
Site-Specific Antibody Conjugation with Payloads beyond Cytotoxins


Abstract
:1. Introduction
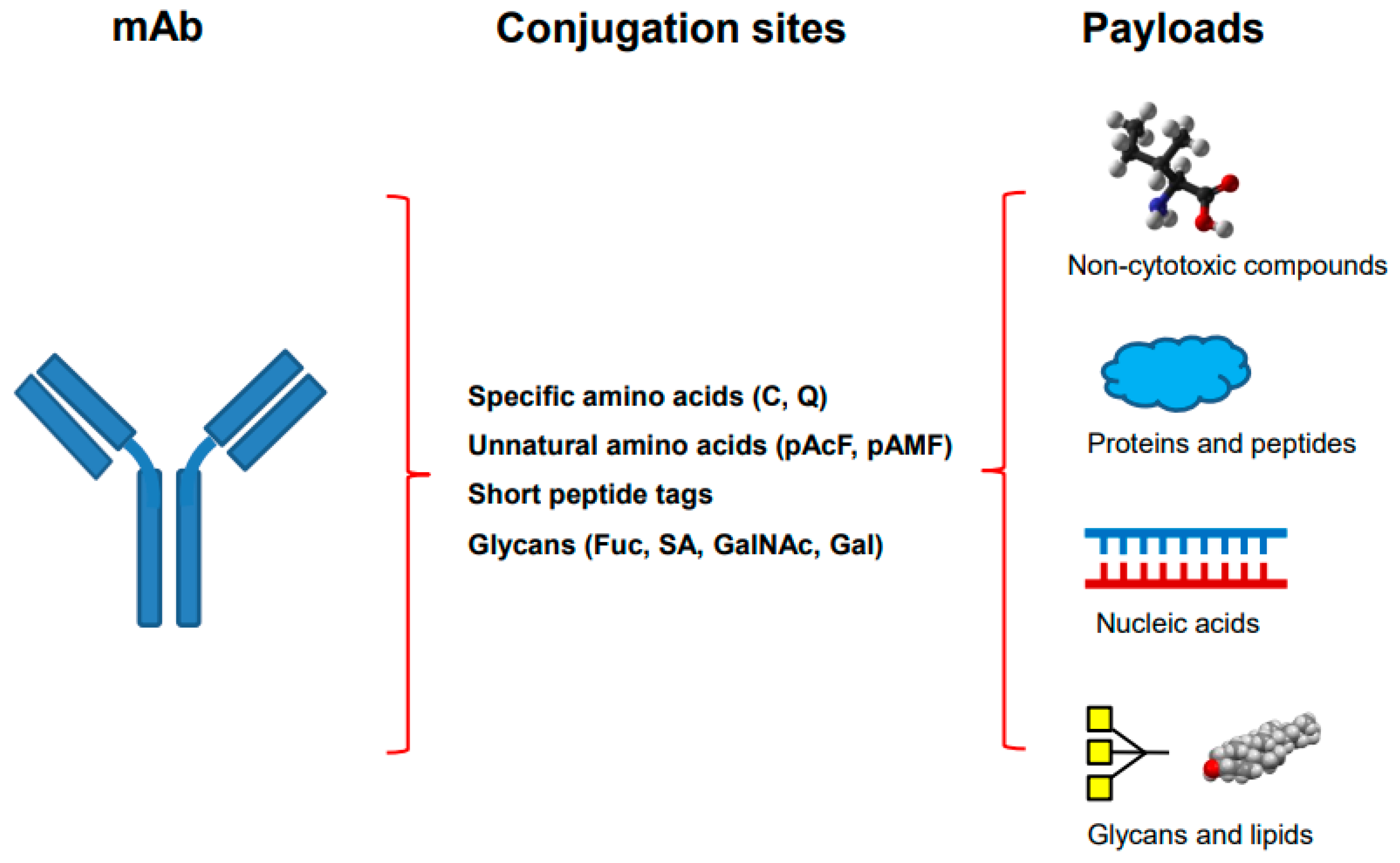
2. Overview of Site-Specific Antibody Conjugation
3. Non-Cytotoxic Compounds as Payloads
3.1. PEG
3.2. Antibiotics
3.3. Immune-Modulating Compounds
3.4. Protein Degraders: PROTAC
3.5. Protein Degraders: LYTAC
3.6. Ligand for Proteins Overexpressed in Cancer
4. Proteins or Peptides as Payloads
4.1. Proteins
4.2. Antibody Fragments
4.3. Peptides or Cyclic Peptides
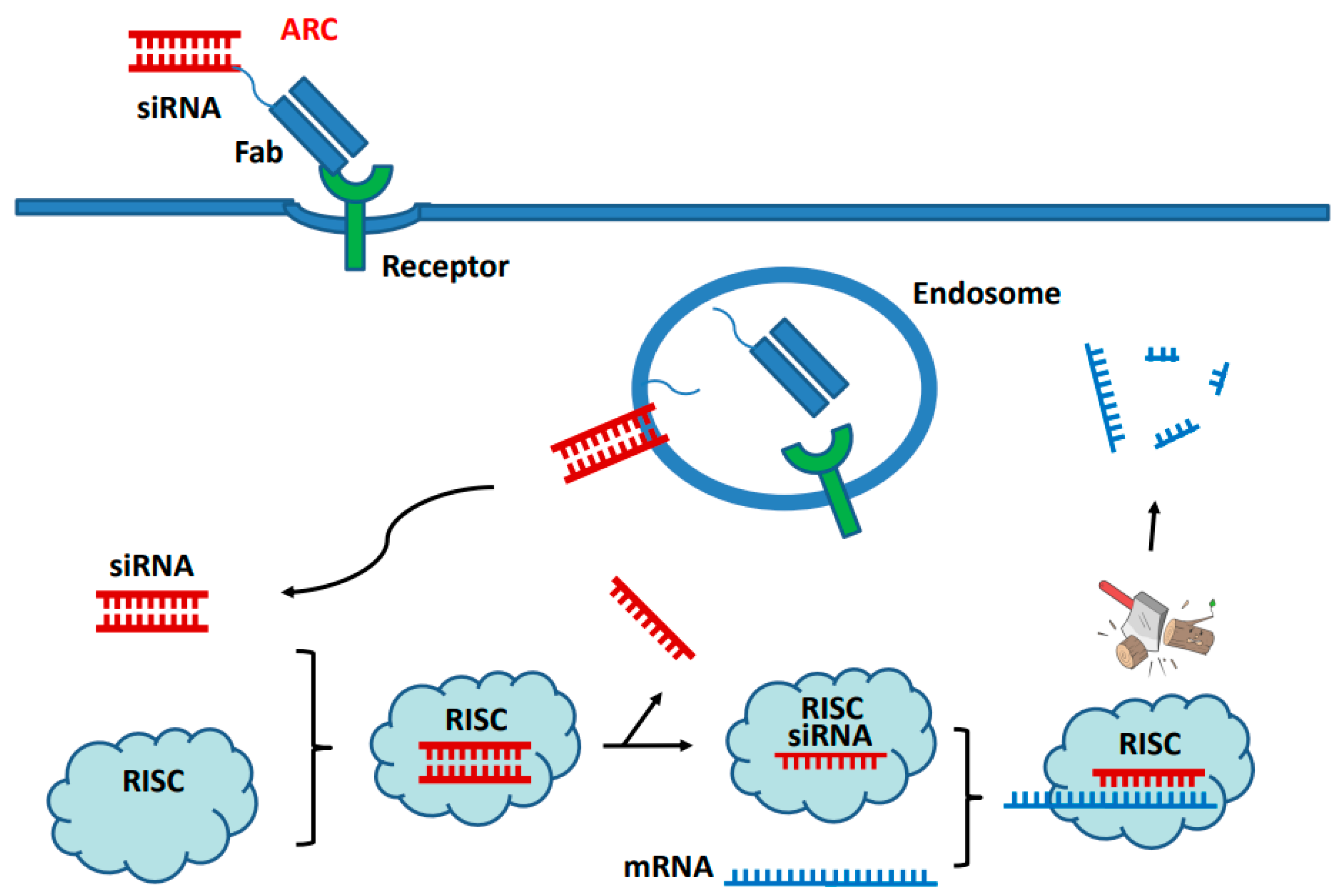
5. Nucleic Acid as Payloads
5.1. Non-Covalent Complex of siRNA with Peptide or Polymer
5.2. Direct Conjugation of siRNA
5.3. Other Nucleic Acid
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Lucas, A.T.; Moody, A.; Schorzman, A.N.; Zamboni, W.C. Importance and Considerations of Antibody Engineering in Antibody-Drug Conjugates Development from a Clinical Pharmacologist’s Perspective. Antibodies 2021, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.T.W.; Harris, P.W.R.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef] [PubMed]
- Dean, A.Q.; Luo, S.; Twomey, J.D.; Zhang, B. Targeting cancer with antibody-drug conjugates: Promises and challenges. mAbs 2021, 13, 1951427. [Google Scholar] [CrossRef]
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduct. Target. Ther. 2022, 7, 39. [Google Scholar] [CrossRef]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody-drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Zhou, Q. Site-Specific Antibody Conjugation for ADC and Beyond. Biomedicines 2017, 5, 64. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Kim, J. Advances in the Development of Site-Specific Antibody-Drug Conjugation. Anti-Cancer Agents Med. Chem. 2015, 15, 828–836. [Google Scholar] [CrossRef]
- Hussain, A.F.; Grimm, A.; Sheng, W.; Zhang, C.; Al-Rawe, M.; Bräutigam, K.; Abu Mraheil, M. Toward Homogenous Antibody Drug Conjugates Using Enzyme-Based Conjugation Approaches. Pharmaceuticals 2021, 14, 343. [Google Scholar] [CrossRef]
- Walsh, S.J.; Bargh, J.D.; Dannheim, F.M.; Hanby, A.R.; Seki, H.; Counsell, A.J.; Ou, X.; Fowler, E.; Ashman, N.; Takada, Y.; et al. Site-selective modification strategies in antibody-drug conjugates. Chem. Soc. Rev. 2021, 50, 1305–1353. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Deweid, L.; Avrutina, O.; Kolmar, H. Recent progress in transglutaminase-mediated assembly of antibody-drug conjugates. Anal. Biochem. 2020, 595, 113615. [Google Scholar] [CrossRef] [PubMed]
- Krüger, T.; Dierks, T.; Sewald, N. Formylglycine-generating enzymes for site-specific bioconjugation. Biol. Chem. 2019, 400, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- Jeffrey, S.C.; Burke, P.J.; Lyon, R.P.; Meyer, D.W.; Sussman, D.; Anderson, M.; Hunter, J.H.; Leiske, C.I.; Miyamoto, J.B.; Nicholas, N.D.; et al. A potent anti-CD70 antibody-drug conjugate combining a dimeric pyrrolobenzodiazepine drug with site-specific conjugation technology. Bioconj. Chem. 2013, 24, 1256–1263. [Google Scholar] [CrossRef]
- Dimasi, N.; Fleming, R.; Zhong, H.; Bezabeh, B.; Kinneer, K.; Christie, R.J.; Fazenbaker, C.; Wu, H.; Gao, C. Efficient Preparation of Site-Specific Antibody-Drug Conjugates Using Cysteine Insertion. Mol. Pharm. 2017, 14, 1501–1516. [Google Scholar] [CrossRef]
- Bryant, P.; Pabst, M.; Badescu, G.; Bird, M.; McDowell, W.; Jamieson, E.; Swierkosz, J.; Jurlewicz, J.; Tommasi, R.; Henseleit, K.; et al. In Vitro and In Vivo Evaluation of Cysteine Rebridged Trastuzumab-MMAE Antibody Drug Conjugates with Defined Drug-to-Antibody Ratios. Mol. Pharm. 2015, 12, 1872–1879. [Google Scholar] [CrossRef]
- Behrens, C.R.; Ha, E.H.; Chinn, L.L.; Bowers, S.; Probst, G.; Fitch-Bruhns, M.; Monteon, J.; Valdiosera, A.; Bermudez, A.; Liao-Chan, S.; et al. Antibody-Drug Conjugates (ADCs) Derived from Interchain Cysteine Cross-Linking Demonstrate Improved Homogeneity and Other Pharmacological Properties over Conventional Heterogeneous ADCs. Mol. Pharm. 2015, 12, 3986–3998. [Google Scholar] [CrossRef]
- Dennler, P.; Chiotellis, A.; Fischer, E.; Bregeon, D.; Belmant, C.; Gauthier, L.; Lhospice, F.; Romagne, F.; Schibli, R. Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody-drug conjugates. Bioconj. Chem. 2014, 25, 569–578. [Google Scholar] [CrossRef]
- Frutos, S.; Hernández, J.L.; Otero, A.; Calvis, C.; Adan, J.; Mitjans, F.; Vila-Perelló, M. Site-Specific Antibody Drug Conjugates Using Streamlined Expressed Protein Ligation. Bioconj. Chem. 2018, 29, 3503–3508. [Google Scholar] [CrossRef]
