
Targeting NF-κB Signaling in Cancer Stem Cells: A Narrative Review


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction into Scope of Review
2. Cancer and Cancer Stem Cells
2.1. Development of Cancer Stem Cells and Cancer Origin
2.2. Micro- and Macroenvironment of Cancers
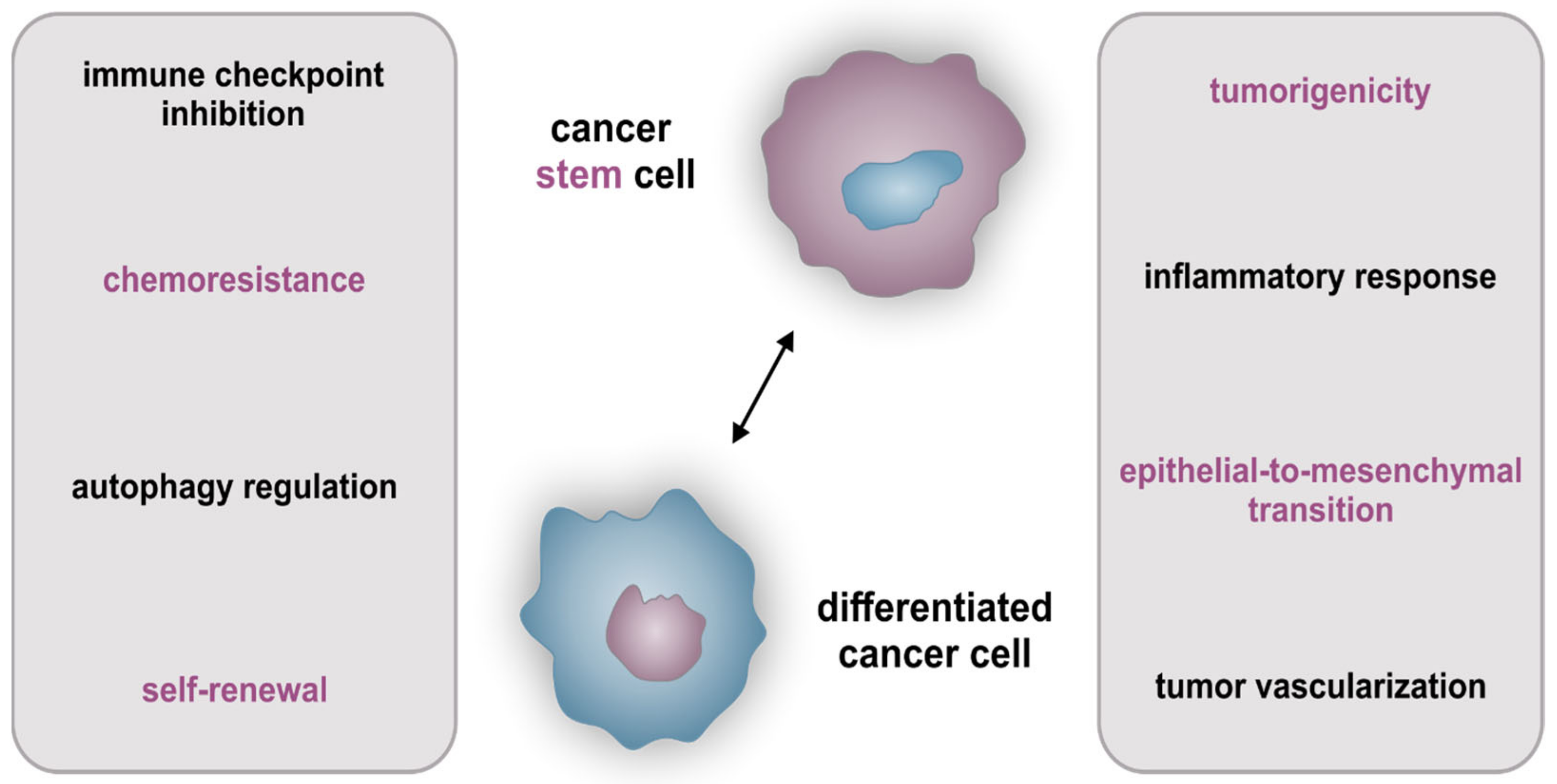
2.3. Cellular Hierarchy and Cancer Stem Cell-Specific Characteristics
2.4. Influence of Tumors’ Heterogeneity on Therapy of Cancer Stem Cells
3. The Transcription Factor NF-κB as a Target in Cancer Stem Cells
3.1. Introduction to Alternative and Classical NF-κB Signaling
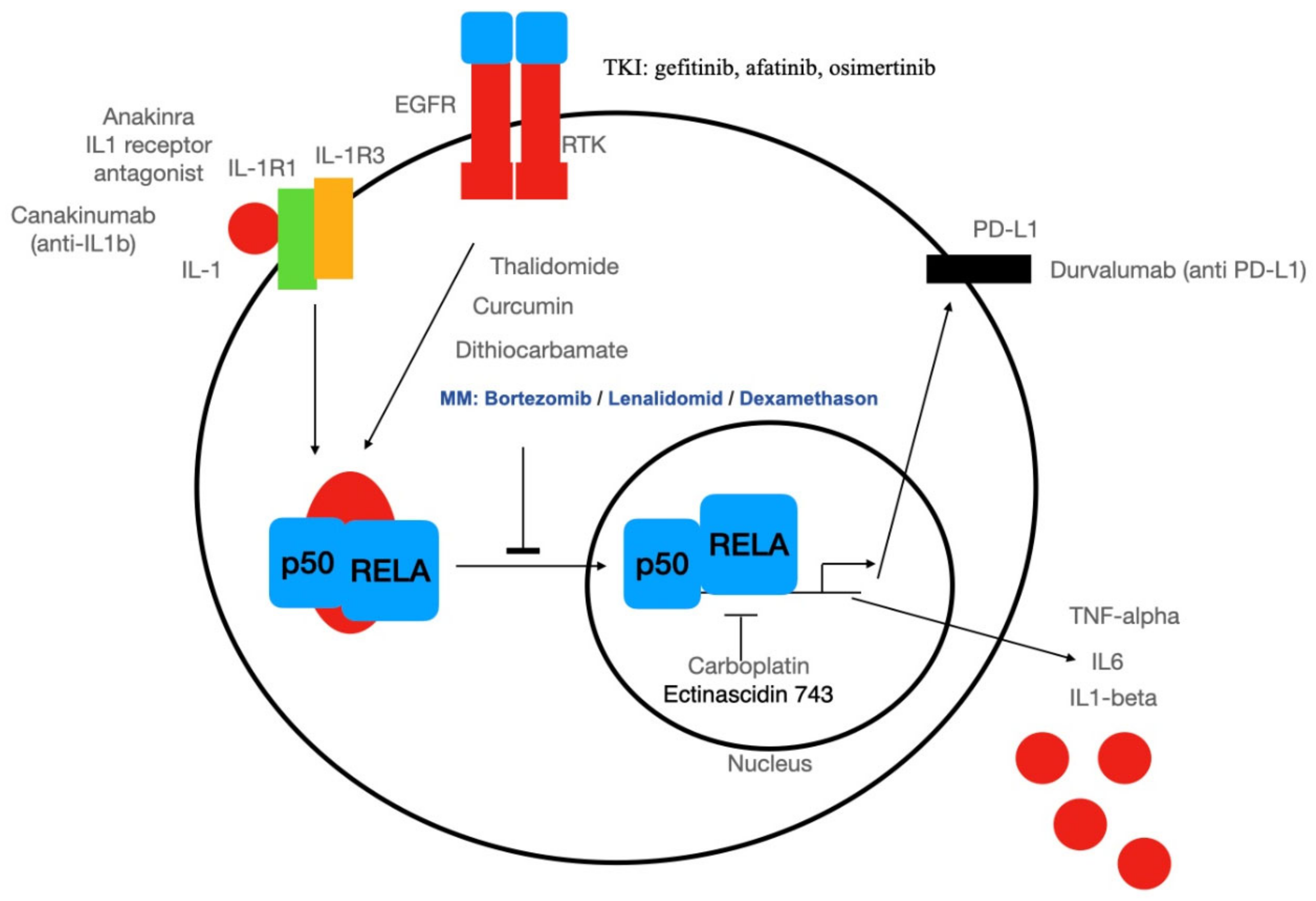
3.2. Systemic Therapy Targeting NF-κB
3.3. Targeted Therapy of Cancer Stem Cells: An Excursion
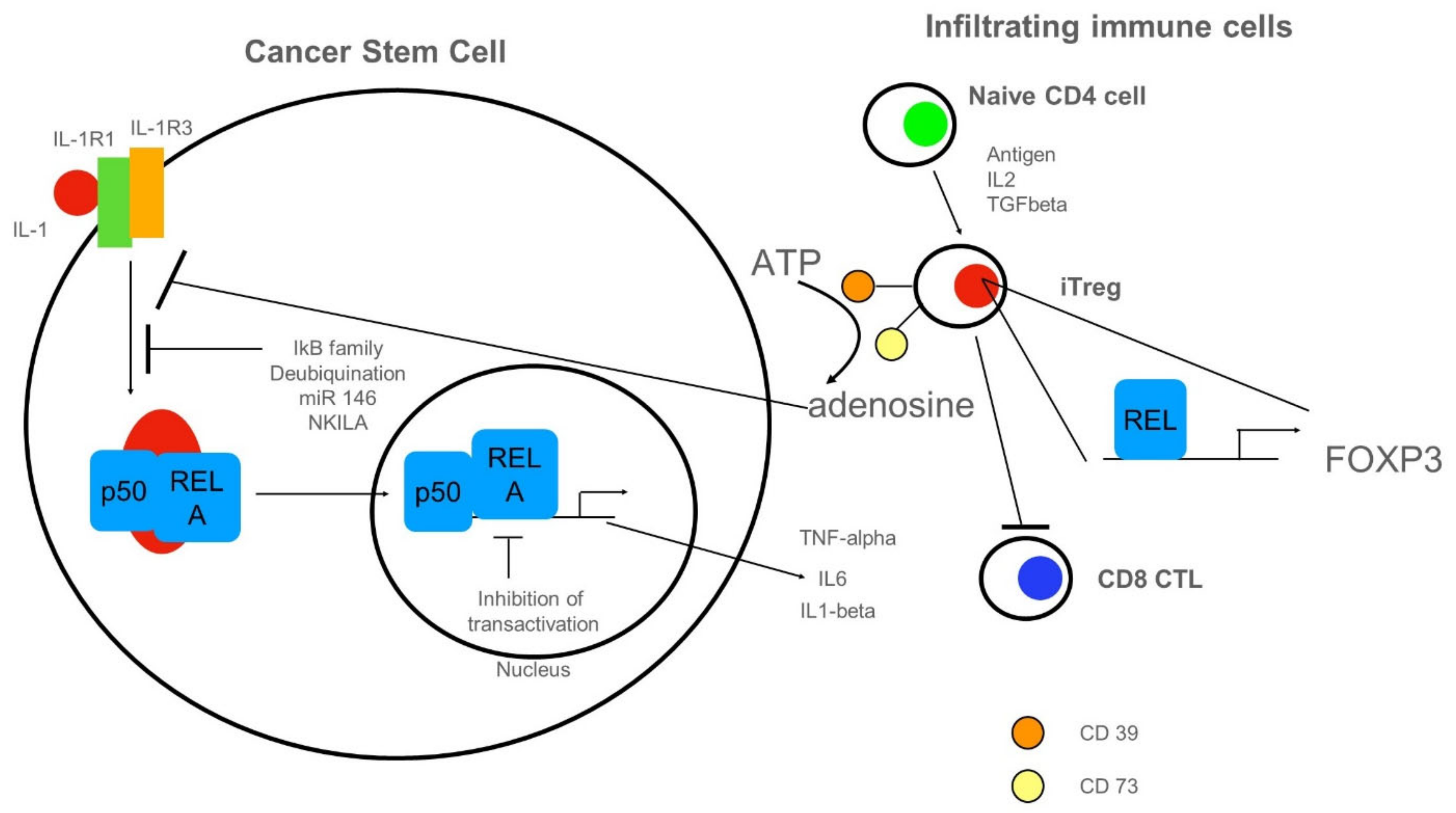
3.4. Anti-Inflammatory Cues Acting on NF-κB in Cancer Stem Cells
4. Conclusions: Lessons Learned for Cancer Stem Cell Therapy
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaltschmidt, C.; Greiner, J.; Kaltschmidt, B. The Transcription Factor NF-κB in Stem Cells and Development. Cells 2021, 10, 2042. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, C.; Banz-Jansen, C.; Benhidjeb, T.; Beshay, M.; Förster, C.; Greiner, J.; Hamelmann, E.; Jorch, N.; Mertzlufft, F.; Pfitzenmaier, J.; et al. A Role for NF-κB in Organ Specific Cancer and Cancer Stem Cells. Cancers 2019, 11, 655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Wyckoff, J.; Wang, W.; Lin, E.Y.; Wang, Y.; Pixley, F.; Stanley, E.R.; Graf, T.; Pollard, J.W.; Segall, J.; Condeelis, J. A Paracrine Loop between Tumor Cells and Macrophages Is Required for Tumor Cell Migration in Mammary Tumors. Cancer Res. 2004, 64, 7022–7029. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Al-Zhoughbi, W.; Huang, J.; Paramasivan, G.S.; Till, H.; Pichler, M.; Guertl-Lackner, B.; Hoefler, G. Tumor Macroenvironment and Metabolism. Semin. Oncol. 2014, 41, 281–295. [Google Scholar] [CrossRef] [Green Version]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.M.; Hiam, K.J.; Burnett, C.E.; Venida, A.; Debarge, R.; Tenvooren, I.; Marquez, D.M.; Cho, N.W.; Carmi, Y.; Spitzer, M.H. Systemic dysfunction and plasticity of the immune macroenvironment in cancer models. Nat. Med. 2020, 26, 1125–1134. [Google Scholar] [CrossRef]
- Vermeulen, L.; de Sousa e Melo, F.; Richel, D.J.; Medema, J.P. The developing cancer stem-cell model: Clinical challenges and opportunities. Lancet Oncol. 2012, 13, e83–e89. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheel, C.; Eaton, E.N.; Li, S.H.-J.; Chaffer, C.L.; Reinhardt, F.; Kah, K.