; et al. Molecular reductions in glucokinase activity increase counter-regulatory responses to hypoglycemia in mice and humans with diabetes. Mol. Metab. 2018, 17, 17–27. [Google Scholar] [CrossRef]
- Dirlewanger, M.; Schneiter, P.H.; Paquot, N.; Jequier, E.; Rey, V.; Tappy, L. Effects of glucocorticoids on hepatic sensitivity to insulin and glucagon in man. Clin. Nutr. 2000, 19, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Shi, Z.Q.; Niwa, M.; Mathoo, J.M.; Vandelangeryt, M.L.; Bilinski, D.; Lewis, G.F.; Vranic, M. Beta-blockade, but not normoglycemia or hyperinsulinemia, markedly diminishes stress-induced hyperglycemia in diabetic dogs. Diabetes 2000, 49, 253–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherwin, R.S.; Shamoon, H.; Hendler, R.; Sacca, L.; Eigler, N.; Walesky, M. Epinephrine and the regulation of glucose metabolism: Effect of diabetes and hormonal interactions. Metabolism 1980, 29, 1146–1154. [Google Scholar] [CrossRef]
- Chan, O.; Inouye, K.; Akirav, E.; Park, E.; Riddell, M.C.; Vranic, M.; Matthews, S.G. Insulin alone increases hypothalamo-pituitary-adrenal activity, and diabetes lowers peak stress responses. Endocrinology 2005, 146, 1382–1390. [Google Scholar] [CrossRef]
- Grunstein, H.S.; James, D.E.; Storlien, L.H.; Smythe, G.A.; Kraegen, E.W. Hyperinsulinemia suppresses glucose utilization in specific brain regions: In Vivo studies using the euglycemic clamp in the rat. Endocrinology 1985, 116, 604–610. [Google Scholar] [CrossRef]
- Fruehwald-Schultes, B.; Kern, W.; Bong, W.; Wellhoener, P.; Kerner, W.; Born, J.; Fehm, H.L.; Peters, A. Supraphysiological hyperinsulinemia acutely increases hypothalamic-pituitary-adrenal secretory activity in humans. J. Clin. Endocrinol. Metab. 1999, 84, 3041–3046. [Google Scholar] [CrossRef]
- Jacobson, L.; Sapolsky, R. The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocr. Rev. 1991, 12, 118–134. [Google Scholar] [CrossRef]
- Palovcik, R.A.; Phillips, M.I.; Kappy, M.S.; Raizada, M.K. Insulin inhibits pyramidal neurons in hippocampal slices. Brain Res. 1984, 309, 187–191. [Google Scholar] [CrossRef]
- Asensio, C.; Muzzin, P.; Rohner-Jeanrenaud, F. Role of glucocorticoids in the physiopathology of excessive fat deposition and insulin resistance. Int. J. Obes. Relat. Metab. Disord. 2004, 28 (Suppl. 4), S45–S52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljung, T.; Andersson, B.; Bengtsson, B.A.; Bjorntorp, P.; Marin, P. Inhibition of cortisol secretion by dexamethasone in relation to body fat distribution: A dose-response study. Obes. Res. 1996, 4, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Rosmond, R.; Dallman, M.F.; Bjorntorp, P. Stress-related cortisol secretion in men: Relationships with abdominal obesity and endocrine, metabolic and hemodynamic abnormalities. J. Clin. Endocrinol. Metab. 1998, 83, 1853–1859. [Google Scholar] [CrossRef]
- Hales, C.N.; Barker, D.J.; Clark, P.M.; Cox, L.J.; Fall, C.; Osmond, C.; Winter, P.D. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 1991, 303, 1019–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, P.M.; Hindmarsh, P.C.; Shiell, A.W.; Law, C.M.; Honour, J.W.; Barker, D.J. Size at birth and adrenocortical function in childhood. Clin. Endocrinol. 1996, 45, 721–726. [Google Scholar] [CrossRef]
- Phillips, D.I.; Barker, D.J.; Fall, C.H.; Seckl, J.R.; Whorwood, C.B.; Wood, P.J.; Walker, B.R. Elevated plasma cortisol concentrations: A link between low birth weight and the insulin resistance syndrome? J. Clin. Endocrinol. Metab. 1998, 83, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Werdermann, M.; Berger, I.; Scriba, L.D.; Santambrogio, A.; Schlinkert, P.; Brendel, H.; Morawietz, H.; Schedl, A.; Peitzsch, M.; King, A.J.F.; et al. Insulin and obesity transform hypothalamic-pituitary-adrenal axis stemness and function in a hyperactive state. Mol. Metab. 2021, 43, 101112. [Google Scholar] [CrossRef]