- Yamazoe, S.; Kotapati, S.; Hogan, J.M.; West, S.M.; Deng, X.A.; Diong, S.J.; Arbanas, J.; Nguyen, T.A.; Jashnani, A.; Gupta, D.; et al. Impact of Drug Conjugation on Thermal and Metabolic Stabilities of Aglycosylated and N-Glycosylated Antibodies. Bioconj. Chem. 2022, 33, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Kyazike, J.; Boudanova, E.; Drzyzga, M.; Honey, D.; Cost, R.; Hou, L.; Duffieux, F.; Brun, M.P.; Park, A.; et al. Site-Specific Antibody Conjugation to Engineered Double Cysteine Residues. Pharmaceuticals 2021, 14, 672. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, S.R.; Jackson, C.P.; Fang, S.; Carlson, D.P.; Guo, Z.; Tumey, L.N. Thiolation of Q295: Site-Specific Conjugation of Hydrophobic Payloads without the Need for Genetic Engineering. Mol. Pharm. 2019, 16, 2795–2807. [Google Scholar] [CrossRef] [PubMed]
- Axup, J.Y.; Bajjuri, K.M.; Ritland, M.; Hutchins, B.M.; Kim, C.H.; Kazane, S.A.; Halder, R.; Forsyth, J.S.; Santidrian, A.F.; Stafin, K.; et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. USA 2012, 109, 16101–16106. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.; Lu, Y.; Manibusan, A.; Sellers, A.; Tran, H.; Sun, Y.; Phuong, T.; Barnett, R.; Hehli, B.; Song, F.; et al. A general approach to site-specific antibody drug conjugates. Proc. Natl. Acad. Sci. USA 2014, 111, 1766–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanBrunt, M.P.; Shanebeck, K.; Caldwell, Z.; Johnson, J.; Thompson, P.; Martin, T.; Dong, H.; Li, G.; D’Hooge, F.; Masterson, L.; et al. Genetically Encoded Azide Containing Amino Acid in Mammalian Cells Enables Site-Specific Antibody-Drug Conjugates Using Click Cycloaddition Chemistry. Bioconj. Chem. 2015, 26, 2249–2260. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, E.S.; Heibeck, T.H.; Gill, A.; Li, X.; Murray, C.J.; Madlansacay, M.R.; Tian, C.; Uter, N.T.; Yin, G.; Rivers, P.J.; et al. Production of site-specific antibody-drug conjugates using optimized non-natural amino acids in a cell-free expression system. Bioconj. Chem. 2014, 25, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Nelson, C.G.; Nair, R.R.; Hazlehurst, L.; Moroni, T.; Martinez-Acedo, P.; Nanna, A.R.; Hymel, D.; Burke, T.R., Jr.; Rader, C.; et al. Stable and Potent Selenomab-Drug Conjugates. Cell Chem. Biol. 2017, 24, 433–442.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okeley, N.M.; Toki, B.E.; Zhang, X.; Jeffrey, S.C.; Burke, P.J.; Alley, S.C.; Senter, P.D. Metabolic Engineering of Monoclonal Antibody Carbohydrates for Antibody-Drug Conjugation. Bioconj. Chem. 2013, 24, 1650–1655. [Google Scholar] [CrossRef]
- Zhou, Q.; Stefano, J.E.; Manning, C.; Kyazike, J.; Chen, B.; Gianolio, D.A.; Park, A.; Busch, M.; Bird, J.; Zheng, X.; et al. Site-specific antibody-drug conjugation through glycoengineering. Bioconj. Chem. 2014, 25, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, T.; Boons, G.J. Preparation of Well-Defined Antibody-Drug Conjugates through Glycan Remodeling and Strain-Promoted Azide-Alkyne Cycloadditions. Angew. Chem. Int. Ed. 2014, 53, 7179–7182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Geel, R.; Wijdeven, M.A.; Heesbeen, R.; Verkade, J.M.; Wasiel, A.A.; van Berkel, S.S.; van Delft, F.L. Chemoenzymatic Conjugation of Toxic Payloads to the Globally Conserved N-Glycan of Native mAbs Provides Homogeneous and Highly Efficacious Antibody-Drug Conjugates. Bioconj. Chem. 2015, 26, 2233–2242. [Google Scholar] [CrossRef]
- Zhu, Z.; Ramakrishnan, B.; Li, J.; Wang, Y.; Feng, Y.; Prabakaran, P.; Colantonio, S.; Dyba, M.A.; Qasba, P.K.; Dimitrov, D.S. Site-specific antibody-drug conjugation through an engineered glycotransferase and a chemically reactive sugar. mAbs 2014, 6, 1190–1200. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Wang, L.X.; Huang, W. Chemoenzymatic synthesis of glycoengineered IgG antibodies and glycosite-specific antibody-drug conjugates. Nat. Protoc. 2017, 12, 1702–1721. [Google Scholar] [CrossRef]
- Wijdeven, M.A.; van Geel, R.; Hoogenboom, J.H.; Verkade, J.M.M.; Janssen, B.M.G.; Hurkmans, I.; de Bever, L.; van Berkel, S.S.; van Delft, F.L. Enzymatic glycan remodeling-metal free click (GlycoConnect™) provides homogenous antibody-drug conjugates with improved stability and therapeutic index without sequence engineering. mAbs 2022, 14, 2078466. [Google Scholar] [CrossRef]