-J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and Autocrine Signals Induce and Maintain Mesenchymal and Stem Cell States in the Breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreso, A.; Dick, J.E. Evolution of the Cancer Stem Cell Model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, M.D.; Burness, M.L.; Wicha, M.S. Therapeutic Implications of Cellular Heterogeneity and Plasticity in Breast Cancer. Cell Stem Cell 2015, 17, 260–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.-F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic Resistance of Tumorigenic Breast Cancer Cells to Chemotherapy. JNCI J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Tehranchi, R.; Woll, P.S.; Anderson, K.; Buza-Vidas, N.; Mizukami, T.; Mead, A.J.; Åstrand-Grundström, I.; Strömbeck, B.; Horvat, A.; Ferry, H.; et al. Persistent Malignant Stem Cells in del(5q) Myelodysplasia in Remission. N. Engl. J. Med. 2010, 363, 1025–1037. [Google Scholar] [CrossRef]
- Grosse-Gehling, P.; Fargeas, C.A.; Dittfeld, C.; Garbe, Y.; Alison, M.R.; Corbeil, D.; Kunz-Schughart, L. CD133 as a biomarker for putative cancer stem cells in solid tumours: Limitations, problems and challenges. J. Pathol. 2013, 229, 355–378. [Google Scholar] [CrossRef]
- Neradil, J.; Veselska, R. Nestin as a marker of cancer stem cells. Cancer Sci. 2015, 106, 803–811. [Google Scholar] [CrossRef]
- Walcher, L.; Kistenmacher, A.-K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.-R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells—Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- Windmöller, B.; Beshay, M.; Helweg, L.; Flottmann, C.; Beermann, M.; Förster, C.; Wilkens, L.; Greiner, J.; Kaltschmidt, C.; Kaltschmidt, B. Novel Primary Human Cancer Stem-Like Cell Populations from Non-Small Cell Lung Cancer: Inhibition of Cell Survival by Targeting NF-κB and MYC Signaling. Cells 2021, 10, 1024. [Google Scholar] [CrossRef] [PubMed]
- Schulte Am Esch, J.S.A.; Windmöller, B.A.; Hanewinkel, J.; Storm, J.; Förster, C.; Wilkens, L.; Krüger, M.; Kaltschmidt, B.; Kaltschmidt, C. Isolation and Characterization of Two Novel Colorectal Cancer Cell Lines, Containing a Subpopulation with Potential Stem-Like Properties: Treatment Options by MYC/NMYC Inhibition. Cancers 2020, 12, 2582. [Google Scholar] [CrossRef] [PubMed]
- Witte, K.E.; Pfitzenmaier, J.; Storm, J.; Lütkemeyer, M.; Wimmer, C.; Schulten, W.; Czaniera, N.; Geisler, M.; Förster, C.; Wilkens, L.; et al. Analysis of Several Pathways for Efficient Killing of Prostate Cancer Stem Cells: A Central Role of NF-κB RELA. Int. J. Mol. Sci. 2021, 22, 8901. [Google Scholar] [CrossRef] [PubMed]