- Andrew, R.; Phillips, D.I.; Walker, B.R. Obesity and gender influence cortisol secretion and metabolism in man. J. Clin. Endocrinol. Metab. 1998, 83, 1806–1809. [Google Scholar] [CrossRef]
- Stewart, P.M.; Boulton, A.; Kumar, S.; Clark, P.M.; Shackleton, C.H. Cortisol metabolism in human obesity: Impaired cortisone → cortisol conversion in subjects with central adiposity. J. Clin. Endocrinol. Metab. 1999, 84, 1022–1027. [Google Scholar] [CrossRef]
- Pasquali, R.; Vicennati, V.; Cacciari, M.; Pagotto, U. The hypothalamic-pituitary-adrenal axis activity in obesity and the metabolic syndrome. Ann. N. Y. Acad. Sci. 2006, 1083, 111–128. [Google Scholar] [CrossRef]
- Vicennati, V.; Pasquali, R. Abnormalities of the hypothalamic-pituitary-adrenal axis in nondepressed women with abdominal obesity and relations with insulin resistance: Evidence for a central and a peripheral alteration. J. Clin. Endocrinol. Metab. 2000, 85, 4093–4098. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, R.; Gagliardi, L.; Vicennati, V.; Gambineri, A.; Colitta, D.; Ceroni, L.; Casimirri, F. ACTH and cortisol response to combined corticotropin releasing hormone-arginine vasopressin stimulation in obese males and its relationship to body weight, fat distribution and parameters of the metabolic syndrome. Int. J. Obes. Relat. Metab. Disord. 1999, 23, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Holt, H.B.; Wild, S.H.; Postle, A.D.; Zhang, J.; Koster, G.; Umpleby, M.; Shojaee-Moradie, F.; Dewbury, K.; Wood, P.J.; Phillips, D.I.; et al. Cortisol clearance and associations with insulin sensitivity, body fat and fatty liver in middle-aged men. Diabetologia 2007, 50, 1024–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lottenberg, S.A.; Giannella-Neto, D.; Derendorf, H.; Rocha, M.; Bosco, A.; Carvalho, S.V.; Moretti, A.E.; Lerario, A.C.; Wajchenberg, B.L. Effect of fat distribution on the pharmacokinetics of cortisol in obesity. Int. J. Clin. Pharmacol. Ther. 1998, 36, 501–505. [Google Scholar] [PubMed]
- Fernandez-Real, J.M.; Grasa, M.; Casamitjana, R.; Pugeat, M.; Barret, C.; Ricart, W. Plasma total and glycosylated corticosteroid-binding globulin levels are associated with insulin secretion. J. Clin. Endocrinol. Metab. 1999, 84, 3192–3196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manco, M.; Fernandez-Real, J.M.; Valera-Mora, M.E.; Dechaud, H.; Nanni, G.; Tondolo, V.; Calvani, M.; Castagneto, M.; Pugeat, M.; Mingrone, G. Massive weight loss decreases corticosteroid-binding globulin levels and increases free cortisol in healthy obese patients: An adaptive phenomenon? Diabetes Care 2007, 30, 1494–1500. [Google Scholar] [CrossRef] [Green Version]
- Penhoat, A.; Chatelain, P.G.; Jaillard, C.; Saez, J.M. Characterization of insulin-like growth factor I and insulin receptors on cultured bovine adrenal fasciculata cells. Role of these peptides on adrenal cell function. Endocrinology 1988, 122, 2518–2526. [Google Scholar] [CrossRef]
- Kinyua, A.W.; Doan, K.V.; Yang, D.J.; Huynh, M.K.Q.; Choi, Y.H.; Shin, D.M.; Kim, K.W. Insulin Regulates Adrenal Steroidogenesis by Stabilizing SF-1 Activity. Sci. Rep. 2018, 8, 5025. [Google Scholar] [CrossRef]
- Altieri, B.; Tirabassi, G.; Della Casa, S.; Ronchi, C.L.; Balercia, G.; Orio, F.; Pontecorvi, A.; Colao, A.; Muscogiuri, G. Adrenocortical tumors and insulin resistance: What is the first step? Int. J. Cancer 2016, 138, 2785–2794. [Google Scholar] [CrossRef] [Green Version]