- Strop, P.; Liu, S.H.; Dorywalska, M.; Delaria, K.; Dushin, R.G.; Tran, T.T.; Ho, W.H.; Farias, S.; Casas, M.G.; Abdiche, Y.; et al. Location matters: Site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem. Biol. 2013, 20, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Beerli, R.R.; Hell, T.; Merkel, A.S.; Grawunder, U. Sortase Enzyme-Mediated Generation of Site-Specifically Conjugated Antibody Drug Conjugates with High In Vitro and In Vivo Potency. PLoS ONE 2015, 10, e0131177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Shui, W.; Carlson, B.L.; Hu, N.; Rabuka, D.; Lee, J.; Bertozzi, C.R. Site-specific chemical modification of recombinant proteins produced in mammalian cells by using the genetically encoded aldehyde tag. Proc. Natl. Acad. Sci. USA 2009, 106, 3000–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabuka, D.; Rush, J.S.; deHart, G.W.; Wu, P.; Bertozzi, C.R. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat. Protoc. 2012, 7, 1052–1067. [Google Scholar] [CrossRef] [Green Version]
- Drake, P.M.; Albers, A.E.; Baker, J.; Banas, S.; Barfield, R.M.; Bhat, A.S.; de Hart, G.W.; Garofalo, A.W.; Holder, P.; Jones, L.C.; et al. Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconj. Chem. 2014, 25, 1331–1341. [Google Scholar] [CrossRef]
- Stevens, A.J.; Brown, Z.Z.; Shah, N.H.; Sekar, G.; Cowburn, D.; Muir, T.W. Design of a Split Intein with Exceptional Protein Splicing Activity. J. Am. Chem. Soc. 2016, 138, 2162–2165. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Lee, Y.; Ko, B.J.; Yoo, T.H. Peptide-Directed Photo-Cross-Linking for Site-Specific Conjugation of IgG. Bioconj. Chem. 2018, 29, 3240–3244. [Google Scholar] [CrossRef]
- Cheng-Sánchez, I.; Moya-Utrera, F.; Porras-Alcalá, C.; López-Romero, J.M.; Sarabia, F. Antibody-Drug Conjugates Containing Payloads from Marine Origin. Mar. Drugs 2022, 20, 494. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Edalatian Zakeri, S.; Bahal, R.; Wiemer, A.J. New Technologies Bloom Together for Bettering Cancer Drug Conjugates. Pharmacol. Rev. 2022, 74, 680–711. [Google Scholar] [CrossRef]
- Junutula, J.R.; Bhakta, S.; Raab, H.; Ervin, K.E.; Eigenbrot, C.; Vandlen, R.; Scheller, R.H. Rapid identification of reactive cysteine residues for site-specific labeling of antibody-Fabs. J. Immunol. Methods 2008, 332, 41–52. [Google Scholar] [CrossRef]
- Stimmel, J.B.; Merrill, B.M.; Kuyper, L.F.; Moxham, C.P.; Hutchins, J.T.; Fling, M.E.; Kull, F.C., Jr. Site-specific conjugation on serine right-arrow cysteine variant monoclonal antibodies. J. Biol. Chem. 2000, 275, 30445–30450. [Google Scholar] [CrossRef] [Green Version]
- Voynov, V.; Chennamsetty, N.; Kayser, V.; Wallny, H.J.; Helk, B.; Trout, B.L. Design and application of antibody cysteine variants. Bioconj. Chem. 2010, 21, 385–392. [Google Scholar] [CrossRef]
- Tumey, L.N.; Li, F.; Rago, B.; Han, X.; Loganzo, F.; Musto, S.; Graziani, E.I.; Puthenvant, J.; Casavant, J.; Marquette, K.; et al. Site Selection: A Case Study in the Identification of Optimal Cysteine Engineered Antibody Drug Conjugates. AAPS J. 2017, 19, 1123–1135. [Google Scholar] [CrossRef]
- Shinmi, D.; Taguchi, E.; Iwano, J.; Yamaguchi, T.; Masuda, K.; Enokizono, J.; Shiraishi, Y. One-Step Conjugation Method for Site-Specific Antibody-Drug Conjugates through Reactive Cysteine-Engineered Antibodies. Bioconj. Chem. 2016, 27, 1324–1331. [Google Scholar] [CrossRef]
- Dragovich, P.S.; Adhikari, P.; Blake, R.A.; Blaquiere, N.; Chen, J.; Cheng, Y.X.; den Besten, W.; Han, J.; Hartman, S.J.; He, J.; et al. Antibody-mediated delivery of chimeric protein degraders which target estrogen receptor alpha (ERα). Bioorganic Med. Chem. Lett. 2020, 30, 126907. [Google Scholar] [CrossRef]
- Dragovich, P.S.; Pillow, T.H.; Blake, R.A.; Sadowsky, J.D.; Adaligil, E.; Adhikari, P.; Bhakta, S.; Blaquiere, N.; Chen, J.; Dela Cruz-Chuh, J.; et al. Antibody-Mediated Delivery of Chimeric BRD4 Degraders. Part 1: Exploration of Antibody Linker, Payload Loading, and Payload Molecular Properties. J. Med. Chem. 2021, 64, 2534–2575. [Google Scholar] [CrossRef]
- Dragovich, P.S.; Pillow, T.H.; Blake, R.A.; Sadowsky, J.D.; Adaligil, E.; Adhikari, P.; Chen, J.; Corr, N.; Dela Cruz-Chuh, J.; Del Rosario, G.; et al. Antibody-Mediated Delivery of Chimeric BRD4 Degraders. Part 2: Improvement of In Vitro Antiproliferation Activity and In Vivo Antitumor Efficacy. J. Med. Chem. 2021, 64, 2576–2607. [Google Scholar] [CrossRef]
- Dragovich, P.S. Degrader-antibody conjugates. Chem. Soc. Rev. 2022, 51, 3886–3897. [Google Scholar] [CrossRef]