- Witte, K.; Hertel, O.; Windmöller, B.; Helweg, L.; Höving, A.; Knabbe, C.; Busche, T.; Greiner, J.; Kalinowski, J.; Noll, T.; et al. Nanopore Sequencing Reveals Global Transcriptome Signatures of Mitochondrial and Ribosomal Gene Expressions in Various Human Cancer Stem-like Cell Populations. Cancers 2021, 13, 1136. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Su, Y.; Mei, Y.; Leng, Q.; Leng, B.; Liu, Z.; Stass, S.A.; Jiang, F. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Lab. Investig. 2010, 90, 234–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Eaton, E.N.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [Green Version]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [Green Version]
- Arrieta, O.; Montes-Servín, E.; Hernandez-Martinez, J.-M.; Cardona, A.F.; Casas-Ruiz, E.; Crispin, J.; Motola, D.; Flores-Estrada, D.; Barrera, L. Expression of PD-1/PD-L1 and PD-L2 in peripheral T-cells from non-small cell lung cancer patients. Oncotarget 2017, 8, 101994–102005. [Google Scholar] [CrossRef] [Green Version]
- Kaltschmidt, B.; Greiner, J.F.W.; Kadhim, H.M.; Kaltschmidt, C. Subunit-Specific Role of NF-κB in Cancer. Biomedices 2018, 6, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greten, F.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.-W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKβ Links Inflammation and Tumorigenesis in a Mouse Model of Colitis-Associated Cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an Active Player in Human Cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Coussy, F.; De Koning, L.; Lavigne, M.; Bernard, V.; Ouine, B.; Boulai, A.; EL Botty, R.; Dahmani, A.; Montaudon, E.; Assayag, F.; et al. A large collection of integrated genomically characterized patient-derived xenografts highlighting the heterogeneity of triple-negative breast cancer. Int. J. Cancer 2019, 145, 1902–1912. [Google Scholar] [CrossRef] [PubMed]
- Brady, L.; Kriner, M.; Coleman, I.; Morrissey, C.; Roudier, M.; True, L.D.; Gulati, R.; Plymate, S.R.; Zhou, Z.; Birditt, B.; et al. Inter- and intra-tumor heterogeneity of metastatic prostate cancer determined by digital spatial gene expression profiling. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Franceschi, S.; Civita, P.; Pasqualetti, F.; Lessi, F.; Modena, M.; Barachini, S.; Morelli, M.; Santonocito, O.; Vannozzi, R.; Pilkington, G.; et al. Multiregional Sequencing of IDH-WT Glioblastoma Reveals High Genetic Heterogeneity and a Dynamic Evolutionary History. Cancers 2021, 13, 2044. [Google Scholar] [CrossRef]