- Tsatsoulis, A. The Role of Insulin Resistance/Hyperinsulinism on the Rising Trend of Thyroid and Adrenal Nodular Disease in the Current Environment. J. Clin. Med. 2018, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Golden, S.H.; Wand, G.S.; Malhotra, S.; Kamel, I.; Horton, K. Reliability of hypothalamic-pituitary-adrenal axis assessment methods for use in population-based studies. Eur. J. Epidemiol. 2011, 26, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Askani, E.; Rospleszcz, S.; Lorbeer, R.; Kulka, C.; von Kruchten, R.; Muller-Peltzer, K.; Hasic, D.; Kellner, E.; Reisert, M.; Rathmann, W.; et al. Association of MRI-based adrenal gland volume and impaired glucose metabolism in a population-based cohort study. Diabetes Metab. Res. Rev. 2022, 38, e3528. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.M.; Toogood, A.A.; Tomlinson, J.W. Growth hormone, insulin-like growth factor-I and the cortisol-cortisone shuttle. Horm. Res. 2001, 56 (Suppl. 1), 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Rabbitt, E.; Brady, T.; Brown, C.; Guest, P.; Bujalska, I.J.; Doig, C.; Newsome, P.N.; Hubscher, S.; Elias, E.; et al. A switch in hepatic cortisol metabolism across the spectrum of non alcoholic fatty liver disease. PLoS ONE 2012, 7, e29531. [Google Scholar] [CrossRef]
- Balachandran, A.; Guan, H.; Sellan, M.; van Uum, S.; Yang, K. Insulin and dexamethasone dynamically regulate adipocyte 11beta-hydroxysteroid dehydrogenase type 1. Endocrinology 2008, 149, 4069–4079. [Google Scholar] [CrossRef] [Green Version]
- Masuzaki, H.; Paterson, J.; Shinyama, H.; Morton, N.M.; Mullins, J.J.; Seckl, J.R.; Flier, J.S. A transgenic model of visceral obesity and the metabolic syndrome. Science 2001, 294, 2166–2170. [Google Scholar] [CrossRef] [Green Version]
- Morton, N.M.; Holmes, M.C.; Fievet, C.; Staels, B.; Tailleux, A.; Mullins, J.J.; Seckl, J.R. Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11beta-hydroxysteroid dehydrogenase type 1 null mice. J. Biol. Chem. 2001, 276, 41293–41300. [Google Scholar] [CrossRef] [Green Version]
- Bujalska, I.J.; Kumar, S.; Stewart, P.M. Does central obesity reflect “Cushing’s disease of the omentum”? Lancet 1997, 349, 1210–1213. [Google Scholar] [CrossRef]
- Tomlinson, J.W.; Walker, E.A.; Bujalska, I.J.; Draper, N.; Lavery, G.G.; Cooper, M.S.; Hewison, M.; Stewart, P.M. 11beta-hydroxysteroid dehydrogenase type 1: A tissue-specific regulator of glucocorticoid response. Endocr. Rev. 2004, 25, 831–866. [Google Scholar] [CrossRef]
- Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Armstrong, M.J.; Borrows, S.; Yu, J.; Wagenmakers, A.J.; Stewart, P.M.; Tomlinson, J.W. Glucocorticoids fail to cause insulin resistance in human subcutaneous adipose tissue In Vivo. J. Clin. Endocrinol. Metab. 2013, 98, 1631–1640. [Google Scholar] [CrossRef] [Green Version]
- Seimon, R.V.; Hostland, N.; Silveira, S.L.; Gibson, A.A.; Sainsbury, A. Effects of energy restriction on activity of the hypothalamo-pituitary-adrenal axis in obese humans and rodents: Implications for diet-induced changes in body composition. Horm. Mol. Biol. Clin. Investig. 2013, 15, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Parra, M.D.; Martinez de Morentin, B.E.; Alfredo Martinez, J. Impact of weight loss on cortisol secretion in obese men with and without metabolic syndrome features. Nutr. Metab. Cardiovasc. Dis. 2006, 16, 28–34. [Google Scholar] [CrossRef]