- Zhang, X.; Ou, C.; Liu, H.; Prabhu, S.K.; Li, C.; Yang, Q.; Wang, L.X. General and Robust Chemoenzymatic Method for Glycan-Mediated Site-Specific Labeling and Conjugation of Antibodies: Facile Synthesis of Homogeneous Antibody-Drug Conjugates. ACS Chem. Biol. 2021, 16, 2502–2514. [Google Scholar] [CrossRef]
- Zhang, X.; Ou, C.; Liu, H.; Wang, L.X. Synthesis and Evaluation of Three Azide-Modified Disaccharide Oxazolines as Enzyme Substrates for Single-Step Fc Glycan-Mediated Antibody-Drug Conjugation. Bioconj. Chem. 2022, 33, 1179–1191. [Google Scholar] [CrossRef]
- Shi, W.; Li, W.; Zhang, J.; Li, T.; Song, Y.; Zeng, Y.; Dong, Q.; Lin, Z.; Gong, L.; Fan, S.; et al. One-step synthesis of site-specific antibody-drug conjugates by reprograming IgG glycoengineering with LacNAc-based substrates. Acta Pharm. Sin. B 2022, 12, 2417–2428. [Google Scholar] [CrossRef]
- Jevševar, S.; Kusterle, M.; Kenig, M. PEGylation of antibody fragments for half-life extension. Methods Mol. Biol. 2012, 901, 233–246. [Google Scholar] [CrossRef]
- Humphreys, D.P.; Heywood, S.P.; Henry, A.; Ait-Lhadj, L.; Antoniw, P.; Palframan, R.; Greenslade, K.J.; Carrington, B.; Reeks, D.G.; Bowering, L.C.; et al. Alternative antibody Fab’ fragment PEGylation strategies: Combination of strong reducing agents, disruption of the interchain disulphide bond and disulphide engineering. Protein Eng. Des. Sel. PEDS 2007, 20, 227–234. [Google Scholar] [CrossRef]
- Pepinsky, R.B.; Walus, L.; Shao, Z.; Ji, B.; Gu, S.; Sun, Y.; Wen, D.; Lee, X.; Wang, Q.; Garber, E.; et al. Production of a PEGylated Fab’ of the anti-LINGO-1 Li33 antibody and assessment of its biochemical and functional properties in vitro and in a rat model of remyelination. Bioconj. Chem. 2011, 22, 200–210. [Google Scholar] [CrossRef]
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Morisaki, J.H.; et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef]
- Zhou, C.; Lehar, S.; Gutierrez, J.; Rosenberger, C.M.; Ljumanovic, N.; Dinoso, J.; Koppada, N.; Hong, K.; Baruch, A.; Carrasco-Triguero, M.; et al. Pharmacokinetics and pharmacodynamics of DSTA4637A: A novel THIOMAB antibody antibiotic conjugate against Staphylococcus aureus in mice. mAbs 2016, 8, 1612–1619. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Pearson, A.D.; Lim, R.K.; Rodgers, D.T.; Li, S.; Parker, H.B.; Weglarz, M.; Hampton, E.N.; Bollong, M.J.; Shen, J.; et al. Targeted Delivery of an Anti-inflammatory PDE4 Inhibitor to Immune Cells via an Antibody-drug Conjugate. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 2078–2089. [Google Scholar] [CrossRef] [Green Version]
- Lim, R.K.; Yu, S.; Cheng, B.; Li, S.; Kim, N.J.; Cao, Y.; Chi, V.; Kim, J.Y.; Chatterjee, A.K.; Schultz, P.G.; et al. Targeted Delivery of LXR Agonist Using a Site-Specific Antibody-Drug Conjugate. Bioconj. Chem. 2015, 26, 2216–2222. [Google Scholar] [CrossRef] [Green Version]
- Pishesha, N.; Harmand, T.; Carpenet, C.; Liu, X.; Bhan, A.; Islam, A.; van den Doel, R.; Pinney, W., 3rd; Ploegh, H.L. Targeted delivery of an anti-inflammatory corticosteroid to Ly6C/G-positive cells abates severity of influenza A symptoms. Proc. Natl. Acad. Sci. USA 2022, 119, e2211065119. [Google Scholar] [CrossRef]
- Maneiro, M.A.; Forte, N.; Shchepinova, M.M.; Kounde, C.S.; Chudasama, V.; Baker, J.R.; Tate, E.W. Antibody-PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chem. Biol. 2020, 15, 1306–1312. [Google Scholar] [CrossRef]
- Pillow, T.H.; Adhikari, P.; Blake, R.A.; Chen, J.; Del Rosario, G.; Deshmukh, G.; Figueroa, I.; Gascoigne, K.E.; Kamath, A.V.; Kaufman, S.; et al. Antibody Conjugation of a Chimeric BET Degrader Enables in vivo Activity. ChemMedChem 2020, 15, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Ahn, G.; Banik, S.M.; Miller, C.L.; Riley, N.M.; Cochran, J.R.; Bertozzi, C.R. LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat. Chem. Biol. 2021, 17, 937–946. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, H.; He, J.; Ou, C.; Donahue, T.C.; Muthana, M.M.; Su, L.; Wang, L.X. Site-Specific Chemoenzymatic Conjugation of High-Affinity M6P Glycan Ligands to Antibodies for Targeted Protein Degradation. ACS Chem. Biol. 2022, 17, 3013–3023. [Google Scholar] [CrossRef]