- Karin, M.; Shalapour, S. Regulation of antitumor immunity by inflammation-induced epigenetic alterations. Cell. Mol. Immunol. 2021, 1–8. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Inducibility of κ immunoglobulin enhancer-binding protein NF-κB by a posttranslational mechanism. Cell 1986, 47, 921–928. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Baltimore, D. IκB: A Specific Inhibitor of the NF-κB Transcription Factor. Science 1988, 242, 540–546. [Google Scholar] [CrossRef]
- Ghosh, G.; Van Duyne, G.; Ghosh, S.; Sigler, P.B. Structure of NF-κB p50 homodimer bound to a κB site. Nature 1995, 373, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.D.; Harrison, S.C. Structure of an IκBα/NF-κB Complex. Cell 1998, 95, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Henkel, T.; Zabel, U.; van Zee, K.; Müller, J.M.; Fanning, E.; Baeuerle, P.A. Intramolecular masking of the nuclear location signal and dimerization domain in the precursor for the p50 NF-κB subunit. Cell 1992, 68, 1121–1133. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [Green Version]
- Renner, F.; Schmitz, M.L. Autoregulatory feedback loops terminating the NF-κB response. Trends Biochem. Sci. 2009, 34, 128–135. [Google Scholar] [CrossRef]
- Stancovski, I.; Baltimore, D. NF-κB Activation: The IκB Kinase Revealed? Cell 1997, 91, 299–302. [Google Scholar] [CrossRef] [Green Version]
- Kaltschmidt, B.; Kaltschmidt, C. NF- B in the Nervous System. Cold Spring Harb. Perspect. Biol. 2009, 1, a001271. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, P. Address in Pathology on chemotherapeutics: Scientific principles, methods, and results. Lancet 1913, 182, 445–451. [Google Scholar] [CrossRef]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef] [PubMed]
- Blakely, C.M.; Pazarentzos, E.; Olivas, V.; Asthana, S.; Yan, J.J.; Tan, I.; Hrustanovic, G.; Chan, E.; Lin, L.; Neel, D.S.; et al. NF-κB-Activating Complex Engaged in Response to EGFR Oncogene Inhibition Drives Tumor Cell Survival and Residual Disease in Lung Cancer. Cell Rep. 2015, 11, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bredel, M.; Scholtens, D.M.; Yadav, A.; Alvarez, A.A.; Renfrow, J.J.; Chandler, J.P.; Yu, I.L.; Carro, M.S.; Dai, F.; Tagge, M.J.; et al. NFKBIADeletion in Glioblastomas. N. Engl. J. Med. 2011, 364, 627–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercurio, F.; Zhu, H.; Murray, B.W.; Shevchenko, A.; Bennett, B.L.; Li, J.W.; Young, D.B.; Barbosa, M.; Mann, M.; Manning, A.; et al. IKK-1 and IKK-2: Cytokine-Activated IκB Kinases Essential for NF-κB Activation. Science 1997, 278, 860–866. [Google Scholar] [CrossRef]
- Kopp, E.; Ghosh, S. Inhibition of NF-κB by Sodium Salicylate and Aspirin. Science 1994, 265, 956–959. [Google Scholar] [CrossRef]
