






Proteomes 2026, 14(2), 16; https://doi.org/10.3390/proteomes14020016 - 7 Apr 2026
Abstract
►
Show Figures
Proteomics represents a fundamental layer for understanding the molecular complexity of solid tumors by quantifying protein abundance and capturing proteoforms and post-translational modifications undetected in genomics or transcriptomics analyses. As mass spectrometry-based technologies and public proteomics repositories have expanded, opportunities for large-scale data
[...] Read more.
Proteomics represents a fundamental layer for understanding the molecular complexity of solid tumors by quantifying protein abundance and capturing proteoforms and post-translational modifications undetected in genomics or transcriptomics analyses. As mass spectrometry-based technologies and public proteomics repositories have expanded, opportunities for large-scale data reuse have grown accordingly. Nevertheless, data availability has not been translated into straightforward reuse: differences in experimental design, acquisition strategies, quantification workflows and metadata quality still limit the reproducibility and cross-study comparability. In this review, proteomics data reuse is defined as the systematic reanalysis and integration of publicly available datasets to support precision oncology applications such as biomarker assessment and antibody–drug conjugate target prioritization. We discuss reuse as an end-to-end analytical process, focusing on data analysis workflows, harmonization strategies, and the impact of heterogeneous experimental and analytical choices on interoperability. The increased application of artificial intelligence in proteomics data integration and reuse is also addressed, highlighting its analytical potential while underscoring the risks of overinterpretation when biological context and data structure are not adequately considered. Using colorectal and prostate cancer as representative examples, we illustrate how proteomics data reuse can support biological discovery and translational research, while critically examining the factors that limit robustness and clinical relevance.
Full article

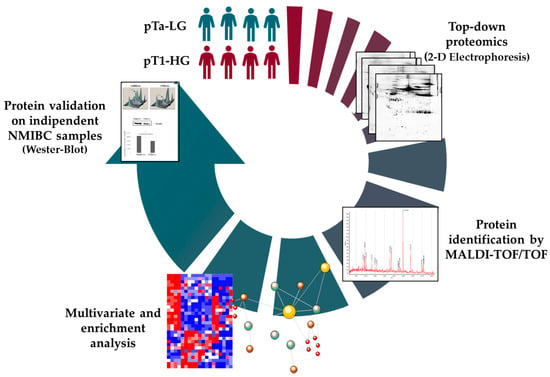
Figure 1





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}