- Kim, C.H.; Axup, J.Y.; Lawson, B.R.; Yun, H.; Tardif, V.; Choi, S.H.; Zhou, Q.; Dubrovska, A.; Biroc, S.L.; Marsden, R.; et al. Bispecific small molecule-antibody conjugate targeting prostate cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 17796–17801. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Thomas, J.D.; Burke, T.R., Jr.; Rader, C. Chemically programmed bispecific antibodies that recruit and activate T cells. J. Biol. Chem. 2012, 287, 28206–28214. [Google Scholar] [CrossRef]
- Chapman, A.P.; Antoniw, P.; Spitali, M.; West, S.; Stephens, S.; King, D.J. Therapeutic antibody fragments with prolonged in vivo half-lives. Nat. Biotechnol. 1999, 17, 780–783. [Google Scholar] [CrossRef]
- Kawamura, A.; Miura, S.; Murayama, T.; Iwata, A.; Zhang, B.; Nishikawa, H.; Tsuchiya, Y.; Matsuo, K.; Tsuji, E.; Saku, K.; et al. Increased expression of monocyte CD11a and intracellular adhesion molecule-1 in patients with initial atherosclerotic coronary stenosis. Circ. J. Off. J. Jpn. Circ. Society. 2004, 68, 6–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [Green Version]
- Negi, A.; Kesari, K.K.; Voisin-Chiret, A.S. Estrogen Receptor-α Targeting: PROTACs, SNIPERs, Peptide-PROTACs, Antibody Conjugated PROTACs and SNIPERs. Pharmaceutics 2022, 14, 2523. [Google Scholar] [CrossRef]
- Garber, K. The PROTAC gold rush. Nat. Biotechnol. 2022, 40, 12–16. [Google Scholar] [CrossRef]
- Dragovich, P.S.; Blake, R.A.; Chen, C.; Chen, J.; Chuh, J.; den Besten, W.; Fan, F.; Fourie, A.; Hartman, S.J.; He, C.; et al. Conjugation of Indoles to Antibodies through a Novel Self-Immolating Linker. Chemistry 2018, 24, 4830–4834. [Google Scholar] [CrossRef]
- Ahn, G.; Banik, S.M.; Bertozzi, C.R. Degradation from the outside in: Targeting extracellular and membrane proteins for degradation through the endolysosomal pathway. Cell Chem. Biol. 2021, 28, 1072–1080. [Google Scholar] [CrossRef]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]
- Zhou, Y.; Teng, P.; Montgomery, N.T.; Li, X.; Tang, W. Development of Triantennary N-Acetylgalactosamine Conjugates as Degraders for Extracellular Proteins. ACS Cent. Sci. 2021, 7, 499–506. [Google Scholar] [CrossRef]
- Agarwal, P.; Kudirka, R.; Albers, A.E.; Barfield, R.M.; de Hart, G.W.; Drake, P.M.; Jones, L.C.; Rabuka, D. Hydrazino-Pictet-Spengler ligation as a biocompatible method for the generation of stable protein conjugates. Bioconj. Chem. 2013, 24, 846–851. [Google Scholar] [CrossRef]
- Goswami, R.K.; Bajjuri, K.M.; Forsyth, J.S.; Das, S.; Hassenpflug, W.; Huang, Z.Z.; Lemer, R.A.; Felding-Habermann, B.; Sinha, S.C. Chemically programmed antibodies targeting multiple alpha(v) integrins and their effects on tumor-related functions in vitro. Bioconj. Chem. 2011, 22, 1535–1544. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Goswami, R.K.; Liu, C.; Sinha, S.C. Chemically Programmed Bispecific Antibody Targeting Legumain Protease and αvβ3 Integrin Mediates Strong Antitumor Effects. Mol. Pharm. 2015, 12, 2544–2550. [Google Scholar] [CrossRef]
- Rader, C.; Sinha, S.C.; Popkov, M.; Lerner, R.A.; Barbas, C.F., 3rd. Chemically programmed monoclonal antibodies for cancer therapy: Adaptor immunotherapy based on a covalent antibody catalyst. Proc. Natl. Acad. Sci. USA 2003, 100, 5396–5400. [Google Scholar] [CrossRef] [Green Version]
- Walseng, E.; Nelson, C.G.; Qi, J.; Nanna, A.R.; Roush, W.R.; Goswami, R.K.; Sinha, S.C.; Burke, T.R., Jr.; Rader, C. Chemically Programmed Bispecific Antibodies in Diabody Format. J. Biol. Chem. 2016, 291, 19661–19673. [Google Scholar] [CrossRef] [Green Version]
- Lacek, K.; Urbanowicz, R.A.; Troise, F.; De Lorenzo, C.; Severino, V.; Di Maro, A.; Tarr, A.W.; Ferrana, F.; Ploss, A.; Temperton, M.; et al. Dramatic potentiation of the antiviral activity of HIV antibodies by cholesterol conjugation. J. Biol. Chem. 2014, 289, 35015–35028. [Google Scholar] [CrossRef] [Green Version]
- Brennan, M.; Davison, P.F.; Paulus, H. Preparation of bispecific antibodies by chemical recombination of monoclonal immunoglobulin G1 fragments. Science 1985, 229, 81–83. [Google Scholar] [CrossRef]
- Xiao, H.; Woods, E.C.; Vukojicic, P.; Bertozzi, C.R. Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2016, 113, 10304–10309. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.A.; Stanczak, M.A.; Mantuano, N.R.; Xiao, H.; Pijnenborg, J.F.A.; Malaker, S.A.; Miller, C.L.; Weidenbacher, P.A.; Tanzo, J.T.; Ahn, G.; et al. Targeted glycan degradation potentiates the anticancer immune response in vivo. Nat. Chem. Biol. 2020, 16, 1376–1384. [Google Scholar] [CrossRef]