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends—An Update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Ziegelbauer, K.; Gantner, F.; Lukacs, N.W.; Berlin, A.; Fuchikami, K.; Niki, T.; Sakai, K.; Inbe, H.; Takeshita, K.; Ishimori, M.; et al. A selective novel low-molecular-weight inhibitor of Iκ B kinase-β (IKK-β ) prevents pulmonary inflammation and shows broad anti-inflammatory activity. J. Cereb. Blood Flow Metab. 2005, 145, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Österlund, C.; Lilliehöök, B.; Ekstrand-Hammarström, B.; Sandstrom, T.; Bucht, A. The nitrogen mustard melphalan activates mitogen-activated phosphorylated kinases (MAPK), nuclear factor-κB and inflammatory response in lung epithelial cells. J. Appl. Toxicol. 2005, 25, 328–337. [Google Scholar] [CrossRef]
- Franks, M.E.; Macpherson, G.R.; Figg, W.D. Thalidomide. Lancet 2004, 363, 1802–1811. [Google Scholar] [CrossRef] [Green Version]
- Haslett, P.A.; Corral, L.G.; Albert, M.; Kaplan, G. Thalidomide Costimulates Primary Human T Lymphocytes, Preferentially Inducing Proliferation, Cytokine Production, and Cytotoxic Responses in the CD8+ Subset. J. Exp. Med. 1998, 187, 1885–1892. [Google Scholar] [CrossRef] [Green Version]
- Davies, F.; Raje, N.; Hideshima, T.; Lentzsch, S.; Young, G.; Tai, Y.-T.; Lin, B.; Podar, K.; Gupta, D.; Chauhan, D.; et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood 2001, 98, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Onder, T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of Selective Inhibitors of Cancer Stem Cells by High-Throughput Screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, M.; Patro, B.S. Salinomycin reduces growth, proliferation and metastasis of cisplatin resistant breast cancer cells via NF-kB deregulation. Toxicol. Vitr. 2019, 60, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Rai, G.; Suman, S.; Mishra, S.; Shukla, Y. Evaluation of growth inhibitory response of Resveratrol and Salinomycin combinations against triple negative breast cancer cells. Biomed. Pharmacother. 2017, 89, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, B.; Lee, H.-G.; Kwon, S.-H.; Cha, S.-D.; Shin, S.-J.; Lee, G.-H.; Bae, I.; Cho, C.-H. Salinomycin inhibits Akt/NF-κB and induces apoptosis in cisplatin resistant ovarian cancer cells. Cancer Epidemiol. 2013, 37, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Ketola, K.; Hilvo, M.; Hyötyläinen, T.; Vuoristo, A.; Ruskeepää, A.-L.; Orešič, M.; Kallioniemi, O.; Iljin, K. Salinomycin inhibits prostate cancer growth and migration via induction of oxidative stress. Br. J. Cancer 2012, 106, 99–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Wang, K.; Zheng, Y.; Zeng, X.; Lim, Y.C.; Liu, T. Co-delivery of Salinomycin and Curcumin for Cancer Stem Cell Treatment by Inhibition of Cell Proliferation, Cell Cycle Arrest, and Epithelial–Mesenchymal Transition. Front. Chem. 2021, 8, 601649. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.C.; Huang, R.; Sakamuru, S.; Shukla, S.J.; Attene-Ramos, M.S.; Shinn, P.; Van Leer, D.; Leister, W.; Austin, C.P.; Xia, M. Identification of known drugs that act as inhibitors of NF-κB signaling and their mechanism of action. Biochem. Pharmacol. 2010, 79, 1272–1280. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Cruzado, L.; Tornin, J.; Rodriguez, A.; Santos, L.; Allonca, E.; Fernandez-Garcia, M.T.; Astudillo, A.; Garcia-Pedrero, J.M.; Rodriguez, R. Trabectedin and Campthotecin Synergistically Eliminate Cancer Stem Cells in Cell-of-Origin Sarcoma Models. Neoplasia 2017, 19, 460–470. [Google Scholar] [CrossRef]
- Singh, S.; Bhat, M.K. Carboplatin induces apoptotic cell death through downregulation of constitutively active nuclear factor-κB in human HPV-18 E6-positive HEp-2 cells. Biochem. Biophys. Res. Commun. 2004, 318, 346–353. [Google Scholar] [CrossRef]
- Caldenhoven, E.; Liden, J.; Wissink, S.; Okret, S.; Van De Stolpe, A.; Raaijmakers, J.; Koenderman, L.; Gustafsson, J.Å.; Van Der Saag, P.T. Negative cross-talk between RelA and the glucocorticoid receptor: A possible mechanism for the antiinflammatory action of glucocorticoids. Mol. Endocrinol. 1995, 9, 401–412. [Google Scholar] [CrossRef] [Green Version]
- Auphan, N.; DiDonato, J.A.; Rosette, C.; Helmberg, A.; Karin, M. Immunosuppression by Glucocorticoids: Inhibition of NF-κB Activity Through Induction of IκB Synthesis. Science 1995, 270, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Kauffman, M. Development of the Proteasome Inhibitor Velcade™ (Bortezomib). Cancer Investig. 2004, 22, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Traenckner, E.; Pahl, H.; Henkel, T.; Schmidt, K.; Wilk, S.; Baeuerle, P. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J. 1995, 14, 2876–2883. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosialls, E.; El Hage, R.; Dos Santos, L.; Gong, C.; Mehrpour, M.; Hamaï, A. Ferroptosis: Cancer Stem Cells Rely on Iron until “to Die for” It. Cells 2021, 10, 2981. [Google Scholar] [CrossRef]
- Mai, T.T.; Hamaï, A.; Hienzsch, A.; Cañeque, T.; Müller, S.; Wicinski, J.; Cabaud, O.; Leroy, C.; David, A.; Acevedo, V.; et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 2017, 9, 1025–1033. [Google Scholar] [CrossRef] [Green Version]