- Giovannini, C.; Ciucci, E.; Clementi, R.; Cugini, P.; Facchinetti, F.; Negri, M. Beta-endorphin, insulin, ACTH and cortisol plasma levels during oral glucose tolerance test in obesity after weight loss. Horm. Metab. Res. 1990, 22, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.T.; Keogh, J.B.; Bornstein, S.R.; Ehrhart-Bornstein, M.; Lewis, J.G.; Clifton, P.M.; Torpy, D.J. Moderate weight loss reduces renin and aldosterone but does not influence basal or stimulated pituitary-adrenal axis function. Horm. Metab. Res. 2007, 39, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Nonino-Borges, C.B.; Martins Borges, R.; Bavaresco, M.; Suen, V.M.; Moreira, A.C.; Marchini, J.S. Influence of meal time on salivary circadian cortisol rhythms and weight loss in obese women. Nutrition 2007, 23, 385–391. [Google Scholar] [CrossRef]
- Purnell, J.Q.; Kahn, S.E.; Samuels, M.H.; Brandon, D.; Loriaux, D.L.; Brunzell, J.D. Enhanced cortisol production rates, free cortisol, and 11beta-HSD-1 expression correlate with visceral fat and insulin resistance in men: Effect of weight loss. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E351–E357. [Google Scholar] [CrossRef]
- Kalra, S.; Gupta, Y.; Patil, S. Sodium-glucose cotransporter-2 inhibition and the insulin: Glucagon ratio: Unexplored dimensions. Indian J. Endocrinol. Metab. 2015, 19, 426–429. [Google Scholar] [CrossRef]
- Ferrannini, E. Sodium-Glucose Co-transporters and Their Inhibition: Clinical Physiology. Cell Metab. 2017, 26, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Schlogl, H.; Stumvoll, M. The Brains behind SGLT2 Inhibition. Diabetes Care 2022, 45, 273–275. [Google Scholar] [CrossRef]
- Higashikawa, T.; Ito, T.; Mizuno, T.; Ishigami, K.; Kuroki, K.; Maekawa, N.; Usuda, D.; Morita, T.; Hamada, K.; Takagi, S.; et al. Effects of tofogliflozin on adrenocorticotropic hormone, renin and aldosterone, and cortisol levels in elderly patients with diabetes mellitus: A retrospective study of a patient cohort. Medicine 2021, 100, e27638. [Google Scholar] [CrossRef]
- Shin, A.C.; Balasubramanian, P.; Suryadevara, P.; Zyskowski, J.; Herdt, T.H.; MohanKumar, S.M.J.; MohanKumar, P.S. Metformin effectively restores the HPA axis function in diet-induced obese rats. Int. J. Obes. 2021, 45, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Chung, J.Y.; Cho, S.K.; Shin, H.W.; Jang, I.J.; Park, J.W.; Yu, K.S.; Cho, J.Y. Antihyperglycemic mechanism of metformin occurs via the AMPK/LXRalpha/POMC pathway. Sci. Rep. 2015, 5, 8145. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Syndrome | Cushing Syndrome | |
|---|---|---|
| Plasma Insulin levels | ↑↑ | ↑ * or ↓ ** |
| insulin resistance | + | + |
| AbdominalObesity | + | + |
| impaired glucose tolerance | + | + |
| Hypertriglyceridemia | + | + |
| Hypertension | + | + |
| HPA Activity | ↑ | ↑ |
| Plasma Cortisol | ↓/N | ↑ |
| 24 h-Free Cortisoluria | ↑ | ↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janssen, J.A.M.J.L. New Insights into the Role of Insulin and Hypothalamic-Pituitary-Adrenal (HPA) Axis in the Metabolic Syndrome. Int. J. Mol. Sci. 2022, 23, 8178. https://doi.org/10.3390/ijms23158178
Janssen JAMJL. New Insights into the Role of Insulin and Hypothalamic-Pituitary-Adrenal (HPA) Axis in the Metabolic Syndrome. International Journal of Molecular Sciences. 2022; 23(15):8178. https://doi.org/10.3390/ijms23158178
Chicago/Turabian StyleJanssen, Joseph A. M. J. L. 2022. "New Insights into the Role of Insulin and Hypothalamic-Pituitary-Adrenal (HPA) Axis in the Metabolic Syndrome" International Journal of Molecular Sciences 23, no. 15: 8178. https://doi.org/10.3390/ijms23158178
APA StyleJanssen, J. A. M. J. L. (2022). New Insights into the Role of Insulin and Hypothalamic-Pituitary-Adrenal (HPA) Axis in the Metabolic Syndrome. International Journal of Molecular Sciences, 23(15), 8178. https://doi.org/10.3390/ijms23158178