- Lu, Z.; Truex, N.L.; Melo, M.B.; Cheng, Y.; Li, N.; Irvine, D.J.; Pentelute, B.L. IgG-Engineered Protective Antigen for Cytosolic Delivery of Proteins into Cancer Cells. ACS Central Sci. 2021, 7, 365–378. [Google Scholar] [CrossRef]
- Kim, C.H.; Axup, J.Y.; Dubrovska, A.; Kazane, S.A.; Hutchins, B.A.; Wold, E.D.; Smider, V.V.; Schultz, P.G. Synthesis of bispecific antibodies using genetically encoded unnatural amino acids. J. Am. Chem. Soc. 2012, 134, 9918–9921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Zhou, Q.; Deshmukh, V.; Phull, H.; Ma, J.; Tardif, V.; Naik, R.R.; Bouvard, C.; Zhang, Y.; Choi, S.; et al. Targeting human C-type lectin-like molecule-1 (CLL1) with a bispecific antibody for immunotherapy of acute myeloid leukemia. Angew. Chem. Int. Ed. Engl. 2014, 53, 9841–9845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazane, S.A.; Axup, J.Y.; Kim, C.H.; Ciobanu, M.; Wold, E.D.; Barluenga, S.; Hutchins, B.A.; Schultz, P.G.; Winssinger, N.; Smider, V.V. Self-assembled antibody multimers through peptide nucleic acid conjugation. J. Am. Chem. Soc. 2013, 135, 340–346. [Google Scholar] [CrossRef] [Green Version]
- Yumura, K.; Akiba, H.; Nagatoishi, S.; Kusano-Arai, O.; Iwanari, H.; Hamakubo, T.; Tsumoto, K. Use of SpyTag/SpyCatcher to construct bispecific antibodies that target two epitopes of a single antigen. J. Biochem. 2017, 162, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Akiba, H.; Takayanagi, K.; Kusano-Arai, O.; Iwanari, H.; Hamakubo, T.; Tsumoto, K. Generation of biparatopic antibody through two-step targeting of fragment antibodies on antigen using SpyTag and SpyCatcher. Biotechnol. Rep. 2020, 25, e00418. [Google Scholar] [CrossRef]
- Touti, F.; Lautrette, G.; Johnson, K.D.; Delaney, J.C.; Wollacott, A.; Tissire, H.; Viswanathan, K.; Shriver, Z.; Mong, S.K.; Mijalis, A.j.; et al. Antibody-Bactericidal Macrocyclic Peptide Conjugates To Target Gram-Negative Bacteria. Chembiochem A Eur. J. Chem. Biol. 2018, 19, 2039–2044. [Google Scholar] [CrossRef] [PubMed]
- van Lith, S.A.M.; van den Brand, D.; Wallbrecher, R.; van Duijnhoven, S.M.J.; Brock, R.; Leenders, W.P.J. A Conjugate of an Anti-Epidermal Growth Factor Receptor (EGFR) VHH and a Cell-Penetrating Peptide Drives Receptor Internalization and Blocks EGFR Activation. Chembiochem A Eur. J. Chem. Biol. 2017, 18, 2390–2394. [Google Scholar] [CrossRef]
- Herce, H.D.; Schumacher, D.; Schneider, A.F.L.; Ludwig, A.K.; Mann, F.A.; Fillies, M.; Kasper, M.A.; Reinke, S.; Krause, E.; Leonhardt, H.; et al. Cell-permeable nanobodies for targeted immunolabelling and antigen manipulation in living cells. Nat. Chem. 2017, 9, 762–771. [Google Scholar] [CrossRef]
- Mao, H.; Hart, S.A.; Schink, A.; Pollok, B.A. Sortase-mediated protein ligation: A new method for protein engineering. J. Am. Chem. Soc. 2004, 126, 2670–2671. [Google Scholar] [CrossRef]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef]
- Keeble, A.H.; Turkki, P.; Stokes, S.; Khairil Anuar, I.N.A.; Rahikainen, R.; Hytönen, V.P.; Howarth, M. Approaching infinite affinity through engineering of peptide-protein interaction. Proc. Natl. Acad. Sci. USA 2019, 116, 26523–26533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, A.A. Treating Disease at the RNA Level with Oligonucleotides. N. Engl. J. Med. 2019, 380, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Tushir-Singh, J. Antibody-siRNA conjugates: Drugging the undruggable for anti-leukemic therapy. Expert Opin. Biol. Ther. 2017, 17, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Dugal-Tessier, J.; Thirumalairajan, S.; Jain, N. Antibody-Oligonucleotide Conjugates: A Twist to Antibody-Drug Conjugates. J. Clin. Med. 2021, 10, 838. [Google Scholar] [CrossRef] [PubMed]
- Benizri, S.; Gissot, A.; Martin, A.; Vialet, B.; Grinstaff, M.W.; Barthélémy, P. Bioconjugated Oligonucleotides: Recent Developments and Therapeutic Applications. Bioconj. Chem. 2019, 30, 366–383. [Google Scholar] [CrossRef]
- Song, E.; Zhu, P.; Lee, S.K.; Chowdhury, D.; Kussman, S.; Dykxhoorn, D.M.; Feng, Y.; Palliser, D.; Weiner, D.B.; Shankar, P.; et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol. 2005, 23, 709–717. [Google Scholar] [CrossRef]