- Labanieh, L.; Majzner, R.G.; Mackall, C.L. Programming CAR-T cells to kill cancer. Nat. Biomed. Eng. 2018, 2, 377–391. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Alhabbab, R.Y. Targeting Cancer Stem Cells by Genetically Engineered Chimeric Antigen Receptor T Cells. Front. Genet. 2020, 11, 312. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Prasad, S.; Gaedicke, S.; Hettich, M.; Firat, E.; Niedermann, G. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget 2014, 6, 171–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Z.; Wu, Y.; Ma, W.; Zhang, S.; Zhang, Y.-Q. Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol. 2015, 16, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Huang, Y.; Jiang, D.-Q.; Cui, L.-Z.; He, Z.; Wang, C.; Zhang, Z.-W.; Zhu, H.-L.; Ding, Y.-M.; Li, L.-F.; et al. Antitumor activity of EGFR-specific CAR T cells against non-small-cell lung cancer cells in vitro and in mice. Cell Death Dis. 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Haist, C.; Schulte, E.; Bartels, N.; Bister, A.; Poschinski, Z.; Ibach, T.C.; Geipel, K.; Wiek, C.; Wagenmann, M.; Monzel, C.; et al. CD44v6-targeted CAR T-cells specifically eliminate CD44 isoform 6 expressing head/neck squamous cell carcinoma cells. Oral Oncol. 2021, 116, 105259. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, S.T.; Israël, A. IκB proteins: Structure, function and regulation. Semin. Cancer Biol. 1997, 8, 75–82. [Google Scholar] [CrossRef]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [Green Version]
- Taganov, K.D.; Boldin, M.; Chang, K.-J.; Baltimore, D. NF- B-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Sun, L.; Liu, Q.; Gong, C.; Yao, Y.; Lv, X.; Lin, L.; Yao, H.; Su, F.; Li, D.; et al. A Cytoplasmic NF-κB Interacting Long Noncoding RNA Blocks IκB Phosphorylation and Suppresses Breast Cancer Metastasis. Cancer Cell 2015, 27, 370–381. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Wang, X.; Gu, J.; Lu, H.; Zhang, F.; Li, X.; Qian, X.; Wang, X.; Lu, L. iTreg induced from CD39+ naive T cells demonstrate enhanced proliferate and suppressive ability. Int. Immunopharmacol. 2015, 28, 925–930. [Google Scholar] [CrossRef]
- Di Gennaro, P.; Gerlini, G.; Caporale, R.; Sestini, S.; Brandani, P.; Urso, C.; Pimpinelli, N.; Borgognoni, L. T regulatory cells mediate immunosuppresion by adenosine in peripheral blood, sentinel lymph node and TILs from melanoma patients. Cancer Lett. 2018, 417, 124–130. [Google Scholar] [CrossRef]
- Grinberg-Bleyer, Y.; Oh, H.; Desrichard, A.; Bhatt, D.M.; Caron, R.; Chan, T.A.; Schmid, R.M.; Klein, U.; Hayden, M.; Ghosh, S. NF-κB c-Rel Is Crucial for the Regulatory T Cell Immune Checkpoint in Cancer. Cell 2017, 170, 1096–1108.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demidem, A.; Lam, T.; Alas, S.; Hariharan, K.; Hanna, N.; Bonavida, B. Chimeric Anti-CD20 (IDEC-C2B8) Monoclonal Antibody Sensitizes a B Cell Lymphoma Cell Line to Cell Killing by Cytotoxic Drugs. Cancer Biother. Radiopharm. 