- Kumar, P.; Ban, H.S.; Kim, S.S.; Wu, H.; Pearson, T.; Greiner, D.L.; Laouar, A.; Yao, J.; Haridas, V.; Habiro, K.; et al. T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell 2008, 134, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Peer, D.; Zhu, P.; Carman, C.V.; Lieberman, J.; Shimaoka, M. Selective gene silencing in activated leukocytes by targeting siRNAs to the integrin lymphocyte function-associated antigen-1. Proc. Natl. Acad. Sci. USA 2007, 104, 4095–4100. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Sun, J.F.; Wang, T.T.; Guo, X.H.; Wei, J.X.; Jia, L.T.; Yang, A.G. Targeted inhibition of hantavirus replication and intracranial pathogenesis by a chimeric protein-delivered siRNA. Antivir. Res. 2017, 147, 107–115. [Google Scholar] [CrossRef]
- Lu, H.; Wang, D.; Kazane, S.; Javahishvili, T.; Tian, F.; Song, F.; Sellers, A.; Barnett, B.; Schultz, P.G. Site-specific antibody-polymer conjugates for siRNA delivery. J. Am. Chem. Soc. 2013, 135, 13885–13891. [Google Scholar] [CrossRef]
- Su, Y.; Yu, L.; Liu, N.; Guo, Z.; Wang, G.; Zheng, J.; Wei, M.; Wang, H.; Yang, A.G.; Qin, W.; et al. PSMA specific single chain antibody-mediated targeted knockdown of Notch1 inhibits human prostate cancer cell proliferation and tumor growth. Cancer Lett. 2013, 338, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Cuellar, T.L.; Barnes, D.; Nelson, C.; Tanguay, J.; Yu, S.F.; Wen, X.; Scales, S.J.; Gesch, J.; Davis, D.; van Brabant Smith, A.; et al. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB-siRNA conjugates. Nucleic Acids Res. 2015, 43, 1189–1203. [Google Scholar] [CrossRef] [PubMed]
- Sugo, T.; Terada, M.; Oikawa, T.; Miyata, K.; Nishimura, S.; Kenjo, E.; Ogasawara-Shimizu, M.; Makita, Y.; Imaichi, S.; Murata, S.; et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release Off. J. Control. Release Soc. 2016, 237, 1–13. [Google Scholar] [CrossRef]
- Zavoiura, O.; Brunner, B.; Casteels, P.; Zimmermann, L.; Ozog, M.; Boutton, C.; Helms, M.W.; Wagenaar, T.; Adam, V.; Peterka, J.; et al. Nanobody-siRNA Conjugates for Targeted Delivery of siRNA to Cancer Cells. Mol. Pharm. 2021, 18, 1048–1060. [Google Scholar] [CrossRef] [PubMed]
- Nanna, A.R.; Kel’in, A.V.; Theile, C.; Pierson, J.M.; Voo, Z.X.; Garg, A.; Nair, J.K.; Maier, M.A.; Fitzgerald, K.; Rader, C. Generation and validation of structurally defined antibody-siRNA conjugates. Nucleic Acids Res. 2020, 48, 5281–5293. [Google Scholar] [CrossRef]
- Konč, J.; Brown, L.; Whiten, D.R.; Zuo, Y.; Ravn, P.; Klenerman, D.; Bernardes, G.J.L. A Platform for Site-Specific DNA-Antibody Bioconjugation by Using Benzoylacrylic-Labelled Oligonucleotides. Angew. Chem. Int. Ed. Engl. 2021, 60, 25905–25913. [Google Scholar] [CrossRef]
- Rosen, C.B.; Kodal, A.L.; Nielsen, J.S.; Schaffert, D.H.; Scavenius, C.; Okholm, A.H.; Voigt, N.V.; Enghild, J.J.; Kjems, J.; Tørring, T.; et al. Template-directed covalent conjugation of DNA to native antibodies, transferrin and other metal-binding proteins. Nat. Chem. 2014, 6, 804–809. [Google Scholar] [CrossRef]
- Kushnarova-Vakal, A.; Äärelä, A.; Huovinen, T.; Virta, P.; Lamminmäki, U. Site-Specific Linking of an Oligonucleotide to Mono- and Bivalent Recombinant Antibodies with SpyCatcher-SpyTag System for Immuno-PCR. ACS Omega 2020, 5, 24927–24934. [Google Scholar] [CrossRef]
- Zhang, K.; Hao, L.; Hurst, S.J.; Mirkin, C.A. Antibody-linked spherical nucleic acids for cellular targeting. J. Am. Chem. Soc. 2012, 134, 16488–16491. [Google Scholar] [CrossRef] [Green Version]
- Mullard, A. Antibody-oligonucleotide conjugates enter the clinic. Nat. Rev. Drug Discov. 2022, 21, 6–8. [Google Scholar] [CrossRef]
- Wiener, J.; Kokotek, D.; Rosowski, S.; Lickert, H.; Meier, M. Preparation of single- and double-oligonucleotide antibody conjugates and their application for protein analytics. Sci. Rep. 2020, 10, 1457. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Techniques | Conjugation Sites | Genetic Engineering | Metabolic Labeling | Chemo-Enzymatic Modification | Selective References |
|---|---|---|---|---|---|
| Specific amino acids | C (Cys), Q (Gln) | + | − | ± | [14,15,16,17,18,19,20,21,22,23] |
| Unnatural amino acids | pAcF, pAMF, Sec, etc. | + | + | ± | [24,25,26,27,28] |
| Glycans | Sialic acid, GalNAc, GlcNAc, Gal, Fuc, etc. | − | ± | + | [29,30,31,32,33,34,35] |
| Short peptide tags | LLQG, LCTPSR, etc. | + | − | + | [36,37,38,39,40,41,42] |
| Categories of Payloads | MOA of Payload | Antibody Formats | Specific Site Used | Conjugation Chemistry | References |
|---|---|---|---|---|---|
| PEG | Prolonged serum half-life | Fab | Engineered Cys in C-terminus | Thiol-mediated conjugation | [57,58,59] |
| Antibiotics | Inhibitor of bacterial RNA polymerase | mAb | LC V205C | THIOMABTM | [60,61] |