1997, 12, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B.; Appelbaum, F.R.; Estey, E.H.; Bernstein, I.D. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood 2012, 119, 6198–6208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Keefe, T.L.; Williams, G.T.; Davies, S.L.; Neuberger, M.S. Mice carrying a CD20 gene disruption. Immunogenetics 1998, 48, 125–132. [Google Scholar] [CrossRef]
- Baud, V.; Karin, M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Ruscitti, P.; Ursini, F.; Cipriani, P.; Greco, M.; Alvaro, S.; Vasiliki, L.; Di Benedetto, P.; Carubbi, F.; Berardicurti, O.; Gulletta, E.; et al. IL-1 inhibition improves insulin resistance and adipokines in rheumatoid arthritis patients with comorbid type 2 diabetes. Medicine 2019, 98, e14587. [Google Scholar] [CrossRef]
- Elaraj, D.M.; Weinreich, D.; Varghese, S.; Puhlmann, M.; Hewitt, S.; Carroll, N.M.; Feldman, E.D.; Turner, E.M.; Alexander, H.R. The role of interleukin 1 in growth and metastasis of human cancer xenografts. Clin. Cancer Res. 2006, 12, 1088–1096. [Google Scholar] [CrossRef] [Green Version]
- Tulotta, C.; Lefley, D.V.; Freeman, K.; Gregory, W.M.; Hanby, A.M.; Heath, P.R.; Nutter-Howard, F.; Wilkinson, J.M.; Spicer-Hadlington, A.R.; Liu, X.; et al. Endogenous Production of IL1B by Breast Cancer Cells Drives Metastasis and Colonization of the Bone Microenvironment. Clin. Cancer Res. 2019, 25, 2769–2782. [Google Scholar] [CrossRef]
- Antonangeli, F.; Natalini, A.; Garassino, M.C.; Sica, A.; Santoni, A.; Di Rosa, F. Regulation of PD-L1 Expression by NF-κB in Cancer. Front. Immunol. 2020, 11, 584626. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Gao, Z.; Li, X.; Dong, L.; Han, W.; Nie, J. Regulation of PD-1/PD-L1 pathway and resistance to PD-1/PD-L1 blockade. Oncotarget 2017, 8, 110693–110707. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaltschmidt, B.; Witte, K.E.; Greiner, J.F.W.; Weissinger, F.; Kaltschmidt, C. Targeting NF-κB Signaling in Cancer Stem Cells: A Narrative Review. Biomedicines 2022, 10, 261. https://doi.org/10.3390/biomedicines10020261
Kaltschmidt B, Witte KE, Greiner JFW, Weissinger F, Kaltschmidt C. Targeting NF-κB Signaling in Cancer Stem Cells: A Narrative Review. Biomedicines. 2022; 10(2):261. https://doi.org/10.3390/biomedicines10020261
Chicago/Turabian StyleKaltschmidt, Barbara, Kaya E. Witte, Johannes F. W. Greiner, Florian Weissinger, and Christian Kaltschmidt. 2022. "Targeting NF-κB Signaling in Cancer Stem Cells: A Narrative Review" Biomedicines 10, no. 2: 261. https://doi.org/10.3390/biomedicines10020261