| Immune-modulating compounds | PDE4 inhibitor for immune suppression | mAb | Unnatural amino acid | Oxime chemistry | [62] |
| Liver LXR agonist for immune suppression | mAb | Unnatural amino acid | Oxime chemistry | [63] | |
| Agonist of glucocorticoid receptor | Nb | C-terminal LPETGG | Sortase A (SrtA) mediated transpeptidation | [64] | |
| Protein degraders | PROTAC-mediated ERa and BRD4 degradation | mAb | Engineered Cys | THIOMABTM | [50] |
| PROTAC-mediated BRD4 degradation | mAb | Hinge Cys | Click chemistry | [65] | |
| mAb | Engineered Cys | THIOMABTM | [51,52,66] | ||
| LYTAC-mediated degradation through ASGPR | mAb | FGly in C-terminus of HC, hinge, CH1 | Hydrazino-iso-Pictet–Spangler reaction and click chemistry | [67] | |
| LYTAC-mediated degradation through M6PR | mAb | N-glycans | Chemoenzymatic reaction | [68] | |
| Ligand for proteins overexpressed in cancer | Chemically programmed bispecific Fab as T-cell engager (Fab-synthetic ligands) | Fab | Unnatural amino acid at HC K138 | Oxime chemistry | [69] |
| Fab | C-terminal Sec in HC | SeH-maleimide chemistry | [70] |
| Categories of Payloads | MOA of Payload | Antibody Formats | Specific Site Used | Conjugation Chemistry | References |
|---|---|---|---|---|---|
| Proteins | Enzyme | mAb | C-terminal FGly in HC | Oxime or Hydrazino-iso-Pictet–Spangler reaction and click chemistry | [88,89] |
| Immunotoxin | mAb | M252 in Fc | Peptide-directed photo-cross-linking | [42] | |
| Cytosolic delivery through toxin | mAb | HC C-terminal LPSTGGK | Sortase A (SrtA)-mediated transpeptidation | [90] | |
| Antibody fragments | Bispecific Fab as T cell engager (Fab-Fab) | Fab | Unnatural amino acid in LC or HC | Click chemistry | [91,92,93] |
| Biparatopic scFv | scFv | C-terminus | SpyTag and SpyCatcher-mediated ligation | [94,95] | |
| Peptides or cyclic peptides | Antimicrobial macrocyclic peptide (ABCs) | mAb | HC C-terminal (GS)6-LPETGGG | Sortase A (SrtA)-mediated transpeptidation | [96] |
| Internalization through CPP | Nb | C-terminal LPETG | Sortase A (SrtA)-mediated transpeptidation | [97] | |
| Cytosolic delivery through cyclic CPP | Nb | C-terminus | Intein-mediated thioester (or expressed protein ligation) | [98] |
| Categories | Antibody Formats | Specific Site Used | Conjugation Chemistry | MOA of siRNA | References |
|---|---|---|---|---|---|
| Non-covalent complex with peptide or polymer | Fab, scFv | Protamine fused | Noncovalent complex | In vitro and in vivo gene knockdown | [106] |
| scFv | Conjugated with Cys oligo-9 arginine | Noncovalent complex | Antiviral in vivo | [107] | |
| scFv | Protamine fused | Noncovalent complex | In vitro gene knockdown | [108] | |
| scFv | Protamine fused | Noncovalent complex | Antiviral in vivo | [109] | |
| Ab, Fab | Conjugated unnatural amino acid with cationic copolymer | Noncovalent complex | In vitro gene knockdown | [110] | |
| scFv | Protamine fused | Noncovalent complex | Anticancer | [111] | |
| Direct conjugation | Ab | Cys | THIOMABTM | In vitro and in vivo gene knockdown | [112] |
| Fab | Cys | Thiol-mediated conjugation | In vivo gene knockdown in muscle | [113] | |
| Nb | Cys | Thiol-mediated conjugation | In vitro gene knockdown | [114] | |
| Ab | Engineered Lys | Lys-β-lactam | In vitro gene knockdown | [115] |
| Methods Targeting Different Sites | Technical Categories | Advantages | Disadvantages |
|---|---|---|---|
| Cys-mediated conjugation | Specific amino acids |
|
|
| Conjugation through Gln295 in deglycosylated Ab using mTG | Specific amino acids |
|
|
| Conjugation through introduced unnatural amino acid (pAcF) | Unnatural amino acid |
|
|
| Modification (GlcNAc) using transglycosidase | Glycan |
|
|
| Modification using co-expressed FGE (Cys in LCTPSR) | Short peptide tag |
|
|
| Conjugation using SrtA-mediated transpeptidation | Short peptide tag |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Q. Site-Specific Antibody Conjugation with Payloads beyond Cytotoxins. Molecules 2023, 28, 917. https://doi.org/10.3390/molecules28030917
Zhou Q. Site-Specific Antibody Conjugation with Payloads beyond Cytotoxins. Molecules. 2023; 28(3):917. https://doi.org/10.3390/molecules28030917
Chicago/Turabian StyleZhou, Qun. 2023. "Site-Specific Antibody Conjugation with Payloads beyond Cytotoxins" Molecules 28, no. 3: 917. https://doi.org/10.3390/molecules28030917
APA StyleZhou, Q. (2023). Site-Specific Antibody Conjugation with Payloads beyond Cytotoxins. Molecules, 28(3), 917. https://doi.org/10.3390/molecules28030917